

Abstract
The study of Human immunodeficiency virus (HIV) in humans and animal models in last 31 years suggested that it is a causative agent of AIDS. This causes serious pandemic public health concern globally. It was reported that the HIV-1 reverse transcriptase (RT) played a critical role in the life cycle of HIV. Therefore, inhibition of HIV-1RT enzyme is one of the major and potential targets in the treatment of AIDS. The enzyme (HIV-1RT) was successfully targeted by non nucleotide reverse transcriptase inhibitors (NNRTIs). But frequent application of NNRTIs led drug resistance mutation on HIV infections. Therefore, there is a need to search new NNRTIs with appropriate pharmacophores. For the purpose, a virtually screened 3D model of unliganded HIV-1RT (1DLO) was explored. The unliganded HIV-1RT (1DLO) was docked with 4-thiazolidinone and its derivatives (ChemBank Database) by using AutoDock4. The best seven docking solutions complex were selected and analyzed by Ligplot. The analysis showed that derivative (5E)-3-(2- aminoethyl)-5-(2- thienylmethylene)-1, 3-thiazolidine-2, 4-dione (CID 3087795) has maximum potential against unliganded HIV-1RT (1DLO). The analysis was done on the basis of scoring and binding ability. The derivative (5E)-3-(2- aminoethyl)-5-(2- thienylmethylene)-1, 3-thiazolidine-2, 4-dione (CID 3087795) indicated minimum energy score and highest number of interactions with active site residue and could be a promising inhibitor for HIV-1 RT as Drug target.
Keywords: HIV-1 reverse transcriptase (RT), Drug Target, ChemBank, AutoDcok4, LIGPLOT
Background
Acquired immunodeficiency syndrome (AIDS) is a global most serious pandemic public health challenges [1]. The study of Human immunodeficiency virus (HIV) in humans and animal models in last 31 years suggested that it is a causative agent of AIDS [2–4]. The HIV-1 reverse transcriptase (RT) had played a critical role in the life cycle of HIV and it was, consequently, an interesting target for anti- HIV drug therapy [5]. The inhibition of HIV-1 RT is one of the major and potential targets in the treatment of AIDS [6–9]. There was a large number of drugs elicit anti-HIV-1 activity by inhibiting RT which are available in the market [10, 11]. The natural compounds and their derivatives are rich source of biologically active pharmacophores. They have been used as lead molecules for treatment of HIV as well as other vital diseases e.g. fungal and bacterial infections, parasitic diseases, Cancer and cardiovascular disease (CVD), Huntington's disease (HD), Alzheimer's and Parkinson's diseases [12–16]. The further modifications in natural compounds (lead molecules for HIV treatment) might improve their anti-HIV ability [17–19]. There are two classes of HIV-1 RT inhibitors: non-nucleoside reverse transcriptase inhibitors, NNRTIs [20–26] and nucleoside/nucleotide reverse transcriptase inhibitors, NRTIs [27–30]. The NNRTIs are chemically diverse group of therapeutic compounds. The NNRTIs bind noncompetitively with active site residues (unique allosteric hydrophobic binding pocket) on the enzyme leading to a conformational change. The conformational change in the structure of enzyme results in decreased affinity for the substrate [31, 32]. The NNRTIs are highly potent, selective, and specific with very low cellular toxicity [33]. It was suggested that NNRTIs could not interfere with normal function of host DNA polymerase. The factual advantages of NNRTIs were departed due to fractious resistance displayed by most of the approved NNRTIs. The emergence of drug resistance mutations among the different therapies, NNRTIs become less effective. To overcome this problem, novel NNRTIs were searched by modifying the existing drug classes with appropriate pharmacophores.
Computational methods are the successful tools in the area of drug designing. The computational tools are simple and nonexpensive which speed up the process of designing novel and potent therapeutic molecules with desired biological activity. Docking is one of the commonly used computational methods for structure based drug designing [34]. Docking consisted of two distinct tasks, firstly it involved in the prediction of bonding geometries for small therapeutic molecules and encouraged them to bind with bonding site on target protein/receptor and secondly, it involved in the estimation of bonding free energy available for newly formed compound/complex. Docking accuracy reflected an algorithm’s ability to ascertain a conformation change and alignment of a ligand relative to protein and to identify the pose exactly [35]. The ability of a scoring method predicted accurately the binding affinity of ligands with a target protein [36]. It gives an idea of how ligands bind with receptors and how much conformational changes could be brought into the receptor structure. Therefore, the quantitative structure activity relationship (QSAR) studies provided rational inputs for molecular modifications in drug designing research and discovery [37, 38]. The objectives of the study are to explore the active site (binding mode), binding energy and interactions with RT proteins (HIV drug targets). The study has been carried out on the derivatives of 4- thiazolidinone and determined the influence of steric, electrostatic, and hydrophobic fields effect on HIV-1 inhibitory activity.
Methodology
Protein selection and preparation:
The PDB structure of unliganded HIV-1 RT (PDB ID=1DLO) at 2.7 Å resolution [39] was retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) (RCSB, www.rcsb.org/). The protein preparation was the first step for molecular docking. The removal of water molecules, metal ions, cofactors, and addition of charges and hydrogen atoms was done in this step using SPDBV (http://spdbv.vital-it.ch/).
Binding Site Prediction:
Binding sites were characterized by CASTp [40, 41], PASS [42], Q-Site finder [43, 44] and compared by extensive literature search. By comparing prediction of CASTp algorithm, PASS and Q-Site Finder, best active sites were selected. CASTp method was used to identify and measure the binding sites, active sites, surface structural pockets (accessible), interior cavities (inaccessible), shape (alpha complex and triangulation), area and volume (solvent and molecular accessible surface) of each pockets and cavities of proteins. CASTp could be used to measure the number, area, circumference of mouth openings of each pocket in solvent and molecular accessible surface [40, 41]. PASS (Putative Active Sites with Spheres) method was used to identify and measure the obscured volume of proteins and to predict the positions of binding sites on proteins based on the size, shape and burial amount of proteins [42]. A method, Q-site finder, was used to predict the ligand binding site on a protein. It involves the binding of hydrophobic probes to proteins, searching probe clusters on the protein with favourable binding energy and arranged them in a order according to the binding energy of each cluster [43]. Pocket finder method was used to detect pockets on the surface of the proteins. The method is based on Ligsite algorithm which scans the probe radius (1.6Å) with a grid resolution 0.9 Å, cubic diagonals and ligands along the proteins [44].
Ligand selection and preparation:
500 derivatives of 4-thiazolidinones were taken as ligand on the basis of structure similarity and functionality as a pharmacophore. These were screened and retrieved from ChemBank (http://chembank.broadinstitute.org/). The structures were drawn by Chemsketch (http://www.acdlabs.com/resources/freeware/chemsketch/) and converted to PDB file by Openbabel software (www.openbabel.org/). Ligand preparation includes addition of hydrogen atoms, neutralization of the charge groups and removal of any miscellaneous structures from the ligand. Prepared and optimized structures of ligand and protein were ultimately used for molecular docking.
Molecular Docking:
The docking of unliganded HIV-1 RT was performed against 500 derivatives of 4-thiazolidinones, retrieved from Chembank database by using Autodock4 (http://autodock.scripps.edu/wiki/AutoDock4). The Python scripts in MGL tools package were used to analyze the docking results. The molecular structures of a protein or substrate could be visualized and analyzed by MGL Tools software [45]. Auto Dock Tools are part of this software which used to predict the smallness of molecule to bind with a known 3D receptor structure of protein. Python Molecular Viewer (PMV) was used to observe a molecule surface at advanced level or 3D structure. PMV has a customizable feature with many command buttons to display the molecular surface. A visual programme called, Vision, could be used to narrate different combinations of computed data and could be predicted a new visualization based on computed data.
Receptor grid generation and molecular docking:
After preparation and optimization of the protein, the grid was generated (6.5Å) on the protein for ligand docking. The structure of HIV-1 RT was a dimer consists of ‘A’ and ‘B’ chain. The catalytic sites for the derivatives of 4-thiazolidinones were present on ‘A’ chain. Hence, grid was generated on ‘A’ chain of HIV-1 RT. Prepared ligands were docked within the grid region. Docking was performed with the help of Autodock4 [46, 47]. Possible favorable interactions (of minimum binding energy and hydrogen bonding) with amino acids at possible target sites were determined by Lamarckian genetic algorithm (LGA) and Genetic Algorithm [35]. Docking was repeatedly performed thrice, with all softwares.
Analyzing the Docking Results:
The search for the best ways is to fit ligand molecules (derivatives of 4-thiazolidinones), into HIV1- RT structure, using Autodock4 resulted in docking files that contained detailed records of docking. The obtained log files were read in ADT (Auto Dock Tool) to analyze the results of docking. The similarity of docked structures was measured by computing the root mean square deviation (RMSD) between the coordinates of the atoms and creating clustering of the conformations based on the RMSD values. The lowest binding energy conformation in all cluster were considered as the most favorable docking pose. Binding energies that are reported represent the sum of the total intermolecular energy, total internal energy and torsional free energy minus the energy of the unbound system. The top seven ligands were selected based on the energy score after virtual screening Table 2 (see supplementary material).
Discussion
Active Site Identification:
After getting the PDB (1DLO) structure from RCSB (http://www.rcsb.org/pdb/home/home.do) the possible binding sites of structure were searched using the CASTp server, PASS programme and Q-site finder also confirmed by extensive literature search. Nine possible binding sites were obtained Table 1 (see supplementary material). These sites were compared with active site of HIV-1 RT and it was found that molecules CID 3087795 and 1656714 were highly interacting with HIV-1 RT.
Virtual screening:
Earlier studies revealed that more than 30 different classes of NNRTIs have some universal features i.e. the overall structure may be considered evocative of a butterfly with hydrophilic centre and two hydrophobic environs [48–52]. The active conformation and virtual screening and the molecular alignment of each derivative of 4-thiazolidinone are done using docking programme, Autodock4 into HIV-1 RT binding pocket. The molecular alignment is done according to the electrostatic and structural properties of the active site of RT. The calculated binding energies, based on the docked structures, agree well with the experimentally observed inhibitory activities. Binding energy calculated from programs like AutoDock, and Dock program etc is reliable. Further, the steric, electrostatic and hydrophobic fields were mapped onto the active binding pocket of HIV-1 RT to better understand these interactions. Prediction of interactions between small molecules and proteins is a crucial step to decipher many biological processes and plays a critical role in drug discovery. The top seven ligand molecules having minimum energy were screened out as the possible inhibitors for HIV-1 RT Table 2 (see supplementary material). All selected molecules having low energy score were selected and ranked. The first rank derivative (CID 3087795) was interacting with active site residues having minimum energy score as follows: -8.69Kcal/Mol with VAL106, -7.60Kcal/Mol with ASP185 and ASP186, -7.39Kcal/Mol with MET184, - 7.13Kcal/Mol with TYR181 and -6.34 Kcal/Mol with ASP 110. The second rank derivative (CID 1442532) has energy score - 7.99Kcal/Mol with MET148, third rank derivative (CID 1187346) has energy score -7.60Kcal/Mol with ASP185, fourth rank derivative (CID 206166) has energy score -7.43Kcal/Mol while fifth rank derivative (CID 1656714) have interactions with ASP185 (-7.34Kcal/Mol), ASP186 (-7.32Kcal/Mol), VAL106 (- 7.30Kcal/Mol), MET184 (-7.24Kcal/Mol), TYR181 (- 6.88Kcal/Mol) and ASP110 (-6.34Kcal/Mol).
With the help of the Ligplot study we have selected CID 3087795 as the possible inhibitor lead molecule, as it has minimum energy score and one of the highest number of interactions with the active site residue, it has 05 hydrophobic and 2 hydrogen bond interactions. (Figure 1 (A) to (D)), represent interactions of the top four ligands drawn by ligplot according to the energy score. The energy score of the ligands is independent of their molecular weight and log P value. The energy score calculated for all selected ligands molecules; log P value and molecular weight Table 2 (see supplementary material). It was found the inhibitor CID 3087795 has minimum energy score which reveals higher binding affinity towards the HIV-1 RT and it was also showing one of the best interactions with the active site residues (Figure 2 (A) to (D)). The other important drug like properties viz. molecular weight and logP value were also found within the limits. When detailed 3D structure of the protein target is available, protein-ligand complexes are obtained with binding affinity and interactions using computational methodology. For the wet laboratory validation of present study, cell lines of HIV-1 RT infected can be maintained and the molecule CID 3087795 can be procured from the ChemBank database. The inhibition of HIV-1 RT activity by (5E) - 3- (2- aminoethyl) - 5- (2- thienylmethylene) - 1, 3- thiazolidine- 2, 4- dione (CID 3087795) can be established by the qualitative and quantities microbiological techniques. Thus, the derivative molecule (CID 3087795) of 4-thiazolidinones will be helpful in enzymatic studies as well as design of specific inhibitors.
Figure 1.
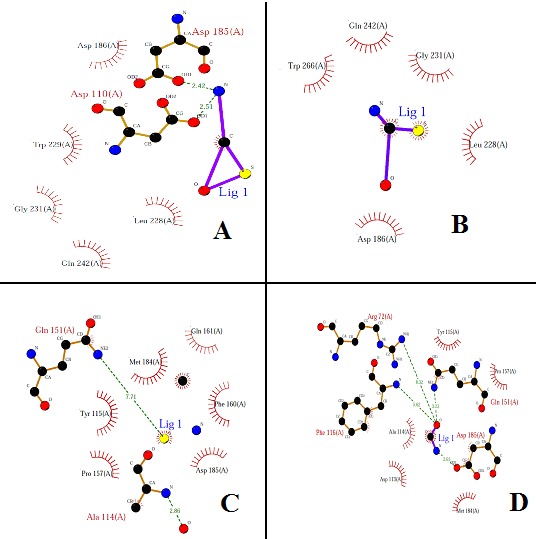
Ligplot showing the protein-ligand interactions of top four ligands, based on energy score (hydrogen bonding and hydrophobic), generated by Ligplot program. (A) CID 3087795, (B) CID 1656714, (C) CID 2062983 and (D) CID 1642811
Figure 2.
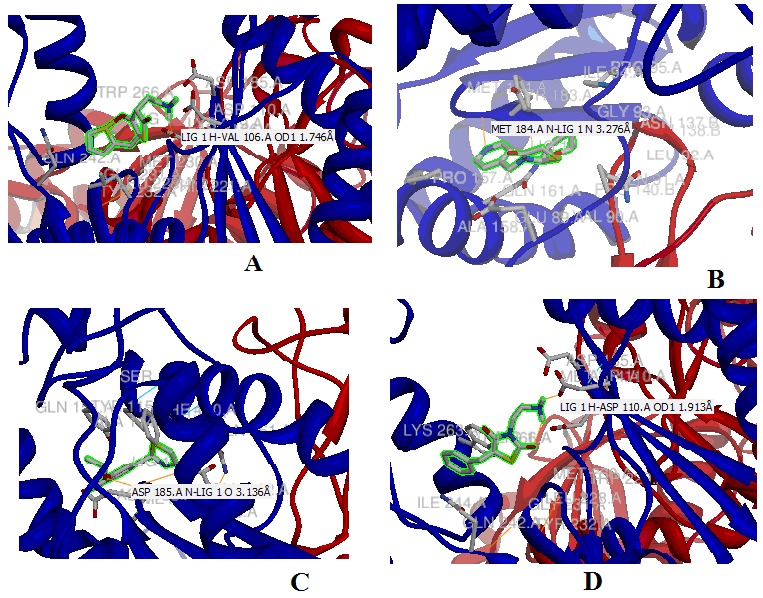
Top four docked molecules showing the protein-ligand interactions, based on energy score (hydrogen bonding and hydrophobic), generated by Chimera. (A) CID 3087795; (B) CID 1442532; (C) CID 1187346 and (D) CID 2061166
Conclusion
Our approach of molecular docking analysis resulted in the identification of potential drug molecules. Hence, in present study, it can be concluded that the molecule 5E - 3- (2- aminoethyl) - 5- (2- thienylmethylene) - 1,3- thiazolidine- 2,4- dione (CID 3087795) has the potential to inhibit the activity of HIV-1 RT and can act as remedy for the treatment of HIV infection. The interaction of molecule with HIV1-RT is similar to the interactions of nevirapine, efavirenz, dihydroxy benzoyl naphthyl hydrazone and delavirdine, some well known drugs in HIV treatment. However, as most of these drugs exhibit considerable side effects it merits search for new drugs. 4- thiazolidinone does not only show interactions with identified active residues that are important for catalytic activity of HIV1- RT but the free energy of binding also ensures that 4- thiazolidinone shows a very strong binding with HIV1-RT at polymerase active site.
Supplementary material
Acknowledgments
The authors are thankful to Department of Biotechnology, Madhav Institute of Technology and Science, Gwalior, for providing computational facility and support.
Footnotes
Citation:Seniya et al, Bioinformation 8(14): 678-683 (2012)
References
- 1.R Tian, et al. Virologica Sinica. 2007;22:476. [Google Scholar]
- 2.SJ O'Brien, JJ Goedert. Curr Opin Immunol. 1996;8:613. doi: 10.1016/s0952-7915(96)80075-6. [DOI] [PubMed] [Google Scholar]
- 3.R Detels. Ann Epidemiol. 2009;19:250. doi: 10.1016/j.annepidem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.TR Frieden, et al. MMWR Morb Mortal Wkly Rep. 2011;60:689. [PubMed] [Google Scholar]
- 5.H Jonckheere. Med Res Rev. 2000;20:129. doi: 10.1002/(sici)1098-1128(200003)20:2<129::aid-med2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 6.A Jacobo-Molina, E Arnold. Biochemistry. 1991;30:6351. doi: 10.1021/bi00240a001. [DOI] [PubMed] [Google Scholar]
- 7.AL Hopkins, et al. J Med Chem. 1996;39:1589. doi: 10.1021/jm960056x. [DOI] [PubMed] [Google Scholar]
- 8.E De Clercq, et al. Int J Biochem Cell Biol. 2004;36:1800. doi: 10.1016/j.biocel.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 9.J Wang, et al. J Am Chem Soc. 2001;123:5221. doi: 10.1021/ja003834q. [DOI] [PubMed] [Google Scholar]
- 10.R Pauwels, et al. Curr Opin Pharmacol. 2004;4:437. doi: 10.1016/j.coph.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 11.E De Clercq. Nature Rev Drug Discov. 2007;6:1001. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 12.RD Yedery, KV Reddy. Eur J Contracept Reprod Health Care. 2005;10:32. doi: 10.1080/13625180500035124. [DOI] [PubMed] [Google Scholar]
- 13.S Nobili, et al. Pharmacol Res. 2009;59:365. doi: 10.1016/j.phrs.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 14.SL Albarracin, et al. Nutr Neurosci. 2012;15:1. [Google Scholar]
- 15.HR Vasanthi, et al. Curr Pharm Des. 2011;17:2170. doi: 10.2174/138161211796957508. [DOI] [PubMed] [Google Scholar]
- 16.B Zhao. Neurochem Res. 2009;34:630. doi: 10.1007/s11064-008-9900-9. [DOI] [PubMed] [Google Scholar]
- 17.D Yu, et al. Med Res Rev. 2007;27:108. doi: 10.1002/med.20075. [DOI] [PubMed] [Google Scholar]
- 18.N Ahmed, et al. Bioorg Med Chem. 2010;18:2872. doi: 10.1016/j.bmc.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 19.SK Chauthe, et al. Bioorg Med Chem. 2010;18:2029. doi: 10.1016/j.bmc.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 20.RA Koup, et al. J Infect Dis. 1991;163:966. doi: 10.1093/infdis/163.5.966. [DOI] [PubMed] [Google Scholar]
- 21.D Richman, et al. Antimicrob Agents chemother. 1991;35:305. doi: 10.1128/aac.35.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.E Arnold, et al. Drug Design Discov. 1996;13:29. [PubMed] [Google Scholar]
- 23.C Tantillo, et al. J Mol Biol. 1994;243:369. doi: 10.1006/jmbi.1994.1665. [DOI] [PubMed] [Google Scholar]
- 24.ED Clercq. Cln Microbiol Rev. 1995;8:200. [Google Scholar]
- 25.G Campiani, et al. Curr Pharm Des. 2002;8:615. [Google Scholar]
- 26.OS Pedersen, EB Pedersen. Antiviral Chem Chemother. 1999;10:285. doi: 10.1177/095632029901000601. [DOI] [PubMed] [Google Scholar]
- 27.H Mitsuya, et al. Proc Natl Acad Sci USA. 1985;82:7096. [Google Scholar]
- 28.R Yarchoan, et al. Science. 1989;245:412. [Google Scholar]
- 29.JA Coates, et al. Antimicrob Agents Chemother. 1992;36:202. doi: 10.1128/aac.36.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ED Clercq. Med Res Rev. 1993;13:229. doi: 10.1002/med.2610130303. [DOI] [PubMed] [Google Scholar]
- 31.R Esnouf, et al. Nat Structural Biology. 1995;2:303. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- 32.H David, H Bill. Rev Med Virol. 2002;12:31. doi: 10.1002/rmv.339. [DOI] [PubMed] [Google Scholar]
- 33.FW Bell, et al. J Med Chem. 1995;38:4929. doi: 10.1021/jm00025a010. [DOI] [PubMed] [Google Scholar]
- 34.J Koska, et al. J Chem Inf Model. 2008;48:1965. doi: 10.1021/ci800081s. [DOI] [PubMed] [Google Scholar]
- 35.J Fuhrmann, et al. J Comput Chem. 2010;31:1911. doi: 10.1002/jcc.21478. [DOI] [PubMed] [Google Scholar]
- 36.SY Huang, X Zou. Int J Mol Sci. 2010;11:3016. [Google Scholar]
- 37.R Garg, et al. Chem Rev. 1999;99:3525. [Google Scholar]
- 38.H Kubinyi. J Cancer Res Clin Oncol. 1990;116:529. doi: 10.1007/BF01637071. [DOI] [PubMed] [Google Scholar]
- 39.Y Hsiou, et al. Structure. 1996;4:853. [Google Scholar]
- 40.J Dundas, et al. Nucl Acids Res. 2006;34:W116. [Google Scholar]
- 41.J Liang, et al. Protein Science. 1998;7:1884. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.GP Brady, PF Stouten. J Computer Aided Mol Des. 2000;14:383. doi: 10.1023/a:1008124202956. [DOI] [PubMed] [Google Scholar]
- 43.AT Laurie, RM Jackson. Bioinformatics. 2005;21:1908. [Google Scholar]
- 44.M Hendlich, et al. J Mol Graph Model. 1997;15:359. doi: 10.1016/s1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
- 45.B Gautam, et al. Bioinformation. 2012;8:134. [Google Scholar]
- 46. http://autodock.scripps.edu/
- 47.AU Khan, et al. Bioinformation. 2011;5:331. [Google Scholar]
- 48.R Silvestri, et al. Bioorg Med Chem Lett. 2000;10:253. [Google Scholar]
- 49.R Silvestri, et al. J Med Chem. 2002;45:1567. doi: 10.1021/jm010904a. [DOI] [PubMed] [Google Scholar]
- 50.D Sriram, et al. Bioorg Med Chem. 2004;12:5865. doi: 10.1016/j.bmc.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 51.TR Bal, et al. Bioorg Med Chem Lett. 2005;15:4451. doi: 10.1016/j.bmcl.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 52.BG Turner, MF Summers. J Mol Biol. 1999;285:1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.