

Abstract
Aflatoxins are polyketide-derived secondary metabolites produced by Aspergillus spp. The toxic effects of aflatoxins have adverse consequences for human health and agricultural economics. The aflR gene, a regulatory gene for aflatoxin biosynthesis, encodes a protein containing a zinc-finger DNA-binding motif. AFLR-Protein three-dimensional model was generated using Robetta server. The modeled AFLR-Protein was further optimization and validation using Rampage. In the simulations, we monitored the backbone atoms and the C-α-helix of the modeled protein. The low RMSD and the simulation time indicate that, as expected, the 3D structural model of AFLR-protein represents a stable folding conformation. This study paves the way for generating computer molecular models for proteins whose crystal structures are not available and which would aid in detailed molecular mechanism of inhibition of aflatoxin.
Keywords: aflR gene, aflatoxin, RMSD
Background
Aflatoxins are a group of mycotoxins with potent toxicity and carcinogenicity toward mammals. They are produced by some strains of Aspergillus flavus, Aspergillus parasiticus, Aspergillus nomius and Aspergillus tamarii and they can be found as contaminants in a wide variety of food and feed commodities. Aflatoxin contamination in agricultural products is a serious problem from the viewpoint of food safety, and also that of economical loss [1, 2]. However, it is difficult to resolve the problem because of the lack of an effective method to control aflatoxin production. The recent advances in cloning several aflatoxin pathway genes revealed that most of these genes are clustered within a 60-kb DNA region in A. parasiticus and A. flavus. [3, 4]. This finding has spawned renewed interest in the study of the regulation of aflatoxin biosynthesis. Previous studies have suggested that one of these genes, aflR, is involved in some aspect of the regulation of aflatoxin biosynthesis. In the present study, 3D-Structure of AFLR-Protein was performed using Robetta Server. The constructed model was further validated by Ramachandran plot. The refined model was again subjected to energy minimization using GROMOS and evaluated for quality assessment.
Methodology
Sequence retrieval alignment and Protein structure prediction:
3D-Structure of AFLR-Protein was performed using Robetta Server. The amino acid sequence of AFLR-Protein was retrieved from NCBI (accession number: AAW32195.1). The Robetta server is an automated protein structure prediction service for ab initio and comparative modeling [5]. Initially server searches for structural homologs using BLAST or PSI-BLAST, [6] and then it breaks down the target sequence into its individual domains, or independently folding units of proteins, by matching the sequence to structural families in the Pfam database. Domains with structural homologs then follow a template-based model (i.e., homology modeling) protocol. The final structure prediction is selected by taking the lowest energy model as determined by a low-resolution Rosetta energy function. The backbone conformation of the modeled structure was calculated by analyzing the phi (F) and psi (ψ) torsion angles using Rampage [7].
Molecular Dynamics Simulations:
The GROMACS 4.5.4 package [8] was used to run Molecular Dynamics simulations with the GROMOS96 43a1 force field and the SPC216 water model. The modeled AFLR-Protein was energy minimized to discard the high-energy intramolecular interactions. The overall geometry and atomic charges were optimized to avoid steric clashes. Then the whole system was gradually heated from 0 to 300 K over 500 ps using the NVT ensemble run with the Berendsen procedure and, subsequently in 500 ps NPT ensemble run at pressure of 1atm. The Parrinello- Rahman pressure coupling has been used. Finally, a 10 MD simulation was carried out to examine the changes and dynamic behaviour of the protein was analyzed by calculating the RMSD and energy.
Protein functional site:
Q-Site Finder [9] server was used to predict the structure based protein functional site.
Discussion
Sequence retrieval alignment and Protein structure prediction:
Robetta server is an automated protein structure prediction service for ab initio and comparative modeling [5]. The server searched for structural homolog using PSI-BLAST, and then it breaks down the target sequence into three individual domains.
Then the Domains with structural homologs follow a template model (3coqA). The final structure prediction was done using 3coqA as template model (Figure 1). Geometric evaluations of the modeled 3D structure were performed using Rampage [7] by calculating the Ramachandran plot (Figure 2). The plot (Figure 2) of our model shows that 98.2% of residues were found in the favored and 1.4% allowed regions and 0.5% were in the outlier region.
Figure 1.
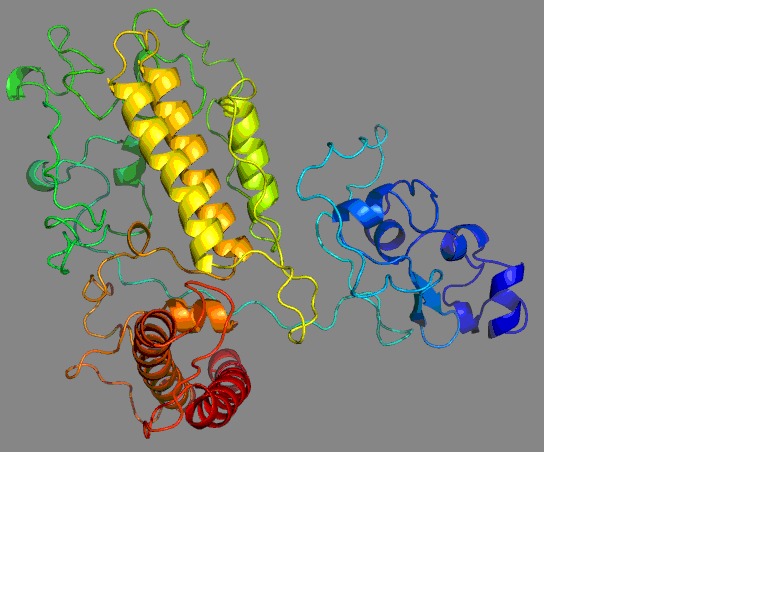
Modeled structure of the AFLR-Protein.
Figure 2.
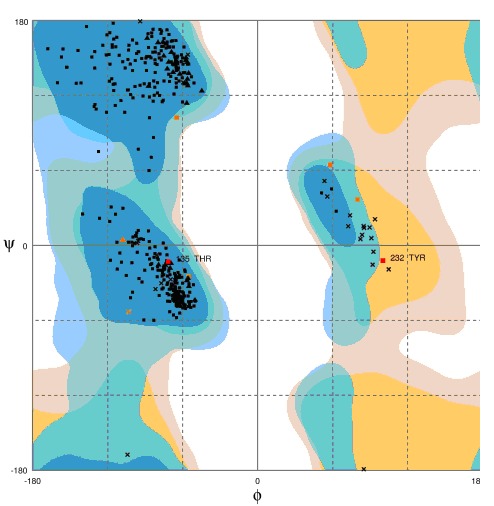
Ramachandran plot for the modeled
Simulations of AFLR-Protein:
Molecular dynamics aimed to explain protein structure and function problems, such as structural stability, folding and conformational flexibility. In the simulations, we monitored the backbone atoms and the C-α-helix of the modeled protein. The RMSD values of the modeled structure’s backbone atoms were plotted as a time-dependent function of the MD simulation. The results support our modeled structure, as they show constant RMSD deviation throughout the whole simulation process. The time dependence of the RMSD of the backbone atoms of the modeled protein during a 10 ns simulation is shown in (Figure 3). The graph clearly indicates that there is a change in the RMSD from 2.0 Å to 4.0 Å in the AFLR-protein during the first 1000 ps, but after that it reaches a plateau. The RMSD values of the backbone atoms in the system tend to converge after 2000 ps, showing fluctuations of around 1 Å. The low RMSD and the simulation time indicate that, as expected, the 3D structural model of AFLR-protein represents a stable folding conformation.
Figure 3.
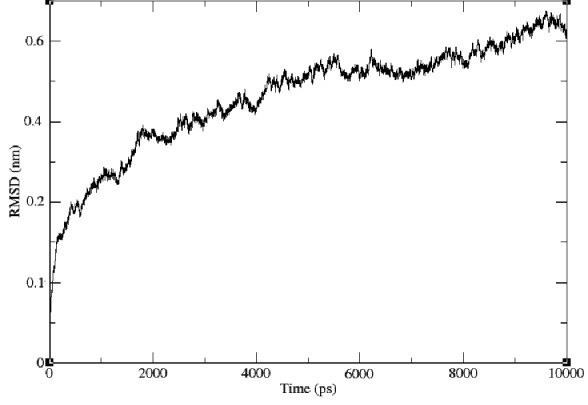
RMSD of the backbone atoms of the modeled protein over a time period of 10 ns
Identification of functional site:
Q-Site Finder server was used for the prediction of functional sites in the modeled AFLR-protein. These servers detected the following putative functional site residues in the modeled protein: LEU-255, THR-270, VAL-273, ILE-274, ASN-277, LYS- 278, THR-281, LEU-307, LEU-310, ALA-311, TYR-313, ALA-314, ALA-317, THR-319, GLN-320, CYS-321, THR-322, SER-323, and THR-324 with significant matches.
Conclusion
The three-dimensional model of AFLR-protein has been generated using Robetta server. The model generated by using the template 3coqA proved to be the best model generated as compared to the other templates. The modeled AFLR-Protein was further optimization and validation using Rampage by calculating the Ramachandran plot. The plot (Figure 1) of our model shows that 98.2% of residues were found in the favored and 1.4% allowed regions and 0.5% were in the outlier region. In the simulations, we monitored the backbone atoms and the C-α-helix of the modeled protein. The low RMSD and the simulation time indicate that, as expected, the 3D structural model of AFLR-protein represents a stable folding conformation. The present study would aid in detailed molecular mechanism of inhibition of aflatoxin in future.
Footnotes
Citation:Kasoju et al, Bioinformation 8(14): 684-686 (2012)
References
- 1.JW Bennett, M Klich. Clin Microbiol Rev. 2003;16:497. doi: 10.1128/CMR.16.3.497-516.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.PJ Cotty, R Jaime-Garcia. Int J Food Microbiol. 2007;119:109. doi: 10.1016/j.ijfoodmicro.2007.07.060. [DOI] [PubMed] [Google Scholar]
- 3.J Yu, et al. Appl Environ Microbiol. 1995;61:2365. doi: 10.1128/aem.61.6.2365-2371.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.F Trail, et al. Appl Environ Microbiol. 1995;61:1665. [Google Scholar]
- 5.DE Kim, et al. Nucleic Acids Res. 2004;32:W526. [Google Scholar]
- 6.SF Altschul, et al. J Mol Biol. 1990;215:403. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 7.SC Lovell, et al. Proteins. 2003;50:437. [Google Scholar]
- 8.D Van Der Spoel, et al. J Comput Chem. 2005;26:1701. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 9.A Laurie, R Jackson. Bioinformatics. 2005;21:1908. [Google Scholar]