

Abstract
The nematodes like root-knot and cyst are plant-parasitic pest found in horticultural and agricultural crops. They do damages in the roots of plants as a result losses million tons of production. High cost of nematicides and environment safety concern has necessitated finding of some alternative methods. Under Integrated Pest Management (IPM) such problems are solving significantly by means of target gene inhibition, agrobacterium mediated transformation etc. One of this strategy use Plant Proteinase Inhibitors (PIs) gene which are used to control the proteolysis mechanism of Pest by inhibiting gut Serine Proteinase (SP). Present work investigates the utility of computer aided methods to study the mechanism of Protein-Protein interactions and thereby inhibition of Serine Proteinase by PIs. Hence 3D models of Serine Proteinase as well as Serine Proteinase Inhibitors (SPIs) generated using homology modeling. Validations of constructed models have been done by PROCHECK, VERIFY3D, ERRAT and PROSA. Prediction of Protein interacting surface patches and site specific protein docking was performed by using ZDOCK Server. Backbone refinement of output protein complexes was executed in Fiber Dock server. Interaction study between SP and SPIs complexes shows their comparative inhibition efficacy, measured in terms of number of hydrogen bonds, Van dar wall attraction and docking energy. This work reported that Vigna marina and Phaseolus oligospermus are having better inhibition efficiency in comparison to other inhibitors.
Keywords: Serine Proteinase, Serine Proteinase Inhibitors, Vigna marina, Phaseolus oligospermus, Integrated Pest Management, Modeling, Docking
Background
Presently sedentary plant parasitic cyst nematodes (Heterodera spp. and Globodera spp.) and root-knot nematodes (Meloidogyne spp.) are the world's most damaging agricultural pests. They reduce crop production and quality of seeds causing billion dollar losses per annum [1]. Crop safety relies predominantly on the use of environmentally toxic synthetic nematicides. Rather than using chemical pesticides on a large scale, other alternative need to dig into that are based on system-oriented science and technology. Such confined systems will decrease inputs of chemicals and will not generate harmful output such as pesticides residues [2]. To carry out this aim it is essential to increase the resistance of plant to pest and nematodes through promising control of Integrated Pest Management (IPM). IPM includes various strategies to control the pest like Mechanical control, Biological controls etc. The crop protection under biological controls principle includes the inhibition of target gene by suitable protein inhibitors [3]. Plant Serine Proteinase Inhibitors (SPIs) are natural defense-related proteins frequently present in seeds and certain plant tissue which prevents plant by herbivores and wounding. SPIs gens are inhibiting the target protease enzymes which are responsible for proteolytic activity in nematodes [4]. Numerous structural biology approaches are facilitating to check insight inhibition mechanism of protease by SPIs. Protein-Protein docking is one of the suited methods to find out the interaction between two proteins. The current work demonstrated the comparative insight of structural inhibition pattern of SPIs with respect to Serine proteinase of nematodes.
Methodology
Molecular model of Serine Proteinase of Heterodera glycines:
The protein sequence Serine Proteinase of Heterodera glycines (SP_HG) was downloaded from National Centre for Biotechnology Information (NCBI) [5].The FASTA sequence of the SP_HG was subjected to PSI-BLAST against PDB database for the selection of best homologous templates [6, 7]. The best template was selected based on similarity, percentage of identity, expectation value, bit scores and query coverage area. A 3D model of SP_GH was generated by Modeller 9v10 [8]. The chain-A of Earthworm Fibrinolytic Enzyme Component A from Eisenia Fetida, PDB: 1M9U was identified as the best template for comparative modeling [9]. This 3D structure was generated by X-ray diffraction studies and resolution was 2.30Å. The Rfactor of the structure was 0.191 and R-free value was 0.236. A bundle of 100 models are formed through random generation and subsequently the good model was obtained.
Molecular models of Plant Serine Proteinase inhibitors:
Following Plant Serine Proteinase Inhibitors (SPIs) of Phaseolus glabellus (SPI_PG), Phaseolus grayanus (SPI_PGn), Phaseolus oligospermus (SPI_PO), Vigna mungo (SPI_VM), Vigna marina (SPI_VMn) sequences having Accession No. CAQ17032, CAM88858, CAO82009, ABD97865, ABD97867 respectively were obtained from NCBI [5]. Each Sequence of proteinase inhibitors was subjected to PSI-BLAST against PDB database for the selection of best homologous templates [6, 7]. The chain-I of The Bowman-Birk type inhibitor from Mung bean, PDB ID: 3MYW and Bowman-Birk Inhibitor from Vigna unguiculata Seeds having, PDB ID: 2G81 were identified as the best model for comparative modeling of Phaselous family and Vigna family inhibitors respectively [10, 11]. Both 3D structures were generated by X-ray diffraction studies and resolution were 2.50Å for 3MYW and 1.55 Å for 2G81. Out of Twenty generated models one good model was obtained for each plant proteinase inhibitors using Modeller 9v10 [8].
Minimization and molecular evaluation:
Energy minimization 3-D model of SP_HG and all SPIs were performed using Amber force field for 10000 steps with ten update interval without fixing any atom in chimera [12, 13]. Stereo- chemistry of all models was evaluated by PROCHEK and WHAT-IF [14, 15]. The overall quality factor of non-bonded interactions between different atoms was calculated by ERRAT [16]. Compatibility of amino acids in each 3D model was checked in VERIFY-3D [17, 18]. X-Ray and NMR spectroscopy structural validation of structures was predicted by PROSA server [19].
Designation of Protein-Protein interacting sites and Docking:
A combine study of various PSI-BLAST results of SPIs showing the specific region of protease binding site of their inhibitor with respect to crystal structures was obtained. Multiple sequence alignment of SPIs sequences with respect to crystal structure helps us to find out protease binding sites shown in (Figure 1). In addition to that we also find out detailed protein interacting sites for all SPIs and SP_HG using online server PPipred [20]. Protein Interface site specific docking of all SPIs with SP_HG was performed using ZDOCK server [21]. ZDOCK output files were subjected in FiberDock server for flexible induced-fit backbone and side chain refinement of the protein complexes [22, 23].
Figure 1.

Multiple sequence alignment of all SPIs with their respective template structures for modeling. Rectangular red box covered residues are having high probability to participate during protein-protein interaction.
Molecular Interaction Study:
The molecular interaction plots between SP_HG and SPIs were generated using Dimplot in LIGPLOT software [24]. The 3D structure and detailed interaction of all proteins complexes were visualized using Accelrys DS Visualizer software [25].
Discussion
Finding of good proteinase inhibitor using structural biology approaches would be helpful for experiment biologist to explore their research in pest management. In this connection we have generated the 3D- models of SPIs and SP_HG. Figure 2 shows the catalytic domains of SP_HG (A) and Serine proteinase inhibitors of Phaseolus Family (B) and Vigna Family (C).The dependability and quality of these models were ensured by evaluating the backbone conformation, angles and bond lengths based on Psi/Phi Ramachandran plot using PROCHECK [14]. Analysis of each model with ProSA web interface brings out that Z-Score and over all residues energies are all well within the limits [15]. ERRAT score shows overall quality factor for non-bonded atomic interactions and higher the score means better the quality of models (accepted range for a high quality model is >50) [16]. VERIFY-3D score indicates good sequenceto- structure agreement because most of the amino acids score for all models have > 0.2 values shown in Table 1 (see supplementary material) [17, 18]. The above said validating parameters assuring the quality and reliability of models. To get insight of structural inhibition mechanism of proteinase inhibitors need to have the protein interacting sites. The protein interacting sites were identified using a web based server PPipred [20]. Most probable interacting residues are common with the crystal structure substrate binding sites which are given in Table 2 (see supplementary material). The Docking of each SPIs with SP_HG deducing a reliable results, in terms of minimum Global, Attractive and Repulsive Van-der wall, Atomic contact and Hydrogen bond energies shown in Table 3 (see supplementary material). In addition to that molecular interaction plots are also certifying the protein-protein interaction between protein complexes. The interaction study inferring that the Serine Proteinase inhibitor of Vigna marina and Phaseolus oligospermus are the potent inhibitors in comparison to others within the same family of SPIs. Molecular interacting plot of SPI_PO with SPI_HG (Figure 3A) shows total 13 hydrogen bonds formation among eight amino acids of SPI_PO and nine residues of SPI_HG. Figure 3B shows the molecular interaction plots between SPI_VMn and SP_HG which having eight hydrogen bonds.The nine residues of SPI_VMn and eight residues of SP_HG are participating in bond formation. The comparative studies between these two inhibitors conclude that SPI_PO is the most capable inhibitor among them. This finding will help to biologist in nematodes control.
Figure 2.
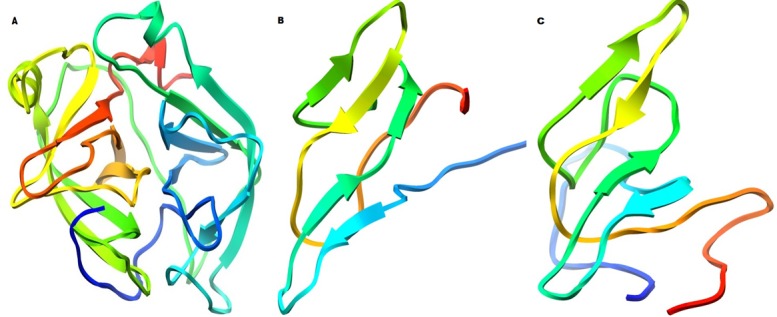
Cartoon representation of catalytic domain of SP_HG (A), Serine Proteinase Inhibitors of Phaseolus Family (B) and Vigna Family (C).
Figure 3.

Molecular Interaction plots of docked complexes of Phaseolus oligospermus (A); (B) Vigna marina, with Serine proteinase inhibitor of Heterodera glycines. Hydrogen bonds with their bond length between Protein interface residues shown in green dotted line
Conclusion
Now a day's protection of crop against Nematodes is being big challenge for the researchers with taking concern of environmental safety and economical balance. Our present work will help to researchers in biological pest control. We have developed high quality 3D models of Plant Serine Proteinase Inhibitors. The comparative In-sillico inhibitory mechanisms of these inhibitors were identified against Serine Proteinase of Heterodera glycines. The inhibitory efficiency of each serine proteinase was calculated on the ground of Number of hydrogen bonds, Van der Waal attraction, and docking energy. Vigna marina and Phaseolus oligospermus proteinase inhibitor forming major hydrogen bonds with minimum docking energy was concluded as potent inhibitors in their family.
Supplementary material
Acknowledgments
We are thankful to Department of Science and Technology, New Delhi, India for supporting us financially in our ongoing project “Development of transgenic Wheat plant against Cereal Cyst nematode (Heterodera Aveane) and Sunnpest (Eurygaster intergrices puton) by using Bioinformatics and Genetic Engineering approaches” Project code: INT/ILTP/A-1.28.
Footnotes
Citation:Prasad et al, Bioinformation 8(14): 673-677 (2012)
References
- 1.JM Nicol, et al. Genomics and Mol Gen of Plant-Nematode Interactions. DOI c 10.1007/978-94-007-0434-3_2. [Google Scholar]
- 2.D Boulter, et al. Biochemistry. 1993;34:1453. [Google Scholar]
- 3.SK Haq, et al. Arch Biochemistry Biophysics. 2004;431:145. doi: 10.1016/j.abb.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 4.H Huma, et al. Biot & Mole Biology Rev. 2007;2:68. [Google Scholar]
- 5. http://www.ncbi.nlm.nih.gov/
- 6. http://www.pdb.org/pdb/home/home.do.
- 7. http://www.ncbi.nlm.nih.gov/blast/Blast.cgi/(PSI)
- 8. http://salilab.org/modeller/
- 9.Y Tang, et al. J Mol Biol. 2002;321:57. [Google Scholar]
- 10.G Lin, et al. Eur J Biochem. 1993;212:549. doi: 10.1111/j.1432-1033.1993.tb17692.x. [DOI] [PubMed] [Google Scholar]
- 11.JA Barbosa, et al. Biophys J. 2007;92:1638. doi: 10.1529/biophysj.106.090555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Junmei, et al. J Comput Chem. 2004;25:1157. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 13.EF Pettersen, et al. J Comput Chem. 2005;25:1605. [Google Scholar]
- 14.RA Laskowski, et al. J Appl Cryst. 1993;26:283. [Google Scholar]
- 15.G Vriend, et al. J Mol Graph. 1990;8:52. [Google Scholar]
- 16.C Colovos, et al. Protein Science. 1993;2:1511. [Google Scholar]
- 17.JU Bowie, et al. Science. 1991;253:164. [Google Scholar]
- 18.R Luthy, et al. Nature. 1992;356:83. [Google Scholar]
- 19. http://prosa.services.came.sbg.ac.at/prosa.php/
- 20. http://bmbpcu36.leeds.ac.uk/ppi_pred/overview.html.
- 21.R Chen, et al. Proteins. 2003;52:80. [Google Scholar]
- 22.E Mashiach, et al. Nucleic Acids Res. 2008;36:W229. [Google Scholar]
- 23.E Mashiach, et al. Nucleic Acids Res. 2008;36:W2296. [Google Scholar]
- 24.AC Wallace, et al. Prot Eng. 1995;8:127. [Google Scholar]
- 25. http://accelrys.com/products/discoverystudio/visualization-download.php.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.