

Abstract
Genome sequencing projects has led to an explosion of large amount of gene products in which many are of hypothetical proteins with unknown function. Analyzing and annotating the functions of hypothetical proteins is important in Staphylococcus aureus which is a pathogenic bacterium that cause multiple types of diseases by infecting various sites in humans and animals. In this study, ten hypothetical proteins of Staphylococcus aureus were retrieved from NCBI and analyzed for their structural and functional characteristics by using various bioinformatics tools and databases. The analysis revealed that some of them possessed functionally important domains and families and protein-protein interacting partners which were ABC transporter ATP-binding protein, Multiple Antibiotic Resistance (MAR) family, export proteins, Helix-Turn-helix domains, arsenate reductase, elongation factor, ribosomal proteins, Cysteine protease precursor, Type-I restriction endonuclease enzyme and plasmid recombination enzyme which might have the same functions in hypothetical proteins. The structural prediction of those proteins and binding sites prediction have been done which would be useful in docking studies for aiding in the drug discovery.
Keywords: Hypothetical proteins, Staphylococcus aureus:, Functional analysis, Bioinformatics tools
Background
Staphylococcus aureus is a gram-positive pathogen that is a major cause of multiple types of infections both in and outside of the hospital setting. These infections range from superficial skin infections to deeper infections of hair follicles, abscesses, and deep tissue infections, and even to systemic infections including those of the heart, lungs, bones, and blood [1]. Although the organism is part of the normal human flora, it can cause infection when there is a break in the skin or mucous membrane that grants it access to the surrounding tissues [2–4]. In the preantibiotic era, these infections were often life threatening, and even today they may give rise to death despite treatment with antibiotics. S. aureus strains can produce a number of different components that may contribute to virulence, including surfaceassociated adhesins, capsular polysaccharides, exoenzymes, and exotoxins. Staphylococcus aureus carries a large repertoire of virulence factors, including over 40 secreted proteins and enzymes that it uses to establish and maintain infections [5, 6]. Some of these virulence factors are known to cause or be associated with specific diseases, for example, endocarditis and osteomyelitis, septic arthritis, and septicemia, toxic shock syndrome toxin (TSST) and toxic shock syndrome; Panton- Valentine leukocidin (PVL) and necrotizing pneumonia and skin diseases [7, 8]; the exfoliative toxins A and B (ETA and ETB) and scalded skin syndrome, impetigo, skin infections, and atopic dermatitis [8, 9]; and the family of staphylococcal enterotoxins A and B(SEA and SEB) and food poisoning [6].
There is a growing need for the automatic annotation of proteins of unknown functional, termed “hypothetical proteins” [10], the structures of which are known [11]. Structural genomics initiatives provide ample structures of hypothetical proteins at an ever increasing rate. However without function annotation, this structural goldmine is of little use to biologists who are interested in particular molecular systems. The structures of many hypothetical proteins are solved in pipelines at structural- genomics centers, which usually lack the resources to engage in thorough functional characterization of each of the solved structures. Moreover, some of the proteins, which are considered to be well annotated, may have additional functions beyond their listed records. About half the proteins in most genomes are candidates for HPs [12]. This group is of utmost importance to complete genomic and proteomic information. Detection of new HPs not only offers presentation of new structures but also new functions. There will be new structures with so far unknown conformations and new domains and motifs will be arising. A series of additional protein pathways and cascades will be revealed, completing our fragmentary knowledge on the mosaic of proteins per se. The network of protein–protein interactions will be increasing logarithmically. New HPs may be serving as markers and pharmacological targets.
Last not least, detection of HP would be of benefit to genomics enabling the discovery of so far unknown or even predicted genes [10]. Hypothetical protein is a protein that is predicted to be expressed from an open reading frame, but for which there is no experimental evidence of translation. Hypothetical proteins constitute a substantial fraction of proteomes of human as well as of other eukaryotes. With the general belief that the majority of hypothetical proteins are the product of pseudogenes, it is essential to have a tool with the ability of pinpointing the minority of hypothetical proteins with a high probability of being expressed [13]. There is so far no classification of hypothetical proteins (HPs) and working terms are replacing definitions of hypothetical proteins. In the strict sense, HPs are predicted proteins, proteins predicted from nucleic acid sequences and that have not been shown to exist by experimental protein chemical evidence. Moreover, these proteins are characterized by low identity to known, annotated proteins. Conserved hypothetical proteins are defined as a large fraction of genes in sequenced genomes encoding those that are found in organisms from several phylogenetic lineages but have not been functionally characterized and described at the protein chemical level [14, 15]. These structures may represent up to half of the potential protein coding regions of a genome.
Methodology
Sequence retrieval:
Randomly retrieved 10 hypothetical protein sequences of Staphylococcus aureus from NCBI [16] and were used in this study. The sequence IDs of those 10 hypothetical proteins were gi|166409299, gi|166409303, gi|166409302, gi|166409301, gi|166409300, gi|390516769, gi|166409293, gi|390516759, gi|390516760 and gi|166409294. To analyze the hypothetical proteins and assign their physicochemical and structural and functional properties, various bioinformatics tools and databases were used.
Physicochemical and functional characterization:
For physicochemical characterization, theoretical Isoelectric point (pI), molecular weight, total number of positive and negative residues, extinction coefficient [17], instability index [18], aliphatic index [19] and grand average hydropathy (GRAVY) [20] were computed using the Expasy's Protparam server [21].
PFAM:
Pfam [22, 23] is a collection of multiple protein-sequence alignments and HMMs, and provides a good repository of models for identifying protein families, domains and repeats. There are two parts to the pfam database: Pfam A, a set of manually curated and annotated models; PfamB, which has higher coverage but is fully automated (with no manual curation). Pfam B HMMs are created from alignments generated by ProDom in the automatic clustering of the protein sequences in SWISS-PROT and TrEMBL.
CDD-BLAST:
CD-Search [24] is NCBI's interface to searching the Conserved Domain Database with protein query sequences. It uses RPSBLAST, a variant of PSI-BLAST, to quickly scan a set of precalculated position-specific scoring matrices (PSSMs) with a protein query [25].
Protein-Protein interactions prediction:
STRING (Search Tool for the Retrieval of Interacting Genes/Proteins,) [26] is a database of known and predicted protein interactions. The interactions include direct (physical) and indirect (functional) associations; they are derived from four sources: Genomic Context, High-throughput Experiments, (Conserved) Co-expression and Previous Knowledge. STRING quantitatively integrates interaction data from these sources for a large number of organisms, and transfers information between these organisms where applicable. The database currently covers 5,214,234 proteins from 1133 organisms [27].
Prediction of transmembrane proteins:
SOSUI server is used to characterize whether the protein is soluble or transmembrane in nature [28].
Stability of proteins:
DISULFIND [29, 30] is a server for predicting the disulfide bonding state of cysteines and their disulfide connectivity starting from sequence alone. Disulfide bridges play a major role in the stabilization of the folding process for several proteins. Prediction of disulfide bridges from sequence alone is therefore useful for the study of structural and functional properties of specific proteins. In addition, knowledge about the disulfide bonding state of cysteines may help the experimental structure determination process and may be useful in other genomic annotation tasks.
Protein structure prediction:
Online server PS2 (PS Square) Protein Structure Prediction Server [31] was used [32–36] which accepts the protein query sequences in FASTA format and uses the strategies of Pair-wise and multiple alignment by combining powers of the programs PSI-BLAST, IMPALA and T-COFFEE in both target – template selection and target–template alignment and finally it constructs the protein 3D structures using integrated modeling package of PS2 using best scored orthologous template.
Q-Site Finder:
Q-Site Finder [37] is a new method of ligand binding site prediction. It works by binding hydrophobic (CH3) probes to the protein, and finding clusters of probes with the most favorable binding energy. These clusters are placed in rank order of the likelihood of being a binding site according to the sum total binding energies for each cluster. Q-Site Finder was shown to identify sites with high precision. The advantage of this is that putative binding sites are identified as closely as possible to the actual binding site. It uses the interaction energy between the protein and a simple Vander Waal's probe to locate energetically favorable binding sites. Energetically favorable probe sites are clustered according to their spatial proximity and clusters are then ranked according to the sum of interaction energies for sites within each cluster [38].
Discussion
The physicochemical properties of hypothetical proteins were tabulated in Table 1 (see supplementary material). The calculated isoelectric point (pI) will be useful because at pI, solubility is least and mobility in an electro focusing system is zero. Isoelectric point (pI) is the pH at which the surface of protein is covered with charge but net charge of protein is zero. At pI, proteins are stable and compact. The computed isoelectric point (pI) will be useful for developing buffer system for purification by isoelectric focusing method. Although Expasy's Protparam computes the extinction coefficient for 276, 278, 279, 280 and 282 nm wavelengths, 280 nm is favored because proteins absorb light strongly there while other substances commonly in protein solutions do not. Extinction coefficient of hypothetical proteins homologue at 280 nm is ranging from 1490 to 77825 M cm with respect to the concentration of Cys, Trp and Tyr. The high extinction coefficient of hypothetical proteins indicates presence of high concentration of Cys, Trp and Tyr. The computed extinction coefficients help in the quantitative study of protein–protein and protein–ligand interactions in solution. The instability index provides an estimate of the stability of protein in a test tube. There are certain dipeptides, the occurrence of which is significantly different in the unstable proteins compared with those in the stable ones. This method assigns a weight value of instability. Using these weight values it is possible to compute an instability index (II). A protein whose instability index is smaller than 40 is predicted as stable, a value above 40 predicts that the protein may be unstable [18]. The instability index value for the hypothetical proteins was found to be ranging from 7.98 to 64.89. The stable proteins were gi|166409302, gi|166409301, gi|166409300, gi|390516769, gi|166409293, gi|390516759 and gi|166409294 and the other proteins were unstable. The aliphatic index (AI) which is defined as the relative volume of a protein occupied by aliphatic side chains (A, V, I and L) is regarded as a positive factor for the increase of thermal stability of globular proteins. Aliphatic index for the hypothetical proteins sequences ranged from 65.36 to 138.39. The very high aliphatic index of the protein sequences indicates that these proteins may be stable for a wide temperature range. The lower thermal stability of proteins was indicative of a more flexible structure when compared to other protein. The Grand Average hydropathy (GRAVY) value for a peptide or protein is calculated as the sum of hydropathy values of all the amino acids, divided by the number of residues in the sequence. GRAVY indices of hypothetical proteins are ranging from - 0.172. This low range of value indicates the possibility of better interaction with water.
Functional analysis of these proteins includes protein domains and family prediction and prediction of trans-membrane regions, disulfide bond and identification of sub-cellular localization sites. Domains can be thought of as distinct functional and/or structural units of a protein. These two classifications coincide rather often, as a matter of fact, and what is found as an independently folding unit of a polypeptide chain also carries specific function. Domains are often identified as recurring (sequence or structure) units, which may exist in various contexts. In molecular evolution such domains may have been utilized as building blocks, and may have been recombined in different arrangements to modulate protein function [24]. The proteins were classified into particular family based on the presence of specific domain in the sequence. Out of 10 hypothetical proteins, 7 proteins possessed specific domains in them which were lactococcin_972, Mob_Pre, L_ocin_972_ABC, DUF 1093 & 1430, COG4652, ABC_MJ0796_Lo1CDE_FtsE, HTH, HTH_MARR, oxido_YhdH and MDR_yhdh_yhfp domains and they were classified as super-families accordingly. Most of these possessed functionally important domains in them except the sequences with id gi|166409301 and gi|166409300 which had domains of unknown function. There were no domains in the other 3 proteins. The presence of these domains in the hypothetical proteins reveals that the proteins might be involved in performing the same function. The domains of the hypothetical proteins and their super-family descriptions were given in Table 2 & Table 3 (see supplementary material).
The study of subcellular localization is important for elucidating protein functions involved in various cellular processes. Knowledge of the subcellular localization of a protein can significantly improve target identification during the drug discovery process. The localization site of the hypothetical proteins selected in this study was predicted by PSORTB and they were tabulated in Table 4 (see supplementary material). Cytoplasmic membranes were found to be preferred site for performing functions in these proteins as they were seen in most of the proteins involved in this study. Multiple localization sites were found in sequences with id gi|166409303, gi|166409301 and gi|390516759 in which the targeting sites might be of anyone of Cytoplasmic, Cytoplasmic membrane, extracellular and cell wall.
Pfam database search made to identify domains and families present in hypothetical proteins Table 5 & Table 6 (see supplementary material). They were Zinc-binding dehydrogenase family, MAR family, TMEM9 and ABCtransporters. SOSUI distinguishes between membrane and soluble proteins from amino acid sequences, and predicts the transmembrane helices for the former. The server SOSUI classified 3 hypothetical proteins as transmembrane proteins having transmembrane helices atleast one in each and maximally six transmembrane regions were found in the protein, gi|166409300. The transmembrane regions type and their length were tabulated in Table 7 (see supplementary material). All the seven other proteins were soluble ones. The transmembrane regions are rich in hydrophobic amino acids. Thus there was higher number of hydrophobic amino acid residues in the transmembrane proteins. When those hypothetical proteins were analyzed for disulphide bridges by DISULPHIND server to predict the thermo stability of the proteins, it revealed no disulphide bonds in any of those proteins which revealed that they were thermally unstable.
Protein-protein interactions (PPI) are essential for almost all cellular functions. Proteins often interact with one another in a mutually dependent way to perform a common function. As an example, the transcription factors interact among themselves to bring about transcription. It is therefore possible to infer the functions of proteins based on their interaction partners. Proteins seldom carry out their function in isolation; rather, they operate through a number of interactions with other biomolecules. Experimental elucidation and computational analysis of the complex networks formed by individual proteinprotein interactions (PPIs) are one of the major challenges in the post-genomic era. Protein-protein interaction databases have become a major resource for investigating biological networks and pathways in cells [39]. The protein with ID gi|166409301 was found to have interaction with ABC transporter ATP binding protein. Gi|166409299 had interaction with arsenate reductase protein which reduces arsenate [As (V)] to arsenite [As (III)] and dephosphorylates tyrosine and thus the protein might involve in the enzymatic function of the protein. Gi|166409302 showed interactions with three proteins which were A) Elongation factor G which promotes the GTPdependent translocation of the nascent protein chain from the A-site to the P-site of the ribosome and also has Vitronectinbinding activity; B) 30S ribosomal protein S5 which plays an important role in translational accuracy with S4 and S12; C) 30S ribosomal protein S7; being one of the primary rRNA binding proteins, it binds directly to 16S rRNA where it nucleates assembly of the head domain of the 30S subunit; it is located at the subunit interface close to the decoding center, probably blocks exit of the E-site tRNA. Gi|166409300 was found to interact with cysteine protease precursor; Cysteine protease is able to degrade elastin, fibrogen, fibronectin and kininogen. It exhibits a strong preference for substrates where arginine is preceded by a hydrophobic amino acid and also promotes detatchment of primary human keratinocytes. Along with other extracellular proteases the protein is involved in the colonization and infection of human tissues.
Gi|166409293 had interaction with carboxy-terminal processing proteinase CtpA. Gi|166409294 showed interaction with quinone oxidoreductase putative YhdH/YhfP. The other interacting proteins were hypothetical proteins. The proteinprotein interacting networks of the hypothetical proteins were given in (Figure 1) & Table 8 (see supplementary material). Thus those hypothetical proteins could have the functions of their interacting proteins. The three dimensional structures of the hypothetical proteins were modeled by PS Square server (Figure 2). Of the eleven hypothetical proteins, PS Square server could model only four proteins. Since there was low sequence identity, the remaining six proteins could not be modeled. The templates used by the server to model those proteins were tabulated in Table 9 (see supplementary material). Identifying the location of ligand binding sites on a protein is of fundamental importance for a range of applications including molecular docking, de novo drug design and structural identification and comparison of functional sites. Active site residues of the hypothetical proteins were given in Table 10 (see supplementary material). The active binding site residues would be helpful for docking with specific ligand to study the binding interactions between them.
Figure 1.
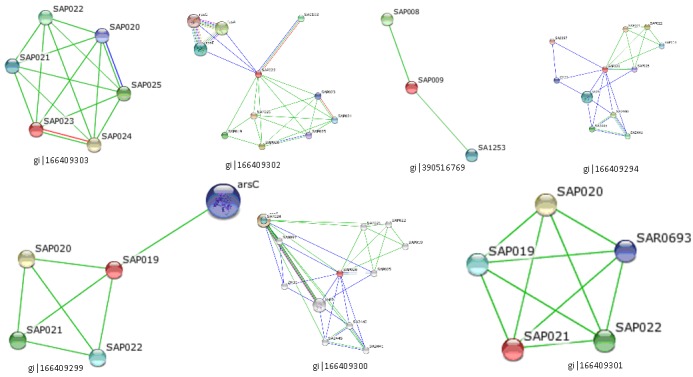
Protein-protein interactions of hypothetical proteins predicted by STRING tool
Figure 2.
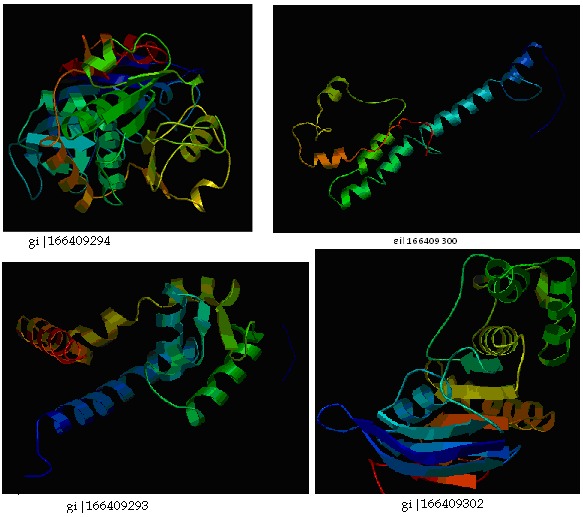
Structures of S. aureus hypothetical proteins modeled by PS SQUARE server
Conclusion
There is a need to annotate and find the structural and functional properties of hypothetical proteins in the pathogenic bacteria Staphylococcus aureus which produce many virulence factors and cause serious infections and disease. We retrieved 10 hypothetical proteins from NCBI database and characterized its physicochemical properties and identified domains and families using various bioinformatics tools and databases. The structures were modeled and their ligand binding sites were identified. The analysis revealed functionally important domains and families which were involved in inducing protein synthesis and multiple antibiotic resistances in the bacteria and also perform enzymatic functions. This also would provide useful solution for drug discovery for those proteins which were involved in multiple antibiotic resistance and disease mechanisms.
Supplementary material
Footnotes
Citation:Mohan & Venugopal, Bioinformation 8(15): 722-728 (2012)
References
- 1.FD Lowy. N Engl J Med. 1998;339:520. [Google Scholar]
- 2.AL Cheung, et al. FEMS Immunol Med Microbiol. 2004;40:1. doi: 10.1016/S0928-8244(03)00309-2. [DOI] [PubMed] [Google Scholar]
- 3.JA Lindsay, MT Holden. Trends Microbiol. 2004;12:378. [Google Scholar]
- 4.DL Goldenberg, JI Reed. N Engl J Med. 1985;312:764. doi: 10.1056/NEJM198503213121206. [DOI] [PubMed] [Google Scholar]
- 5.BA Diep, et al. J Infect Dis. 2006;19:1495. [Google Scholar]
- 6.JP Arbuthnott, et al. Soc Appl Bacteriol Symp Ser. 1990;19:107S. doi: 10.1111/j.1365-2672.1990.tb01802.x. [DOI] [PubMed] [Google Scholar]
- 7.Y Gillet, et al. Lancet. 2002;359:753. [Google Scholar]
- 8.G Lina, et al. Clin Infect Dis. 1997;25:1369. [Google Scholar]
- 9.E Capoluongo, et al. J Dermatol Sci. 2001;26:145. doi: 10.1016/s0923-1811(00)00171-7. [DOI] [PubMed] [Google Scholar]
- 10.G Lubec, et al. Prog Neurobiol. 2005;77:90. [Google Scholar]
- 11.I Friedberg. Brief Bioinform. 2006;7:225. doi: 10.1093/bib/bbl004. [DOI] [PubMed] [Google Scholar]
- 12.FC Minion, et al. J Bacteriol. 2004;186:7123. doi: 10.1128/JB.186.21.7123-7133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D Claus, et al. BMC Bioinformatics. 2009;10:289. doi: 10.1186/1471-2105-10-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MY Galperin, EV Koonin. Nucleic Acids Res. 2004;32:5452. doi: 10.1093/nar/gkh885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MY Galperin. Funct Genomics. 2001;2:14. [Google Scholar]
- 16. http://www.ncbi.nlm.nih.gov/
- 17.SC Gill, PH Von Hippel. Anal Biochem. 1989;182:319. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 18.K Guruprasad, et al. Prot Eng. 1990;4:155. [Google Scholar]
- 19.AJ Ikai, et al. J Biochem. 1980;88:1895. [PubMed] [Google Scholar]
- 20.J Kyte, RF Doolottle. J Mol Biol. 1982;157:105. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 21. http://us.expasy.org/tools/protparam.html.
- 22.A Bateman, et al. Nucleic Acids Res. 2000;28:263. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. http://pfam.sanger.ac.uk/
- 24. http://www.ncbi.nlm.nih.gov/Structurevcdd/wrpsb.cgi/
- 25.BA Marchler, et al. Nucleic Acids Res. 2011;39:225. doi: 10.1093/nar/gkq769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. http://string.embl.de/
- 27.D Szklarczyk, et al. Nucleic Acids Res. 2011;39:D561. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. http://bp.nuap.nagoya-u.ac.jp/sosui/sosui_submit.html.
- 29.A Ceroni, et al. Nucleic Acids Res. 2006;34:W177. [Google Scholar]
- 30. http://disulfind.dsi.unifi.it/
- 31. http://www.ps2.life.nctu.edu.tw/
- 32.Chen-CC, et al. Nucleic Acids Res. 2006;34:W152. doi: 10.1093/nar/gkl187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.SF Altschul, et al. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.AA Schaffer, et al. Nucleic Acids Res. 2001;29:2994. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.C Notredame, et al. J Mol Biol. 2000;302:205. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 36.B Wendy, et al. Nucleic Acids Res. 2000;28:19. doi: 10.1093/nar/28.19.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.AT Laurie, RM Jackson. Bioinformatics. 2005;21:1908. [Google Scholar]
- 38. http://www.modelling.leeds.ac.uk/qsitefinder/
- 39.S Peri, et al. Genome Res. 2003;13:2363. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.