

Abstract
Snakebites are a major neglected tropical disease responsible for as many as 95000 deaths every year worldwide. Viper venom serine proteases disrupt haemostasis of prey and victims by affecting various stages of the blood coagulation system. A better understanding of their sequence, structure, function and phylogenetic relationships will improve the knowledge on the pathological conditions and aid in the development of novel therapeutics for treating snakebites. A large dataset for all available viper venom serine proteases was developed and analysed to study various features of these enzymes. Despite the large number of venom serine protease sequences available, only a small proportion of these have been functionally characterised. Although, they share some of the common features such as a C-terminal extension, GWG motif and disulphide linkages, they vary widely between each other in features such as isoelectric points, potential N-glycosylation sites and functional characteristics. Some of the serine proteases contain substitutions for one or more of the critical residues in catalytic triad or primary specificity pockets. Phylogenetic analysis clustered all the sequences in three major groups. The sequences with substitutions in catalytic triad or specificity pocket clustered together in separate groups. Our study provides the most complete information on viper venom serine proteases to date and improves the current knowledge on the sequence, structure, function and phylogenetic relationships of these enzymes. This collective analysis of venom serine proteases will help in understanding the complexity of envenomation and potential therapeutic avenues.
Background
Snakebite is a major neglected public health issue particularly among agricultural communities living in rural regions throughout the world [1, 2]. An estimated 2.5 million people are bitten by snakes each year and these are estimated to result in up to as high as 95000 deaths worldwide [2, 3]. Snake venoms are complex mixtures of enzymatic [4, 5] and non enzymatic proteins [6], together with other components such as carbohydrates, lipids, nucleosides and metals [4, 7]. Snake venom serine proteases are major components and have been identified mainly in the venoms of snakes belonging to the viperidae family with a few occurring in members of the elapidae, colubridae and hydrophidae families [8]. Many snake venom serine proteases exert their effects through the ability to disrupt the normal haemostasis of envenomed prey and victims [9]. Indeed, viper venom serine proteases (VVSPs) affect various stages of the blood coagulation system, activate platelets and directly act upon fibrinogen. These include pro-coagulant enzymes such as thrombin-like enzymes which clot fibrinogen (fibrinogenolytic), factor V activators, kininogenases and platelet aggregators, and anti-coagulant enzymes such as fibrinolytic enzymes, plasminogen activators and protein C activators [10].
A detailed understanding of the components of snake venoms is important both for acquiring a more complete understanding of the pathology of envenomation and also to aid in the development of improved treatments for snakebites. Moreover, several venom enzymes, including VVSPs have proved to have potential as therapeutics for various human haemostatic disorders [11]. Despite their high sequence similarity, VVSPs differ widely in their functions. Accelerated evolution [12], exon switching [13] and point mutations [14] have been reported to be involved in the generation of novel VVSPs and adaption to different geographical locations and available prey. Due to their physiological and medical importance, understanding of VVSP sequences, structures, functions and phylogenetic relationships represent research priorities. In this article, we report the collection of largest dataset of available VVSP sequences from public databases and literature and the detailed analysis of their sequence, structure, function and phylogenetic relationships.
Methodology
Collection of VVSP sequences:
Basic local alignment search tool (BLAST) [15] was used to search the NCBI protein database using several VVSP amino acid sequences. The NCBI and Swiss-Prot protein databases were searched with individual viper names and several keywords including serpentes, colubroidea, viperidae, viperinae and crotalinae to extract all VVSP sequences. Some published protein sequences, which had not been submitted to any of the sequence databases were collected directly from the literature. Several VVSP sequences were incomplete or contained internal deletions (longer than 10 amino acids), and were therefore excluded from our dataset. For each complete VVSP sequence the viper name, accession number, name (and alternative names if available) of the protein, experimentally determined functions and associated references were collected. Where two or more sequences had 100% sequence identity as determined by the EMBOSS pairwise alignment tool [16], duplicates were removed. Each sequence was allocated a reference code, determined using the viper’s scientific name followed by up to three letters representing the protein name (e.g. CDD-SP, where CDD represent the snake name, Crotalus durissus durissus and SP represent the protein name, serine protease). Where, the similar first letter of two or more species occurs within a genus, the second letter of species name was used in lower case (e.g. CAd-CR).
Sequence analysis:
Multiple sequence alignment analysis of all the VVSP sequences was performed using the ClustalW2 multiple sequence alignment tool [17]. To aid comparison of the VVSP sequences with other serine proteases, the sequence of bovine α- chymotrypsinogen (NCBI accession number: P00766) was included in the alignment. This enabled the commonly used chymotrypsinogen numbering pattern to be identified in the VVSP sequences. The alignment file was viewed using the Jalview-multiple sequence editor tool [18]. In some of the VVSP sequences (obtained from cDNA sequencing), the N-terminal signal and activation peptides were identified in comparison to previous literature and these were removed manually before the complete analysis. NetNGlyc tool was used to predict Nglycosylation sites, and compute pI/MW tool from Expasy bioinformatics portal was used to predict the isoelectric points and molecular weight of VVSPs. The number of Nglycosylation sites and isoelectric points of VVSPs in every snake were clustered using CIMminer software [19].
Phylogenetic analysis:
To generate the phylogenetic tree, sequences were aligned using ClustalW within MEGA 4 [20] using a gap opening penalty of 10 and a gap extension penalty of 0.1 for the initial pairwise alignment and a gap opening penalty of 3 and gap extension penalty of 1.8 for the multiple alignment and the Gonnet protein weight matrix. The phylogenetic tree was generated within MEGA 4 using the neighbour-joining method and the Jones-Taylor-Thornton substitution model. The bootstrap test was performed using 2000 replications. The sequence of bovine α-chymotrypsinogen was used as an outgroup.
Structural modeling:
Structural models of VVSPs were created using the IntFOLD server [21]. Models were obtained for each sequence using the structure of rat trypsin (PDB code: 1co9) as a best matched template. Models were visualised and compared with each other and the structures of bovine α-chymotrpysin (PDB code: 1YPH) and rat trypsin (PDB code: 1CO9) using PyMOL molecular graphics system, version 1.3 (DeLano Scientific).
Results and Discussion
Collection of VVSP sequences:
The collection of available VVSP sequences from the protein databases represented an initial step towards developing a comprehensive dataset of all VVSPs. BLAST was used to search the NCBI protein database using the amino acid sequences of several VVSPs in order to obtain all available sequences of VVSPs. In addition, the NCBI and Swiss-Prot databases were searched using list of viper names and several key words. There were several snake venom serine protease sequences from other than viperiade family members but since our main focus was on VVSPs, non-viper sequences were removed from the dataset. In total 196 complete sequences (after removing the duplicates and partial sequences) were obtained including 11 complete sequences extracted from relevant literature Table 1(see supplementary material). Of the 196 available proteins only a small proportion (49 out of 196 or 25%) of VVSPs has been experimentally characterised. Some (9) VVSPs have been shown to have more than one function and others have not been screened for more than one particular function of interest. Some VVSPs have been designated as thrombin-like enzymes or KN (which might mean kininogenase), but no information was available regarding the basis of this classification. For several available VVSP sequences, associated published article or information regarding respective functions and other characteristics were not available. Structural information for the VVSPs is currently limited with structures solved for only 5 (2.6% in total) VVSPs known.
Our comprehensive dataset of VVSPs has been acquired using semi-automated methods and to our knowledge, this is the most complete collection to date of VVSP sequences. The absence of some sequences from any of the sequence databases makes it impossible to acquire this completely automatically. The use of different naming conventions by various authors also makes it difficult to obtain the VVSP sequences using keyword searches. The VVSP sequences along with other serine protease sequences from reptiles (other than viper snakes and lizards) grouped together in BLAST analysis, indicating them to have higher sequence similarities to each other than to nonreptile serine proteases.
General characteristics of VVSP sequences:
To analyse the various features of VVSPs, all sequences were aligned together along with bovine α-chymotrypsinogen. Initial multiple sequence alignments of VVSPs demonstrated that many sequences occurred in precursor form, containing signal (18 amino acids) and activation peptides (6 amino acids) at the N-terminus. These were removed from all VVSP sequences and only the mature protein sequences were aligned for sequence analysis (Figure S1- this figure is available from authors). The bovine α-chymotrypsinogen-based sequence numbering was used throughout the study to allow easier discussion with non- VVSP sequences.
Activation of VVSPs:
Most of eukaryotic proteases (e.g. digestive enzymes) are synthesised as precursors and become mature enzymes at their target sites following the cleavage of signal/activation peptides. As snake venoms are synthesised from the venom glands and the venom facilitates the immobilization and digestion of prey, they are also synthesised as inactive zymogens with signal and activation peptides [12]. These signal and activation peptides are only present in the sequences which were obtained through cDNA sequencing and were absent in any of the VVSP sequences obtained through Edman N-terminus sequencing methods from the purified proteins. A previous study [21] reported that venom proteins are stored at an acidic pH in the lumen of main secretary gland where they are maintained in their zymogen state to prevent unwanted proteolytic activity in the venom gland. During venom extraction or a bite, the venom flows through the rostral portion of accessory gland which is rich in serous secretions (contain proteolytic enzymes) that result in activation of proteolytic enzymes [22].
Isoelectric point of VVSPs:
The isoelectric point of each VVSP sequence was predicted in order to analyse the distribution of VVSPs with varying characteristics within and among other snake venoms. The predicted isoelectric point of each VVSP varies between 4.6 and 9.4 (Figure 1). Depending upon the number of VVSP sequences known in each venom, the distribution of VVSPs vary. The majority of VVSPs present in all the snake venoms analysed lie between isoelectric points of 5 and 7 or 8 and 9. This analysis indicates that snakes produce a mixture of serine proteases with varying isoelectric points and these may result in alteration of their functional properties to adapt various substrates in diverse preys.
Figure 1.
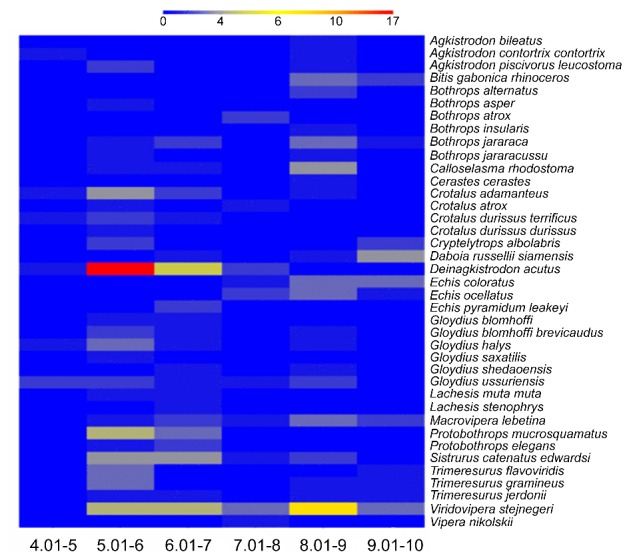
Number of VVSPs with different isoelectric points in each snake. The isoelectric point of all VVSPs was predicted and clustered together for every snake based on the number of VVSPs found with varying isoelectric points. Blue colour indicates the low number (0) and the red colour indicates highest number (17) of VVSPs.
N-glycosylation sites in VVSPs:
Since glycosylation in two of the VVSPs (DA-ASI and DA-ASII) has been demonstrated to affect the substrate specificity of these enzymes [23], the potential N-glycosylation sites were predicted for all VVSP sequences. The number of predicted Nglycosylation sites in VVSPs varies between 0 and 6 (Figure 2). This analysis indicates that although some (16) of VVSPs are not glycosylated, many of the VVSPs have one (68) or more (112) potential glycosylation sites. Glycosylation of VVSPs may represent a general feature that contributes to the determination of substrate specificity similar to DA-ASI and DA-ASII. Moreover, the addition of glycosylation moieties will increase the molecular weights of native VVSPs in actual venom. The difference in molecular weights of VVSP homologs in different species is well reported in literature [14].
Figure 2.
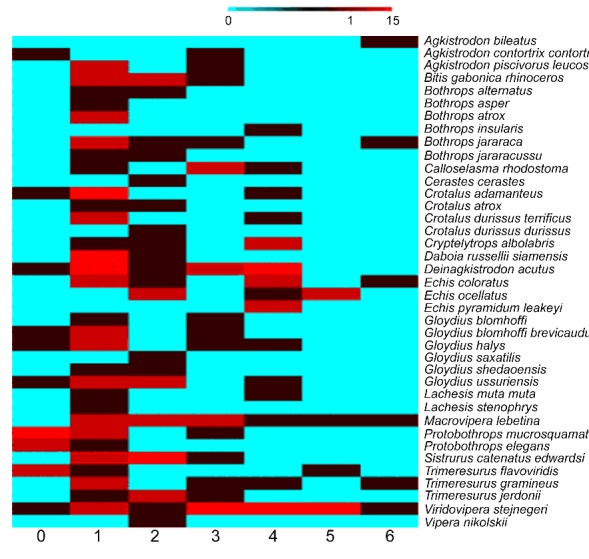
Number of VVSPs with different N-glycosylation sites in each snake. The potential N-glycosylation sites of all VVSPs were predicted and clustered together for every snake based on the number of VVSPs found with varying numbers of predicted sites. Blue colour indicates the low number (0) and the red colour indicates highest number (15) of VVSPs.
GWG motif:
The conversion of inactive zymogens into active enzymes is a key step to initiate the activity of several digestive and clotting enzymes at their target sites. This conversion is usually takes place by proteolytic cleavage at specific sites and by altering the structural fold of active enzymes. Fehlhammer et al. [24] reported that a GWG motif between residues 140 and 142 in chymotrypsinogen was an important site for the conversion of a flexible zymogen structure into a fixed rigid orientation in the activated chymotrypsin. This motif is absolutely conserved in all VVSPs analysed (Figure S1), suggesting that it may play roles in the conversion of inactive to active VVSPs.
C-terminal extension:
The sequence alignment shows that the length of most of the mature VVSPs is between 228 and 239 amino acid residues. Compared to chymotrypsinogen, all the VVSPs lack 15 amino acids at the N-terminus of the mature proteins which indicates the removal of signal and activation peptides. While most (179) of the VVSPs contain a C-terminal extension of 7 amino acids, some (17) contain smaller or larger extension (Figure S1). This C-terminus feature is not present in chymotrypsin, trypsin, thrombin and other known mammalian serine proteases, suggesting this to be a unique feature of VVSPs. This C-terminal extension contains a cysteine residue which may form a disulphide linkage with C91 in all the VVSPs to offer additional stability for maintaining their structures as determined for VSVPA [25].
Stability of VVSPs:
Salt bridge:
Salt bridges are usually formed between an acidic and a basic residue to stabilise the structure. A common salt bridge in trypsin and VVSPs was reported between the residues V/I16 and D194 [25, 26]. This is formed after the activation of the enzyme from its precursor form and is involved in stabilisation of the active site structure. All VVSPs have either valine or isoleucine at position 16 (except one VVSP, EP-SP1 which has a deletion at this position) and aspartate at position 194 (except VS-KN10 which has valine at this position) (Figure S1). Thus with the exception of these two VVSPs, all others are likely to form a salt bridge between the residues in these positions in order to increase the stability of the enzymes.
Disulphide linkages:
The trypsin-like serine proteases contain 10 cysteine residues which normally form 5 disulphide linkages. But in the experimentally analysed VVSPs 12 cysteine residues are found, and in the structure of VS-VPA they have been shown to form 6 disulphide linkages [25]. These are C22-C157, C42-C58, C91- C245e, C136-C201, C168-C182 and C191-C220. It is likely that similar disulphide bonds may form in all other VVSPs where these cysteine residues present at corresponding position. The disulphide linkage C91-C245e is a unique characteristic to VVSPs [25]. In VVSP sequence alignment (Figure S1), although 7 of the cysteine residues [C22, C136, C157, C168, C182, C191 and C220] are absolutely conserved in all VVSPs, 13 VVSPs have substitutions for one of the other 5 cysteine residues Table 2 (see supplementary material). C42 is substituted in 4 VVSPs (CC-CR, VS-KN5, VS-KN6 and VS-KN14); C58 is substituted in one (DA-TL4); C91 is present in all VVSPs except for two (BAIBH and GBB-SL); C245e was substituted in three VVSPs, EOSP2, CAd-SP5 and EC-SP6, and a deletion was found in EO-SP5 at this position. C201 was substituted in two VVSPs, DA-DK and DA-TL7. In some VVSPs, the C91 has been aligned at the positions of 90 or 92, but these form disulphide linkages as normal in modelled structures (data not shown). This analysis shows that in some of the VVSPs, specific disulphide linkages cannot be formed due to the substitutions. It is entirely not clear how these substitutions and subsequent loss of corresponding disulphide linkages would affect the architecture of the VVSP structures and their function. GBB-SL which lacks C91-C245e, however, has been functionally characterised to have α and β fibrinogenolytic activities [information obtained from the NCBI protein database].
Functional characteristics of VVSPs:
Catalytic triad:
Trypsin-like serine proteases share a catalytic triad which includes H57, D102 and S195 (bovine α-chymotrypsinogen numbering) [25]. These residues are conserved in majority of the VVSPs (Figure S1) and so they would be expected to be catalytically active. 21 VVSPs have substitutions for one or two of the catalytic triad residues. Substitutions to the catalytic triad residues have previously been observed in a small number of VVSPs [12, 14, 27, 28]. The majority of these sequences have been identified at transcript level only; prior to this study only four serine proteases (BG-RHIN2 & 3, BAI-BH and TJ-SPH) with catalytic triad substitutions have been identified within the venom of snakes [14, 28, 29]. Functional analysis of one of these proteins (TJ-SPH) has confirmed this to be functionally inactive [28], but, interestingly, another VVSP, bhalternin (BAI-BH) with substitutions at positions 57 (N) and 102 (T) has been shown to be functionally active [29]. It is therefore hard to conclude if these VVSPs would be functionally active or inactive with substitutions within the catalytic triad. The serine proteases with catalytic triad substitutions are commonly called serine protease homologues [28]. Table 3 (see supplementary material) shows the VVSPs with catalytic triad substitutions. The most frequently substituted residue within the catalytic triad is H57, which has been substituted by R in 15 sequences, by N in 3 sequences and by Q in one sequence. Ten of the sequences have substitutions for the catalytic S195; in five VVSPs this has been substituted by N, one protein has a S195P substitution, three have a S195T substitution and one has alanine at this position. In contrast, D102 is conserved in all the sequences except one, BAI-BH which has threonine at this position.
Structural models of these VVSPs show that they are generally similar to each other, consistent with the sequence similarity of the enzymes, but they differ at their catalytic site. For example, rhinocerases, BG-RHIN2 and 3 contain catalytic triad substitutions at positions 57 and 195, but show overall similarity to the predicted structures of other rhinocerases, BGRHIN4 and 5 which contain normal catalytic triad (Figure 3A). They show differences at their catalytic site (Figure 3B). D102 is at almost identical positions in rhinocerases 2 and 4, and bovine chymotrypsin. The catalytic serine is at a very similar position in rhinocerase 4 and chymotrypsin, and the substituted N in BG-RHIN2 is also similarly located. However the substitution of the long arginine side chain instead of histidine at position 57 in BG-RHIN2 has resulted in a significant change to the orientation of the side chain in the modelled structure (Figure 3B). Similarly other VVSPs with different amino acid substitutions show differences in their catalytic sites.:
Figure 3.
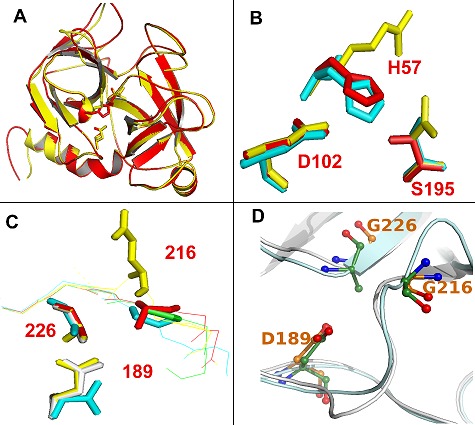
Structural models of VVSPs. Structural models of rhinocerases 2 (BG-RHIN2) and 4 (BG-RHIN4) were created using the IntFOLD server [17] using the structure of rat trypsin (PDB code: 1co9) as a template. (A) Schematic diagram showing the overall similarities in structure between BG-RHIN2 (yellow) and BG-RHIN4 (red). The side chain atom positions for the catalytic triad residues are included. (B) Detailed view of the amino acids corresponding to the catalytic triad residues in BGRHIN2 (yellow), BG-RHIN4 (red) and chymotrypsin (PDB code: 1yph; cyan). BG-RHIN2 has substitutions for the serine and histidine residues. (C) Detailed view of the main constituents of the S1 specificity pocket in BG-RHIN2 (yellow), BG-RHIN4 (red), chymotrypsin (cyan) and trypsin (pdb code: 1co9; green). In chymotrypsin these residues are: S189 at the base of the specificity pocket, with G216 and G226 at the sides. In trypsin D189 is at the base of the pocket, with G216 and G226. All images were generated using PyMOL. (D) Detailed view of the main constituents of the S1 specificity pocket in BG-RHIN2 (green) and protein C activator (PDB code: 2AIQ; purple).
Proteins with substitutions to the catalytic triad are present in many enzyme families; indeed it has been estimated that up to 15% of the members of all encoded enzyme families may have lost their catalytic activity [30]. Siigur et al. [12] suggested that the critical substitutions in the VVSP, ML-P2 from the venom of Macrovipera lebetina could be generated via trans-splicing of the primary gene transcript, exon-shuffling or unequal crossing over at the genome level. We have however, shown previously that these substitutions may have occurred at multiple levels [14]. Although one of the VVSPs with a catalytic triad substitution was proved to be functionally active [29], another was shown to be inactive [28]. No further VVSP of this nature has been functionally characterised. So it is not entirely clear if these proteins are functionally active in the venom.
In many cases in other biological systems, inactive homologues are believed to have acquired alternative functions, such as competing with and antagonising the active proteases, or otherwise regulating their function. Within invertebrates, serine protease homologues have been shown to be involved in various defence responses [31]. It has however, been suggested that some invertebrate serine protease homologues are unlikely to bind peptide substrates by a canonical protease-like mechanism, though other potential protein binding sites have been suggested [32]. Within snake venom, catalytically inactive phospholipase A2 such as myotoxins II and IV from the venom of Bothrops asper are known to act as toxins and are thought to bind to their target membrane substrates in order to reduce their permeability control and cause subsequent necrosis [33]. Corey et al. [34] reported that trypsin with a H57R substitution had 190000 fold lower activities than the wild type enzyme at pH 8.0 but had 38 fold higher activities than the wild type at pH 10.1. Clearly, experiments to determine the function of the serine protease homologues within snake venom need to be performed. It is possible that they may affect the physiology of victims or prey by binding irreversibly to substrates involved in blood coagulation and preventing their normal function.
Specificity of VVSPs:
The primary specificity pocket of serine proteases (also called as S1 binding site) accommodates the side chain of the P1 residue (the residue preceding the scissile bond) of substrates and confer specificity towards substrates. For example, chymotrypsin contains S189, G216 and G226 and is specific for the large hydrophobic side chains (such as F, Y and W) [35]. Trypsin and thrombin contain D189, G216 and G226 and are specific for positively charged amino acids such as R and K [35]. Elastase contains S189, V216 and T226 and is specific for small hydrophobic side chains like alanine [35]. In addition to the catalytic triad residues, there are also differences found in the residues present in the primary specificity pockets of several VVSPs (Figure S1). In total 161 VVSPs have D189, G216 and G/A226 and so they may confer specificity towards either or both positively charged amino acids such as R and K at P1 position of substrates similar to trypsin and thrombin.
The remaining 35 VVSPs, however, have different combinations of specificity pocket residues Table 4 (see supplementary material). Although seven VVSPs contain aspartate at position189, they have substitutions in the other two sites and thus, they might restrict entry for substrates. For example, in the venom of Bitis gabonica rhinoceros, BG-RHIN2 and 3 have D189, which might indicate specificity for basic residues but have E instead of G at position 216 and A instead of G at position 226 (Figure 3C & Figure 3D). These substitutions are likely to restrict access to the specificity pocket, thus the binding specificity of BG-RHIN2 and 3 is not clear. In contrast, two other VVSPs (BJa-HP3 and AP-PA), contain D189-R216-G226 in their specificity pocket. So the presence of arginine at position 216 may create a basic environment in the pocket. The functions of these VVSPs are further critical as they contain D at 189 and R at 216. A further two VVSPs (TG-SP3 and VS-KN8) contain D189-G216-N/V226 in their specificity pocket. Although they can confer specificity towards basic amino acids in their substrates, the presence of N or V at position 226 will have influence in their specificity.
The remaining 28 VVSPs contain substitution mainly for position 189 Table 4 (see supplementary material). Thus, these enzymes may not confer specificity for arginine at P1 position of the substrates. For example, 13 VVSPs have G189-G216-G226 in their specificity pocket. This is an unusual substitution in trypsin-like serine proteases, which is similar to the human kallikrein KLK9 whose specificity is unknown [36]. The substitution might be expected to increase the size of the specificity pocket; M. lebetina α and β-fibrinogenases (ML-AF and ML-BF) also have G189 [37] and Siigur et al. [38] have reported that ML-AF hydrolysed the Y16 – L17, F24 – F25 and F25 – Y26 bonds of the insulin B chain, suggesting that enzymes with G189 can cleave substrates with large hydrophobic residues at the P1 position. Some VVSPs such as BG-RHIN4 and 5 also have a G to A substitution at position 216 which may narrow the specificity pocket slightly. There are also other combinations of residues present in small number of VVSPs whose functions are unknown.
Comparison of the positions and orientations of residues in the S1 specificity pockets of BG-RHIN2 and 4 with those of chymotrypsin and trypsin suggest that, the bottom of the specificity pocket is very similar in BG-RHIN2 and trypsin, while the glycine in BG-RHIN4 is uncharged and protrudes even less into the pocket (Figure 3C). At position 226, the glycines and alanines in all enzymes are very similarly located. In contrast, the substitution of the large negatively charged glutamic acid at position 216 clearly has a significant effect on the S1 pocket in BG-RHIN2 when the modelled structure was compared to model of BG-RHIN4 (Figure 3C). When BGRHIN2 model was superimposed on the determined structure of protein C activator (AC-AN) [39], G217 of BG-RHIN2 is situated at the same position as G216 of AC-AN (Figure 3D). So it is not entirely clear if negatively charged glutamate or glycine would occupy this position in the real structure of BG-RHIN2. However the primary specificity pocket is not the sole determinant of specificity. For serine proteases involved in coagulation, the importance of additional regions of the structure e.g. exosites in recognition of substrates is becoming increasingly recognised [40]. Thus the precise specificity of these enzymes can only be determined experimentally. The potential role of exosites in binding to substrates also strengthens our suggestion above that the serine proteases homologues which lack the catalytic triad may still be capable of interfering with the coagulation cascade through exositemediated interactions.
Oxyanion hole:
A further interesting feature of VVSP sequences is that G193, which is generally highly conserved in serine proteases and is involved in the formation of oxyanion hole and in inhibitor binding [41], is substituted in 57 sequences: R in 25, A in 7, V in 6, F in 6, Q in 4, H in 3, C in 2, Y in 2, S in 1 and T in 1 VVSP (Figure S1). As for VS-VPA, a venom plasminogen activator from Trimeresurus stejnegeri, which has F at position 193 [41], bulky residues substituted for G193 may reduce the sensitivity of the proteins to inhibitors such as bovine pancreatic trypsin inhibitor as well as reduce their interaction with substrates. Similarly other residues including charged residues such as arginine and glutamine may influence the formation of oxyanion hole and inhibitor or substrate binding.
RGD motif:
The arginine-glycine-aspartate (RGD) motif acts as a cell adhesion signal and is present in cellular signal transduction proteins. It has also been reported that proteins containing RGD motifs may induce apoptosis [42]. Lee et al. [43] identified a RGD motif in brevinase (GB-BV) and suggested that it might be involved in cellular recognition and in inhibition of platelet aggregation through binding to integrin αIIbβ3. In our analysis we have identified the RGD motif at the same position as GBBV (between 192 and 194), in four other VVSPs: GB-HY, GH-H, GU-TC and GSh-SH (Figure S1). VVSPs containing the RGD motif could be involved in inducing cell death in order to cause destruction of cells in the host system. They may also inhibit platelet aggregation. As RGD motifs were found only in VVSPs from venoms of the Gloydius genus so far, this may be a unique feature of these snake venoms.
Phylogenetic analysis of VVSPs:
Phylogenetic analysis was performed to analyse the evolutionary and functional relationships of VVSP sequences. The phylogenetic tree generated for 196 VVSP sequences is shown in (Figure 4). The sequence of bovine α- chymotrypsinogen was displayed as an out-group in the tree. Wang et al. [44] generated a phylogenetic tree with 41 VVSP sequences using the Phylip-neighbor method. From their tree, three categories of VVSPs were identified, which they named kininogenases (KN), clotting enzymes (CL) and plasminogen activators (PA). These names described the known functions of some members of each of the categories. They also suggested that the KN category might be divided into 2 subcategories, with one representing GBH and another representing PMM1- PMM5 and that this division might become more apparent if more sequences, particularly from true vipers were added. This classification was not apparent in our analysis with 196 sequences, which cluster together in 3 major groups (named I, II and III).
Figure 4.

Phylogenetic tree of VVSPs. 196 VVSPs sequences from were included for the generation of a phylogenetic tree together with bovine α-chymotrypsinogen (NCBI accession number: P00766) as an out-group. The alignment was generated using ClustalW within MEGA 4 using a gap opening penalty of 10 and a gap extension penalty of 0.1 for the initial pairwise alignment, gap opening penalty of 3 and gap extension penalty of 1.8 for the multiple alignment and the Gonnet protein weight matrix. The phylogenetic tree was generated from this within MEGA 4 using the neighbour-joining method and the Jones- Taylor-Thornton substitution model. The bootstrap test was done using 2000 replications. The VVSPs from true vipers (viperinae sub family) are shown in red. # and * indicate the sequences with catalytic triad and specificity pocket substitutions respectively. The experimentally determined functions of VVSPs are shown in green letters with short codes; AF- α fibrinogenase, BF- β fibrinogenase, ABF- αβ fibrinogenase, KN- kininogenase, CPI- capillary permeability increasing enzyme, FVA- factor V activator, FXIIIA- factor XIII activator, CE- clotting enzyme, PA- plasminogen activator, PLA- platelet activator, ABGF- αβγ fibrinogenase, PTAprothrombin activator, FXA- factor X activator, FVIIIA- factor VIII activator, FVIIA- factor VII activator, PCA- protein C activator, FIXA- factor IX activator and HL- haemolytic enzyme
The sequences from true vipers (viperinae subfamily) are mostly clustered together in group II and III (shown as red letters in Figure 4). Moreover, the VVSPs with catalytic triad substitutions (shown as # in the Figure 4) cluster together in group III accept two VVSPs which cluster in I and II. Similarly, the sequences with substituted primary specificity pocket are clustered in groups I and II, with an exception of one VVSP which cluster in group III (shown as * in the Figure 4). This suggests that the VVSPs from true vipers differ evolutionarily from the pit vipers. Moreover, the evolution of VVSPs with substitutions in the catalytic triad and specificity pocket is not a random process in any particular snake; instead it might be a controlled mechanism to develop novel VVSPs with differing characteristic features.
The experimentally determined functions of some VVSPs are indicated (as green letters in Figure 4) in the tree. Since majority of the functionally characterised VVSPs are capable of performing more than one function, the classification of VVSPs based on their function is difficult. Moreover, several VVSPs have been characterised only for the functions of interest and thus, they were not screened for other functions which they may perform. This analysis indicates that the classification of VVSPs based on some of the functionally characterised VVSPs as previously reported [44], may not appropriate with all the available VVSPs. For example, the VVSPs with kininogenase activities are clustered in all three groups in the tree. Similarly the coagulant enzymes such as α-fibrinogenase (AF), β- fibrinogenase (BF) and αβ-fibrinogenase (ABF) are present randomly in all clusters. Some enzymes such as plasminogen or factor V activators are clustered together in a particular group in the tree, although only a very small number of VVSPs with those functions are identified to date. Moreover, the number of VVSPs performing additional functions such as platelet aggregator, factor VIII/X/XIII activator, an enzyme increases capillary permeability and albuminolytic/ myonecrotic/ heamolytic enzymes are limited and thus it is not apparent to predict the functions of other VVSPs which cluster together with these enzymes.
Conclusion
To our knowledge this study represents the most complete information about VVSPs available to date and this will serve as a dataset for further analysis in these enzymes. VVSPs share several sequence features such as catalytic triad and disulphide linkages with other common serine proteases for example, chymotrypsin, trypsin and thrombin. Some features, such as the C-terminal extension of 7 residues and an additional disulphide linkage, are unique to VVSPs. However, within the VVSP family there are many sequence variations which might be responsible for the variation in specificity of these enzymes. The phylogenetic analysis of VVSPs shows three clusters of sequences and the classification of these enzymes based on only a few functionally characterised enzymes may not be appropriate with a larger dataset. Our analysis showed that the naming or prediction of function for each cluster may not be possible as previously suggested when we include more sequences as most of them are of unknown functions and some of them are only partially characterised. A greater level of functional characterisation is needed to obtain more complete functionally known VVSPs for better understanding of their sequence, function and evolutionary relationships. Posttranslational modification predictions showed that most of the VVSPs were predicted to be N-glycosylated. The functions of these modifications in VVSPs are yet to be clearly identified but, in several other proteins it has been shown that glycosylation affects the function of the protein. Structural analysis of VVSPs has helped to identify the functionally important residues; catalytic triad, primary specificity pocket and disulphide linkages. However only 5 determined structures of VVSPs are available to date and thus more crystallisation work should be performed to obtain more structures. The dataset presented here with complete information of all known VVSPs to date will form a basis for further research to explore more about the sequence, structure, function and phylogenetic relationships of these enzymes in order to develop improved therapeutics for the treatment of snakebites.
Supplementary material
Footnotes
Citation:Vaiyapuri et al, Bioinformation 8(16): 763-772 (2012)
References
- 1.J White. Ther Drug Monit. 2000;22:65. doi: 10.1097/00007691-200002000-00014. [DOI] [PubMed] [Google Scholar]
- 2.JP Chippaux, et al. Bull World Health Organ. 1998;76:515. [PMC free article] [PubMed] [Google Scholar]
- 3.A Kasturiratne, et al. PLoS Med. 2008;5:e218. doi: 10.1371/journal.pmed.0050218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.S Vaiyapuri, et al. PLoS Negl Trop Dis. 2010;4:e796. doi: 10.1371/journal.pntd.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.S Vaiyapuri, et al. PLoS One. 2010;5:e9687. doi: 10.1371/journal.pone.0009687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.S Vaiyapuri, et al. J Biol Chem. 2012;287:26235. doi: 10.1074/jbc.M112.381483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.SD Aird, et al. Toxicon. 2002;40:335. [Google Scholar]
- 8.SM Serrano, RC Maroun. Toxicon. 2005;45:1115. [Google Scholar]
- 9.J Meier, K Stocker. Crit Rev Toxicol. 1991;21:171. doi: 10.3109/10408449109089878. [DOI] [PubMed] [Google Scholar]
- 10.FS Markland, et al. Toxicon. 1998;36:1749. doi: 10.1016/s0041-0101(98)00126-3. [DOI] [PubMed] [Google Scholar]
- 11.RP Atkinson, et al. Cerebrovasc Dis. 1998;1:23. [Google Scholar]
- 12.E Siigur, et al. Gene. 2001;263:199. [Google Scholar]
- 13.R Doley, et al. BMC Evol Biol. 2009;9:146. doi: 10.1186/1471-2148-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.S Vaiyapuri, et al. PLoS One. 2011;6:e21532. doi: 10.1371/journal.pone.0021532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.SF Altschul, et al. J Mol Biol. 1990;215:403. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 16.P Rice, et al. Trends Genet. 2000;16:276. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 17.MA Larkin, et al. Bioinformatics. 2007;23:2947. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 18.AM Waterhouse, et al. Bioinformatics. 2009;25:1189. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.U Scherf, et al. Nat Genet. 2000;24:236. [Google Scholar]
- 20.S Kumar, et al. Brief Bioinform. 2008;9:299. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DB Roche, et al. Nucleic Acids Res. 2011;39:W171. [Google Scholar]
- 22.SP Mackessy, LM Baxter. Zoologischer Anzeiger. 2006;245:174. [Google Scholar]
- 23.Z Zhu, et al. Acta Crystallogr D Biol Crystallogr. 2003;59:547. doi: 10.1107/s0907444902023375. [DOI] [PubMed] [Google Scholar]
- 24.H Fehlhammer, et al. J Mol Biol. 1977;111:415. doi: 10.1016/s0022-2836(77)80062-4. [DOI] [PubMed] [Google Scholar]
- 25.MA Parry, et al. Structure. 1998;6:1195. [Google Scholar]
- 26.I Massova, et al. Bioorganic & Medicinal Chemistry Letters. 1997;7:3139. [Google Scholar]
- 27.IM Francischetti, et al. Gene. 2004;337:55. [Google Scholar]
- 28.J Wu, et al. Toxicon. 2008;52:277. doi: 10.1016/j.toxicon.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 29.O Costa Jde, et al. Toxicon. 2010;55:1365. [Google Scholar]
- 30.B Pils, J Schultz. J Mol Biol. 2004;340:399. doi: 10.1016/j.jmb.2004.04.063. [DOI] [PubMed] [Google Scholar]
- 31.J Ross, et al. Gene. 2003;304:117. [Google Scholar]
- 32.K Fischer, et al. J Mol Biol. 2009;390:635. doi: 10.1016/j.jmb.2009.04.082. [DOI] [PubMed] [Google Scholar]
- 33.Y Angulo, B Lomonte. Toxicon. 2009;54:949. doi: 10.1016/j.toxicon.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 34.DR Corey, CS Craik. J Am Chem Soc. 1992;114:1784. [Google Scholar]
- 35.H Czapinska, J Otlewski. Eur J Biochem. 1999;260:571. doi: 10.1046/j.1432-1327.1999.00160.x. [DOI] [PubMed] [Google Scholar]
- 36.AM LeBeau, et al. Biochemistry. 2009;48:3490. doi: 10.1021/bi9001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.E Siigur, et al. Thromb Haemost. 2003;89:826. [PubMed] [Google Scholar]
- 38.J Siigur, et al. Procedia Chemistry. 2010 [Google Scholar]
- 39.MT Murakami, RK Arni. J Biol Chem. 2005;280:39309. doi: 10.1074/jbc.M508502200. [DOI] [PubMed] [Google Scholar]
- 40.S Krishnaswamy. J Thromb Haemost. 2005;3:54. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- 41.S Braud, et al. J Biol Chem. 2000;275:1823. doi: 10.1074/jbc.275.3.1823. [DOI] [PubMed] [Google Scholar]
- 42.CD Buckley, et al. Nature. 1999;397:534. doi: 10.1038/17409. [DOI] [PubMed] [Google Scholar]
- 43.JW Lee, et al. Biochem Biophys Res Commun. 1999;260:665. doi: 10.1006/bbrc.1999.0977. [DOI] [PubMed] [Google Scholar]
- 44.YM Wang, et al. Biochem J. 2001;354:161. doi: 10.1042/0264-6021:3540161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.