

Abstract
Bestrophin, an integral membrane protein existing in basolateral region of the retina is a propitious target for drug discovery. Mutations in the Bestrophin protein cause Best Vitelliform Macular Dystrophy (BVMD) leading to retinal damages and loss of visual acuity. Owing to the lack of three dimensional structure and related structural homologs in the protein data bank, we modeled the bestrophin protein using Robetta ab initio method. Further, no treatment is available for the disease. In this situation, anthocyanins from natural sources are reported to combat retinal damages. Hence, we identified anthocyanins from Syzygium cumini fruit skin using Electrospray Ionization tandem mass spectrometry. These compounds were docked into the predicted bestrophin model to study the interactions within the active site. The results may provide a valuable insight into the structure of bestrophin and efficacy of anthocyanins in molecular docking studies.
Abbreviations
PTP - Putative transmembrane proteins, VMD - Vitelliform macular dystrophy, BVMD - Best's vitelliform macular dystrophy, RPE - Retinal pigment epithelium, ESI-MS/MS - Electrospray Ionization Tandem Mass Spectrometry, UNIPROT - Universal Protein Resource, PSIPRED - Protein secondary structure prediction, TMH - Transmembrane Helices, SCFS - Syzygium cumini fruit skin DP - Declustering Potential IFD - Induced Fit Docking.
Keywords: Bestrophin, Ab initio, Anthocyanin, Induced Fit Docking
Background
PTP's are attributed a cardinal role in numerous physiological functions [1]. Owing to difficulty in crystallizing, computational methods are employed to predict the PTPs as the basis for structure based drug design.
Bestrophin being a PTP is encoded in the gene VMD2. Mutations in the VMD2 gene causes BVMD, an inherited progressive macular dystrophy, first identified in 1905 by a German ophthalmologist Frederich Best. BVMD advances with juvenile onset causing loss of visual acuity due to atrophic macular changes or choroidal neovascularization associated with sub retinal hemorrhages and fibrosis [2]. Early stages of the disease are characterized by abnormal depositions of lipofuscin-like material at the level of the RPE classically resembling an egg yolk [3]. Disintegration of the central yellow lesions progressively leads to vision loss at a later stage. The elemental cause of the Best disease is the BEST1 gene (previously called VMD2, currently as hbest1), identified in 1998 [4].
Determining the three-dimensional structures of PTPs remains a challenge, although they constitute 15-30% of the entire genome. Bestrophin being a plasma membrane protein has a molecular weight of 68 kDa and consists of 585 amino acids [5]. Computationally, bestrophin is predicted as a transmembrane protein with four membrane-spanning α helical domains, while presence of atleast five helices are reported by few studies [6]. An ecumenical outcome suggests that Bestrophin functions as a chloride ion channel [7]. The homology modeling of Asp-rich domain of hbest1 identified two calcium binding sites, yet the complete structural details of the transmembrane protein are not available [8]. Obtaining the protein structure is crucial to understand protein function which eventually leads to drug designing [9, 10]. Hence the hbest1 protein was modeled using the Robetta web server (http://robetta.bakerlab.org) [11].
Natural compounds are tangible as therapeutic drug targets. More significantly, anthocyanins belonging to the flavonoid group are beneficial in curing visual acuity [12]. Anthocyanins are water-soluble pigments, pre-eminent in a variety of plants mainly imparting colour to flowers and fruits [13]. Innumerable reports exist for the rich resource of anthocyanins in the fruits of Syzygium cumini (L.) Skeels (Black Plum) [14, 15]. Our study also involves quantification of anthocyanins from the fruit skin by ESI – MS/MS Mass Spectrometer. Small molecular structures of the separated anthocyanins were obtained from publicly available chemical databases and used for molecular docking with hbest1. According to the literature, Resveratrol and Niflumic acid found to be useful in combating retinal damages was also used in our docking studies [16].
Methodology
Retrieval of Target Sequence:
The FASTA format of human Bestophin-1 (hbest1) with Accession number O76090 was obtained from the UNIPROT, database [17]. The protein sequence contains 585 amino acid residues.
Secondary structure Prediction:
The secondary structure of hbest1 was predicted using PSIPRED v3.0 [18, 19]. Neural nets are used to convert PsiBlast profile data to secondary structure propensities. A putative secondary structure is obtained for each residue associated with a confidence value for the prediction.
Prediction of Transmembrane regions:
The predictors available are inaccurate in predicting the ends of TMHs, or TMHs of unusual length [20]. MemBrain, a predictor based on machine learning approach was used to predict the TMHs of hbest1.
Ab Initio modeling:
Pertaining to lack of suitable structural template for hbest1, we resorted to Ab initio modeling of the hbest1 using Robetta, a full chain Protein prediction server. The primary sequence was submitted to the server, and the generated models were received by email.
Structure Validation:
Five structures were obtained from Robetta server. The qualities of the models were evaluated using the ProQ webserver [21]. The models were validated using PROCHECK program [22].
Experimental:
Fruit sample:
Mature fruits of Syzygium cumini were collected from Sirumalai hills of Dindigul, Tamilnadu, India and identified by Dr P. Eganathan, M. S. Swaminathan Research Foundation, Chennai. Rotten, damaged fruits were removed; skin was peeled from fruits without pulp and shade dried under room temperature (31°C) until complete disappearance of moisture. Dried skin was stored for further analysis.
Solvents and Reagents:
Analytical grade chemicals and solvents purchased from Sigma Aldrich (Saint Louis, MO) were used in the present investigations.
Preparation of extract for ESI- MS/MS analysis:
SCFS was ground to a fine powder using a Thomas Wiley Machine (Model 5 USA) at room temperature. Subsequently, 50g of powdered plant material was extracted with 1.5 L methanol using soxhlet apparatus at 65°C for 4 hours consecutively. The solvent was removed in vacuo using a Buchi Heating Bath (B-490) rotavapor to yield dried methanol (purple color) extract. The extract was filtered and evaporated to dryness in a vacuum at 40°C with a rotary evaporator.
ESI - MS/MS Analysis:
Crude methanol extract of SCFS was diluted with methanol and filtered with 0.22μm nylon membrane filter and subjected to ESI-MS/MS analysis. Anthocyanin identification was performed on a 3200 QTRAP instrument (ABSciex Instruments, Singapore) equipped with Linear Ion Trap Quadrupole Mass Spectrometer and electrospray ionization (ESI) source. Mass parameters DP was adjusted to get the maximum sensitivity. Data was generated by Analyst 1.4.1 software. The MS-MS conditions are : positive ion mode; gas (N2), curtain gas was set to 15 psi, heater gas and nebulizer gas were set to 10 psi and source temperature maintained at ambient.The positively identified compounds (cyanidin,petunidin,malvidin) were subjected to MS-MS analysis to study the fragment patterns and were found to match with that of the earlier reported compounds.
Docking:
The active site residues are Cys69, Cys42, Cys23, Phe80, Phe84, Val86, Pro77, Leu82, Tyr85, Gly83, Arg92, Trp94, Trp93 [7, 23]. Cyanidin 3, 5 Diglucoside, Malvidin 3, 5 diglucoside Petunidin 3, 7 diglucoside were docked with hbest1. In addition, other compounds included in docking were peonidin, delphinidin (other anthocyanins). Niflumic acid [24] and resveratrol [25] were included as positive controls in our docking studies for standardization.
Preparation of protein and ligands:
Glide (Grid-based Ligand Docking with Energetics) software, developed by Schrodinger running on Red Hat Enterprise Linux 5 (RHEL 5) workstation, was used for Protein, Ligand preparation and Induced Fit Docking.
The hbest1 structure was prepared with Protein Preparation module Wizard of Glide software. Water molecules were removed, the hydrogens atoms were added to the protein and all atom force filed (OPLS -2001) charges and atom types were assigned.
The CID files of the ligands Cyanidin 3,5 Diglucoside (CID 441688), Delphinidin 3,5 glucoside (CID 10100906), Malvidin 3,5 diglucoside (CID 441765), Petunidin 3,7-diglucoside (CID 44256973), Peonidin 3,5-diglucoside (CID 44256843) Niflumic acid (CID 4488), Resveratrol (CID 445154) were acquired from the NCBI Pubchem database [26]. Using the Impact module of glide the ligands were minimized with 1000 cycles of steepest gradient and 5000 cycles of conjugate gradient.
IFD protocol:
Induced fit docking combines Glide and Prime modules to arrive at accurate prediction of ligand binding modes and concomitant structural changes in the receptor [27]. Systematic and Simulation methods are adopted by glide for searching poses and ligand flexibility. Incremental construction for searching is employed by the systematic method, with Gscore being the empirical scoring function [28]. The calculation of GScore in Kcal/mol is: G-Score = H bond + Lipo+ Metal + Site + 0.130 Coul + 0.065vdW – Bury P – RotB. Where Hbond= Hydrogen bonds, Lipo = hydrophobic interactions, Metal – metal binding term, Site = Polar interactions in the binding site, vdW = Vander Waals forces, Coul = coulombic forces, Bury P= penalty for buried polar group, RotB= freezinf rotatable bonds. The prepared protein was docked with the minimized ligands. The active sites in the protein hbest1 were selected to be docked with the ligand. IFD was performed and best conformations were selected based on Glide Score, Glide energy, and Glide emodel scores.
Hydrophobic interactions:
Ligplot was used for analyzing the hydrophobic contacts of the protein and ligand [29]. The best pose for each ligand was submitted to Ligplot server and results obtained.
Results & Discussion
The percentage of secondary structures predicted by PSIPRED showed that 35.90% of the total structures were alpha helices, 3.07% were beta sheets and 59.48% were random coils within the target sequence. The protein was predicted to have 16 helices, 18 coils and 4 beta sheets (Figure 1A). The percentage of coils is more than the helices in the sequence. The C-terminal region of the hbest1 is composed of about 150 residues indicating the presence of a randomly coiled region. The Nterminal region, predicted to have signal peptide region has also a coiled region. Secondary structure prediction provides valuable information about the content of the protein which provides insights into the tertiary structure of hbest1.
Figure 1.
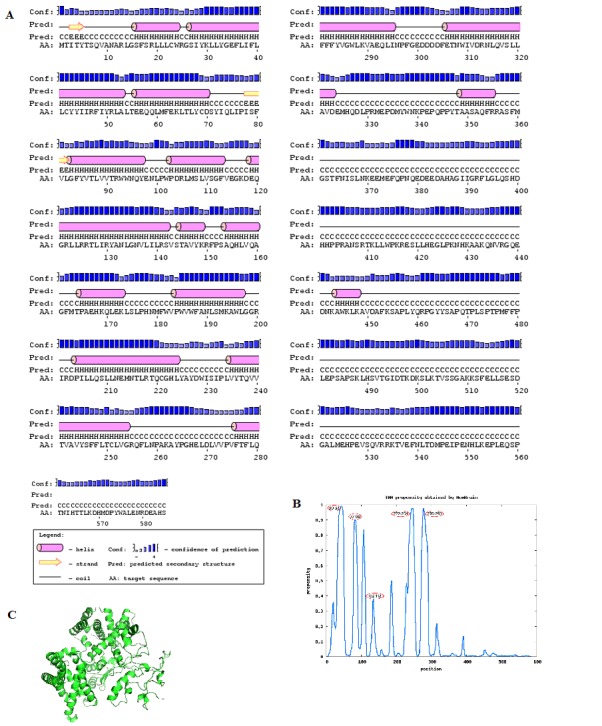
(A) Secondary structure prediction of hbest1 sequence using PSI-PRED server; (B) Graph representing propensities of TM helices. Numbers circled in red are the positions of TM helices; (C) Robetta modeled structure of hbest1
Prediction of TMHs in helical membrane proteins provides valuable information about the protein topology when the high resolution structures are not available. The predictors available are inaccurate in predicting the ends of TMHs, or TMHs of unusual length [20]. The prediction accuracy of MemBrain is 97.9%. N-terminal signal peptides were also detected. Five Transmembrane helices were predicted by MemBrain (Figure 1B).
The models obtained for hbest1 from Robetta server were evaluated by the ProQ program with the following results: model-1 (ProQ-LG=1.267, ProQ-MX=0.080), model-2 (ProQLG= 1.088, ProQ-MX=0.075), model-3 (ProQ-LG=1.171, ProQMX= 0.061), model-4 (ProQ-LG=1.852, ProQ-MX=0.062) , model- 5 (ProQ-LG=2.310, ProQ-MX=0.104). The cutoffs to finalize the best model are ProQ-LG > 1.5 or ProQ-MX >0.1 Model-5 is in complete agreement with the cutoffs providing a good modeled structure (Figure 1C).
Validation of the model by PROCHECK presented a Ramachandran plot analysis rendering 98.1% residues in the most favoured regions, 1.9% in the additionally allowed regions indicating the efficacy of Ab initio modeling. Secondary structural elements of the predicted hbest1 were found to be almost similar to the PSIPRED server results of the primary sequence.
Docking studies suggest the ability of the three anthocyanins to bind to active and additional active sites around hbest1. The binding poses and interactions were analyzed by Glide Table 1 (see supplementary material) & (Figure 2). The glide energy, glide score and glide emodel score for Cyanidin 3, 5 glucoside were -64.34 Kcal/mol and -11.80 Kcal/mol -71.60 Kcal/mol respectively. The compound interacts with Leu 82, Gln59, Arg105, and Arg47 that are the active sites within 20 Å distance of the literature cited residues. Hydrophobic contacts are with Phe82, Phe48, and Thr66. Malvidin 3, 5 diglucoside generated glide energy of -71.61 kcal/mol, glide score of -5.15 Kcal/mol and glide emodel score of -92.42 Kcal/mol. Interacting residues are Arg92, Arg461, Arg534, and Thr 87. Arg92 is the active site in interaction with Malvidin. Residues Ile129, Thr 91, Pro152, Asn 136 are in the hydrophobic pockets of docked structure. Petunidin 3, 7 diglucoside provided glide energy of -68.39 Kcal/mol, a glide score of -7.98 Kcal/mol and glide emodel score of -94.47 Kcal/mol. Asn136, Thr87, Arg461, are the residues in interaction besides Arg92 and Trp94 that are the two active sites involved in the interactions. Hydrophobic interactions comprise of Pro152 and Val90. Peonidin 3, 5 diglucoside resulted in a glide energy of -58.82 Kcal/mol, glide score of -8.47 Kcal/mol and glide emodel score of -85.17 Kcal/mol. Hydrogen bond interactions were exhibited with Asn133, Arg 534, and Gln460. Trp94, Val90, Pro406 residues were found in hydrophobic contacts. A glide energy of -77.17 Kcal/mol, glide score of -9.18 and a glide emodel score of - 107.57 Kcal/mol was generated by delphinidin 3,5 diglucoside. Hydrogen bond interactions include Gly159, Glu382, Ile49, Arg461, and Gln 96, while hydrophobic interactions involve active sites at Arg92, and Trp93. All anthocyanins have favourable binding potential with the involvement of the active site or neighboring residues.
Figure 2.
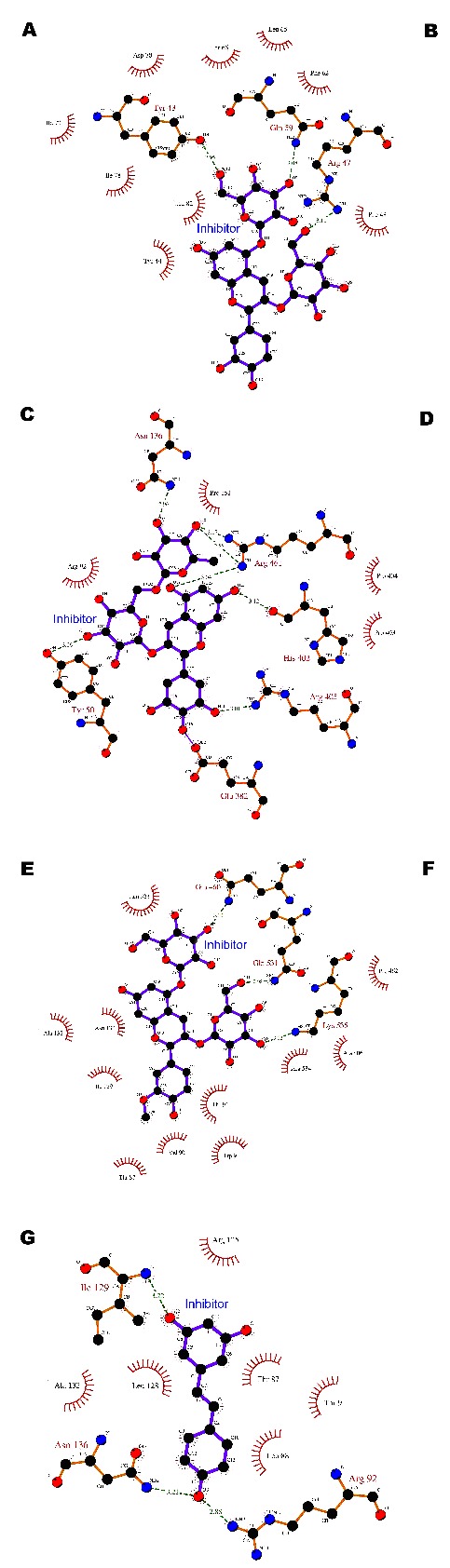
Protein-ligand interaction 2D map of hbest1 and inhibitors (A-G) using Ligplot diagram. Green dotted lines are Hydrogen bond interactions and red semicircles are amino acid residues of the protein showing hydrophobic interactions. (A) cyanidin 3,5 diglucoside; (B) Malvidin 3,5 diglucoside; (C) Petunidin 3, 7 diglucoside; (D) Delphidin 3, 5 diglucoside; (E) Peonidin 3, 5 diglucoside; (F) Niflumic acid; (G) Resveratrol.
Resveratrol rendered glide energy of -30.89 Kcal/mol, glide score of -8.25 Kcal/mol and glide emodel score of -48.71 with one interaction at Tyr131. Similarly the glide energy for Niflumic acid was -38.19 Kcal/mol, glide score of -8.80 and glide emodel score being -49.34 Kcal/mol. Niflumic acid has three interactions including an active site interaction at Arg92.
ESI-MS/MS, an accomplished method [30] for determining anthocyanins shows the presence of cyanidin, petunidin, malvidin in the methanolic extract of SCFS (Figure 3). In accordance to earlier studies [31, 32], the chromatographic profile displays the domination of anthocyanins in the sample. The identified compounds were Malvidin diglucoside at peak 655, Cyanidin 3, 5 diglucoside at peak 611 and, peak 641 corresponds to Petunidin 3, 7 diglucoside. Anthocyanins from other berries, amenable for oral intake, are reported to cure visual acuity [33]. Our study indicates the potential of the three anthocyanins as inhibitors for BVMD, whereas, pronounced results are not obtained with other compounds. The inherent capability of anthocyanins to protect the retinal pigment epithelium from age related macular degeneration has been reported [34]. The tangibility of anthocyanins as antioxidants has been substantiated by many authors [33, 36]. In conclusion, we imply that anthocyanins are safe [37] to use for BVMD and similar retinopathies.
Figure 3.
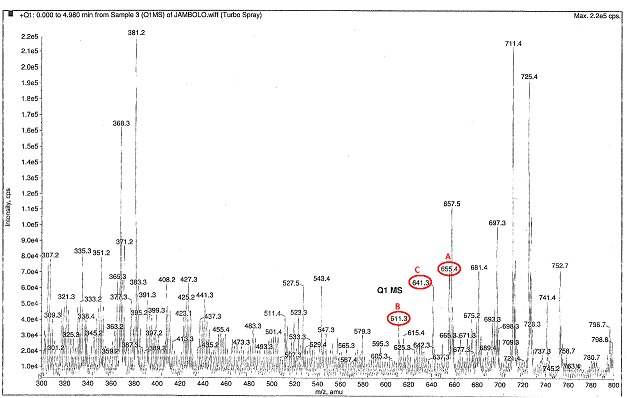
ESI-MS/MS chromatographic profile of anthocyanins separated from Szygium cumini fruit skin. Compounds circled in red are: (A) Malvidin 3, 5 diglucoside, (B) Cyanidin 3, 5 diglucoside and (C) Petunidin 3, 7 diglucoside.
Conclusion
Mutations in bestrophin causes age related macular degeneration eventually leading to loss of central vision. Currently no specific drug is available. Anthocyanin compounds are known to exert a positive effect in the treatment of visual acuity. Our present work provides an understanding into the structure of hbest1, identification of anthocyanins in the Syzygium cumini fruit skin, and docking of hbest1 with the compounds. The efficacy of anthocyanins demonstrates that natural compounds may serve as ideal therapies for diseases affecting the retinal pigment epithelium.
Supplementary material
Acknowledgments
The authors thank Bioinformatics Infrastructure Facility (DBTBIF), University of Madras for providing computational facilities.
Footnotes
Citation:Priya et al, Bioinformation 8(16): 742-748 (2012)
References
- 1.Y Arinaminpathy, et al. Drug Discov Today. 2009;14:1130. doi: 10.1016/j.drudis.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.F Krämer, et al. Eur J Hum Genet. 2000;8:286. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- 3.A Marquardt, et al. Hum Mol Genet. 1998;7:1517. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- 4.K Petrukhin, et al. Nat Genet. 1998;19:241. [Google Scholar]
- 5.K White, et al. Hum Mutat. 2000;15:301. [Google Scholar]
- 6.T Tsunenari, et al. J Biol Chem. 2003;278:41114. doi: 10.1074/jbc.M306150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.H Sun, et al. Proc Natl Acad Sci. 2002;9:4008. [Google Scholar]
- 8.A Kranjc, et al. PLoS One. 2009;4:e4672. doi: 10.1371/journal.pone.0004672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D Baker, A Sali. Science. 2001;294:93. [Google Scholar]
- 10.J Skolnick, et al. Nat Biotechnol. 2000;18:283. [Google Scholar]
- 11.D Chivian, et al. Proteins. 2003;53:524. [Google Scholar]
- 12.H Matsumoto, et al. J Agric Food Chem. 2001;49:1546. doi: 10.1021/jf001246q. [DOI] [PubMed] [Google Scholar]
- 13.JM Kong, et al. Phytochemistry. 2003;64:923. [Google Scholar]
- 14.JM Veigas, et al. Food Chemistry. 2007;105:619. [Google Scholar]
- 15.F Aqil, et al. Nutr Cancer. 2012;64:428. doi: 10.1080/01635581.2012.657766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arreola Jorge, Pérez‐Cornejo Patricia. Adv Mole Cell Biol. 2006;38:181. [Google Scholar]
- 17.A Bairoch, R Apweiler. Nucleic Acids Res. 2000;28:45. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.K Bryson, et al. Nucleic Acids Res. 2005;33:W36. [Google Scholar]
- 19.DT Jones. J Mol Biol. 1999;292:195. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 20.H Shen, JJ Chou. PLoS One. 2008;3:e2399. doi: 10.1371/journal.pone.0002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.B Wallner, A Elofsson. Protein Sci. 2003;12:1073. doi: 10.1110/ps.0236803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.RA Laskowski, et al. J Appl Cryst. 1993;26:283. [Google Scholar]
- 23.HC Hartzell, et al. Physiol Rev. 2008;88:639. doi: 10.1152/physrev.00022.2007. [DOI] [PubMed] [Google Scholar]
- 24.Z Qu, HC Hartzell. J Biol Chem. 2001;276:18423. doi: 10.1074/jbc.M101264200. [DOI] [PubMed] [Google Scholar]
- 25.A Pintea, et al. J Ocul Pharmacol Ther. 2011;27:315. doi: 10.1089/jop.2010.0144. [DOI] [PubMed] [Google Scholar]
- 26. http://pubchem.ncbi.nlm.nih.gov/
- 27.W Sherman, et al. J Med Chem. 2006;49:534. [Google Scholar]
- 28.RA Friesner, et al. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 29.AC Wallace, et al. Protein Eng. 1995;8:127. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 30.JI Nakajima, et al. J Biomed Biotechnol. 2004;5:241. doi: 10.1155/S1110724304404045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.ES de Brito, et al. J Agric Food Chem. 2007;55:9389. doi: 10.1021/jf0715020. [DOI] [PubMed] [Google Scholar]
- 32.L Li, et al. J Agric Food Chem. 2009;57:826. doi: 10.1021/jf803407q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.PH Canter, E Ernst. Surv Ophthalmol. 2004;49:38. doi: 10.1016/j.survophthal.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 34.PE Milbury, et al. Invest Ophthalmol Vis Sci. 2007;48:2343. doi: 10.1167/iovs.06-0452. [DOI] [PubMed] [Google Scholar]
- 35.JM Kong, et al. Phytochemistry. 2003;64:923. [Google Scholar]
- 36.J Dai, RJ Mumper. Molecules. 2010;15:7313. doi: 10.3390/molecules15107313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D Bagchi, et al. Mol Cell Biochem. 2006;281:197. doi: 10.1007/s11010-006-1030-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.