

Abstract
Engineered proteins are attractive affinity scaffolds for molecular imaging and drug delivery. Although exquisite binding specificity and affinity can be engineered, many proteins exhibit off-target uptake, particularly in the kidneys and liver, from physiologic effects. We quantified the ability to alter renal and hepatic uptake via hydrophilic and charge mutations. As a model protein, we used the 10th type III domain of human fibronectin, which has been engineered to bind many targets and has been validated for molecular imaging. We screened rational mutants, identified by structural and phylogenetic analyses, to yield eight mutations that collectively substantially increase protein hydrophilicity. Mutation of two parental clones yielded four domains with a range of hydrophilicity. These proteins were labeled with 64Cu, injected intravenously into nu/nu mice (n = 3–5 each) and evaluated by positron emission tomography. Renal uptake strongly correlated with hydrophilicity (Pearson's correlation coefficient = 0.97), ranging from 29 ± 11 to 100 ± 22% ID/g at 1 h. Hepatic uptake inversely correlated with hydrophilicity (Pearson's correlation coefficient = −0.92), ranging from 30 ± 7 to 3 ± 1% ID/g. Thus, renal and hepatic uptake are directly tunable through hydrophilic mutation, identifiable by structural and phylogenetic analyses. To investigate charge, we mutated acidic and basic residues in both parental clones and evaluated 64Cu-labeled mutants in nu/nu mice (n = 5–7). Selected charge removal reduced kidney signal: 78 ± 13 to 51 ± 8%ID/g (P < 0.0001) for the hydrophilic clone and 32 ± 10 to 21 ± 3 (P = 0.0005) for the hydrophobic clone. Elucidation of hydrophilicity and charge enabled modulation of background signal thereby enhancing the utility of protein scaffolds as translatable targeting agents for molecular imaging and therapy.
Keywords: biodistribution, charge, fibronectin, hydrophilicity, protein engineering
Introduction
Engineered proteins are emerging as affinity scaffolds for the development of molecular imaging agents (Miao et al., 2010) and therapeutics. These scaffolds enable excellent tumor-to-background contrast and rapid imaging because of fast clearance. Yet, high renal retention hinders many scaffolds including the nanobody (Gainkam et al., 2008; Huang et al., 2008b), the affibody (Tolmachev et al., 2009) and its two-helix derivative (Ren et al., 2009), the agouti-related cystine knot peptide (Jiang et al., 2010), the designed ankyrin repeat domain (Zahnd et al., 2010), and the type III fibronectin domain (Hackel et al., 2012). Some engineered proteins also exhibit problematic liver uptake. These phenomena could confound imaging proximal to these organs and limit dosage. We sought to modulate renal and hepatic retention by quantifying the impact of charge and hydrophilicity on protein biodistribution.
There has been anecdotal evidence in the literature on the influences of hydrophilicity and charge on biodistribution. Kidney and liver uptake have exhibited varying effects from hydrophobicity (Supplementary Fig. S1). Although some studies indicate a decrease in kidney uptake with increased hydrophobicity (Giglio et al., 2011), others indicate the inverse trend (Ono et al., 2002) or lack a correlation (Koehler et al., 2010). For the liver, several small molecule and peptide studies exhibit elevated signal with increased hydrophobicity (Giglio et al., 2011), whereas an albumin study indicates the inverse trend (Ono et al., 2002), and two studies lack a correlation (Ono et al., 2002; Kunstler et al., 2010).
Renal uptake has also observed varying influence from charge modification. Basic residues have been implicated in elevated renal retention. Arginine-rich cystine knot peptides exhibit greater kidney signal than serine-rich knots (75%ID/g and 18%ID/g, respectively) (Kimura et al., 2012). Affibodies with zero, one or three lysines in the mercaptoacetyl-based technitium chelator had 18 ± 4, 33 ± 2 and 127 ± 9%ID/g in the kidney at 4 h (Tran et al., 2008). Polycationic synthetic polypeptides exhibited greater kidney signal (96%ID/g) than amphoteric (32%ID/g) and polyanionic (6%ID/g) polypeptides (Pimm et al., 1995). Yet, mutation of two arginine residues to glutamic acids did not reduce the renal retention of a fibronectin domain (Hackel et al., 2012). In agreement with this result, but in contradiction to the branched polypeptide study, acidic residues have also been shown to increase renal retention. Glutamic acid-rich cystine knot peptides exhibit greater kidney signal than serine-rich knots (72 and 18%ID/g, respectively) (Kimura et al., 2012). Bombesin peptide analogs with zero, one and two glutamic acids had 0.6 ± 0.1, 0.9 ± 0.1 and 3.5 ± 0.4%ID/g in the kidney at 1.5 h (García Garayoa et al., 2008). Affibodies with zero, one or three glutamic acids in the chelator had 6 ± 1, 9 ± 1 and 95 ± 23%ID/g in the kidney at 4 h (Tran et al., 2007).
We sought to systematically quantify protein biodistribution over a range of hydrophilicity and charge distributions. As a model protein scaffold, we use the 10th type III domain of fibronectin a 10-kDa β-sandwich. Fibronectin domains have been engineered for picomolar to nanomolar affinity binding to many targets (Koide et al., 2007; Lipovsek, 2011) and have been validated for molecular imaging in murine xenograft tumor models (Hackel et al., 2012). In this work, we engineered mutant fibronectin domains with a range of hydrophilicities and charge distributions. Biodistribution was evaluated via small animal positron emission tomography (PET) imaging in a nude mouse model. Quantitative elucidation of these protein parameters enabled substantial reduction of background signal thereby enhancing the utility of protein scaffolds as a robust source of translatable molecular imaging agents.
Materials and methods
DNA modifications
The gene for the 92-amino acid epidermal growth factor receptor (EGFR)-binding fibronectin domain clone E6.2.6 (Hackel et al., 2010) was shuttled into a modified pET24 plasmid (Hackel et al., 2012) using NheI and BamHI restriction enzyme sites. Thus, the corresponding Ala-Ser and Gly-Ser dipeptides flank the protein, as does a C-terminal six-histidine tail. The resulting clone is herein referred to as A′. The plasmid for clone EI3.4.3′ (Hackel et al., 2012) was previously described (Hackel et al., 2010) and is herein referred to as D′. Site-directed mutagenesis was performed using the QuikChange mutagenesis kit (Stratagene). Multi-mutant genes were created by sequential mutagenesis or overlap extension polymerase chain reaction of eight oligonucleotides collectively encoding the full fibronectin domain gene. All plasmids were verified by DNA sequencing.
Protein production and analysis
BL21(DE3) Escherichia coli were transformed with plasmid and grown overnight in lysogeny broth (LB) medium. One milliliter of overnight culture was added to 200 ml of LB medium, grown for 3 h, and induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside for 3 h. Cells were pelleted, resuspended in 1 ml of lysis buffer (50 mM sodium phosphate, pH 8.0, 500 mM sodium chloride, 5% glycerol, 5 mM CHAPS, 25 mM imidazole, and complete ethylenediaminetetraacetic acid-free protease inhibitor cocktail (Roche)), freeze/thawed, and sonicated. The insoluble fraction was removed by centrifugation at 12 000g for 10 min. Fibronectin domain was purified by immobilized metal affinity chromatography using HisTrap spin columns (GE Healthcare). Purified protein was acidified with trifluoroacetic acid, passed through a 0.2-μm filter, and analyzed by reverse-phase high-performance liquid chromatography (RP-HPLC) on an analytical C18 column with a 10–90% gradient of acetonitrile in water with 0.1% trifluoroacetic acid. Standard protein was routinely analyzed to ensure consistent HPLC conditions. Retention times for replicate runs differed by <0.1 min. Expression yield consistency was examined by replicate production. Average variability was 25% with a maximum of 42%, which is sufficient for the coarse-grained use of yield herein.
For imaging experiments, 1-l cultures were similarly processed except immobilized metal affinity chromatography was performed via fast protein liquid chromatography with a HisTrap column and was followed by RP-HPLC on a semi-preparative C18 column. Protein mass was verified by matrix-assisted laser desorption/ionization-time of flight-mass spectrometry (MALDI-TOF-MS). Protein was lyophilized, resuspended in dimethyl sulfoxide, and reacted for 20 min. with the N-hydroxysuccinimide ester of 1,4,7,10-tetra-azacyclododecane-N,N′,N′,N‴-tetraacetic acid (DOTA) with 2% triethylamine. DOTA-fibronectin domain was purified by RP-HPLC. Conjugation was verified by MALDI-TOF-MS.
Small animal PET imaging
Animal experiments were conducted in accordance with federal and institutional regulations under a protocol approved by the Stanford University Institutional Animal Care and Use Committee. For tumor imaging, 5 million A431 human epidermoid carcinoma cells were subcutaneously injected into the shoulder of 8-week-old female nu/nu mice. Xenografted tumors were grown to ∼5–10 mm diameter. Radioactive 64CuCl2 was neutralized and incubated with ∼40 μM DOTA-fibronectin domain in 100 mM sodium acetate, pH 5.5 at 37° for 1 h. 64Cu-DOTA-fibronectin domain was purified by RP-HPLC followed by rotary evaporation of solvent and dilution in phosphate buffered saline (PBS). Anesthetized female nu/nu mice were injected via the tail vein with ∼2 MBq of 64Cu-DOTA-fibronectin domain. Five-minute static PET scans were acquired at the indicated times with a microPET rodent R4 scanner (Siemens). Signals in the kidneys and liver were quantified with AsiProVM 6.3.3.0. All data are presented as mean ± standard deviation.
Wild-type fibronectin domain analysis
Solvent accessible surface area (SASA) for each amino acid was calculated from the nine available fibronectin domain structures: 1TTG (Main et al., 1992), 1FNA (Dickinson et al., 1994), 2OBG (Koide et al., 2007), 2QBW (Huang et al., 2008a), 3K2M (Wojcik et al., 2010), 3CSG (Gilbreth et al., 2008), 3CSB (Gilbreth et al., 2008), 3CH8 (Huang et al., 2009) and 2OCF (Koide et al., 2002). The ratio of the SASA in the structure relative to the SASA in a random coil peptide was measured using GetArea (Fraczkiewicz and Braun, 1998) and averaged for all nine structures.
Phylogenetic sequence alignment was performed as described (Hackel et al., 2010). Fibronectin sequences from chimpanzee (XP_516072), cow (P07589), dog (XP_536059), horse (XP_001489154), human (NP_997647), mouse (NP_ 034363), opossum (XP_001368449), platypus (XP_00150 9150), rat (NP_062016) and rhesus monkey (XP_001083548) were aligned using ClustalW, and the relative frequency of each amino acid was calculated at each position.
Affinity measurement
A431 human epidermoid carcinoma cells and Balb3T3 murine fibroblasts were cultured in high glucose Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum. Cells were incubated at 37° in humidified air with 5% CO2. Cells were detached using trypsin/ethylenediaminetetraacetic acid, washed with PBS with 0.1% bovine serum albumin and placed on ice to prevent EGFR internalization. Cells were labeled with varying concentrations of purified fibronectin domain followed by fluorescein-conjugated anti-His6 antibody (Abcam). The mean fluorescein signal was quantified by flow cytometry. The equilibrium dissociation constant, Kd, was calculated by minimizing the sum of squared errors assuming a single-site binding model.
Results
Analysis of an alternative EGFR-targeted fibronectin domain
The fibronectin domain initially tested for molecular imaging, D′, has modest hydrophobicity and is balanced by seven basic and seven acidic residues. An alternative EGFR-targeted fibronectin domain, A′, is more hydrophobic and negatively charged with only four basic residues and eight acidic residues. PET imaging of nude mice reveals substantially different biodistribution of these two fibronectin domains with 79% sequence identity (Fig. 1). Relative to D′, A′ exhibits reduced renal uptake (29 ± 11 and 76 ± 13%ID/g for A′ and D′ respectively, P = 0.0003) but elevated hepatic uptake (30 ± 7 and 1.6 ± 0.3%ID/g, P = 0.0009). To ascertain the impact of charge and hydrophobicity on these differences, mutants of each clone were engineered to modify each parameter independently.
Fig. 1.

PET imaging of A′ and D′. A431 epidermoid carcinoma cells were xenografted into nude mice. ∼2 MBq of 64Cu-fibronectin domain was injected via the tail vein. Five-minute static scans were acquired at 1, 2, 4 and 24 h post-injection. (A) Coronal slices are presented for 1 h post-injection for D′ (left) and A′ (right). T indicates tumor; K, kidneys; L, liver. (B) Signals in the kidney and liver were quantified with AsiProVM. Value and error bars represent mean and standard deviation (n = 5).
Identification of sites for hydrophilic mutation
Sites to potentially improve protein hydrophilicity were identified using five primary criteria: impact on target binding, charge, solvent exposure, structural integrity and phylogenetic occurrence of hydrophilic residues (Table I). The 24 sites in the three solvent-exposed loops (BC loop: D23 through Y31; DE loop: G52 through T56; FG loop G77 through K86) were excluded from mutation because of their impact on target binding in engineered fibronectin domains. Ten charged residues were excluded from mutation so that the resulting mutants could be evaluated based on hydrophilicity rather than charge effects. Likewise, despite the hydrophilicity of charged amino acids, no charged residues were introduced by mutation. Twenty-four amino acids had a SASA <20% relative to random coil and were excluded from mutation under the assumptions that these amino acids may be critical to the stability of the protein core and mutation of a non-exposed side-chain may have minimal impact on hydrophilicity of the folded protein. Although exposed, all five prolines and three of four glycines were excluded from mutation because of the unique structural rigidity and flexibility provided by these residues. G40 was considered for mutation because of its location in the solvent-exposed CD loop (T39 through V45) and its limited phylogenetic conservation (see below).
Table I.
Amino acid frequency and solvent accessible surface area
| Site | WT | A | C | D | E | F | G | H | I | K | L | M | N | P | Q | R | S | T | V | W | Y | Z | SASA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Charged (10) | |||||||||||||||||||||||
| 3 | D | 1 | 0 | 44 | 0 | 0 | 2 | 0 | 0 | 0 | 5 | 0 | 5 | 20 | 0 | 0 | 16 | 3 | 5 | 0 | 0 | 0 | 36 |
| 6 | R | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 5 | 0 | 0 | 0 | 5 | 0 | 18 | 15 | 37 | 6 | 0 | 0 | 0 | 46 |
| 7 | D | 0 | 0 | 28 | 2 | 0 | 13 | 0 | 0 | 0 | 0 | 0 | 22 | 7 | 11 | 7 | 11 | 0 | 0 | 0 | 0 | 0 | 62 |
| 9 | E | 7 | 0 | 7 | 7 | 2 | 5 | 7 | 4 | 7 | 0 | 0 | 0 | 0 | 16 | 22 | 6 | 4 | 0 | 0 | 7 | 0 | 52 |
| 33 | R | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 20 | 6 | 6 | 0 | 0 | 0 | 6 | 55 | 0 | 0 | 6 | 0 | 0 | 0 | 24 |
| 38 | E | 1 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 6 | 5 | 0 | 0 | 70 | 0 | 0 | 12 | 0 | 0 | 0 | 0 | 0 | 45 |
| 47 | E | 0 | 0 | 0 | 43 | 6 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 6 | 26 | 0 | 1 | 6 | 6 | 0 | 0 | 0 | 39 |
| 63 | K | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 1 | 8 | 6 | 6 | 5 | 6 | 25 | 8 | 11 | 17 | 0 | 0 | 0 | 0 | 68 |
| 67 | D | 0 | 0 | 13 | 51 | 0 | 0 | 0 | 2 | 6 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 12 | 4 | 0 | 6 | 0 | 47 |
| 93 | R | 2 | 0 | 0 | 6 | 0 | 0 | 0 | 6 | 14 | 0 | 0 | 0 | 0 | 1 | 6 | 19 | 32 | 14 | 0 | 0 | 0 | 62 |
| Buried (24) | |||||||||||||||||||||||
| 5 | P | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 82 | 0 | 0 | 7 | 1 | 0 | 0 | 0 | 0 | 8 |
| 8 | L | 7 | 0 | 0 | 6 | 1 | 0 | 2 | 6 | 0 | 51 | 7 | 0 | 10 | 0 | 0 | 0 | 5 | 6 | 0 | 0 | 0 | 13 |
| 10 | V | 0 | 0 | 0 | 0 | 43 | 0 | 0 | 6 | 0 | 6 | 0 | 6 | 0 | 6 | 0 | 1 | 0 | 30 | 0 | 0 | 0 | 8 |
| 18 | L | 0 | 0 | 0 | 0 | 14 | 0 | 6 | 35 | 0 | 26 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 11 | 0 | 0 | 0 | 2 |
| 20 | I | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 44 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 47 | 0 | 0 | 0 | 1 |
| 22 | W | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 |
| 32 | Y | 0 | 0 | 0 | 0 | 16 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 75 | 0 | 0 |
| 34 | I | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 37 | 0 | 19 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 38 | 0 | 0 | 0 | 0 |
| 35 | T | 0 | 0 | 12 | 1 | 0 | 6 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | 25 | 6 | 32 | 6 | 0 | 6 | 0 | 12 |
| 36 | Y | 7 | 6 | 0 | 6 | 0 | 0 | 6 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | 24 | 6 | 31 | 0 | 13 |
| 37 | G | 2 | 0 | 0 | 6 | 0 | 12 | 11 | 7 | 3 | 8 | 0 | 1 | 0 | 2 | 10 | 19 | 16 | 4 | 0 | 0 | 0 | 14 |
| 50 | V | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 19 | 0 | 12 | 0 | 0 | 24 | 0 | 0 | 0 | 1 | 40 | 0 | 0 | 0 | 16 |
| 57 | A | 32 | 6 | 0 | 0 | 0 | 0 | 0 | 13 | 0 | 0 | 6 | 6 | 0 | 0 | 0 | 0 | 0 | 18 | 0 | 19 | 0 | 4 |
| 59 | I | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 33 | 0 | 37 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 17 | 0 | 0 | 0 | 1 |
| 62 | L | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 98 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| 68 | Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 94 | 0 | 7 |
| 70 | I | 1 | 0 | 0 | 0 | 6 | 6 | 0 | 32 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | 44 | 0 | 6 | 0 | 2 |
| 71 | T | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 6 | 6 | 0 | 0 | 1 | 0 | 8 | 0 | 42 | 21 | 6 | 0 | 8 | 0 | 18 |
| 72 | V | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 20 | 0 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 54 | 0 | 6 | 0 | 0 |
| 74 | A | 63 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | 12 | 6 | 0 | 0 | 0 | 2 |
| 75 | V | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 41 | 0 | 0 | 0 | 6 | 0 | 6 | 0 | 41 | 0 | 0 | 0 | 11 |
| 76 | T | 0 | 0 | 0 | 12 | 0 | 0 | 8 | 0 | 31 | 0 | 0 | 23 | 0 | 6 | 6 | 1 | 7 | 0 | 0 | 6 | 0 | 8 |
| 88 | I | 11 | 0 | 0 | 1 | 0 | 0 | 0 | 15 | 0 | 43 | 0 | 0 | 0 | 1 | 0 | 0 | 6 | 24 | 0 | 0 | 0 | 18 |
| 94 | T | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 1 | 92 | 0 | 0 | 0 | 0 | 5 |
| Structural (8) | |||||||||||||||||||||||
| 15 | P | 12 | 0 | 29 | 14 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 25 | 6 | 0 | 6 | 0 | 6 | 0 | 0 | 0 | 78 |
| 41 | G | 1 | 0 | 6 | 6 | 1 | 41 | 0 | 3 | 7 | 8 | 1 | 7 | 0 | 0 | 0 | 13 | 6 | 0 | 0 | 0 | 0 | 57 |
| 44 | P | 0 | 0 | 7 | 14 | 1 | 0 | 7 | 0 | 0 | 0 | 0 | 2 | 28 | 8 | 22 | 1 | 6 | 0 | 0 | 4 | 0 | 90 |
| 51 | P | 4 | 0 | 6 | 0 | 0 | 12 | 6 | 3 | 3 | 0 | 0 | 1 | 31 | 0 | 7 | 18 | 6 | 3 | 0 | 0 | 0 | 74 |
| 61 | G | 0 | 0 | 6 | 0 | 0 | 61 | 1 | 0 | 0 | 6 | 0 | 26 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 84 |
| 64 | P | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 81 | 0 | 6 | 0 | 0 | 6 | 0 | 0 | 0 | 69 |
| 65 | G | 8 | 0 | 1 | 0 | 0 | 79 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 6 | 0 | 0 | 0 | 0 | 56 |
| 87 | P | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 12 | 1 | 0 | 0 | 74 | 0 | 1 | 1 | 0 | 6 | 0 | 0 | 0 | 33 |
| Hydrophilic (3) | |||||||||||||||||||||||
| 42 | N | 1 | 0 | 12 | 6 | 0 | 24 | 1 | 0 | 0 | 3 | 0 | 5 | 17 | 12 | 2 | 5 | 9 | 3 | 0 | 0 | 0 | 99 |
| 46 | Q | 13 | 0 | 0 | 0 | 0 | 6 | 0 | 5 | 24 | 19 | 0 | 0 | 0 | 6 | 8 | 0 | 6 | 8 | 0 | 6 | 0 | 69 |
| 91 | N | 1 | 0 | 14 | 3 | 0 | 0 | 0 | 2 | 6 | 0 | 0 | 4 | 0 | 17 | 14 | 6 | 19 | 14 | 0 | 0 | 0 | 58 |
| Mid-hydrophilic; rare N, Q (10) | |||||||||||||||||||||||
| 2 | S | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 27 | 0 | 26 | 0 | 0 | 18 | 0 | 0 | 8 | 0 | 21 | 0 | 0 | 0 | 69 |
| 14 | T | 0 | 0 | 11 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | 1 | 12 | 6 | 0 | 8 | 55 | 0 | 0 | 0 | 0 | 64 |
| 17 | S | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 6 | 0 | 1 | 1 | 0 | 0 | 0 | 58 | 27 | 6 | 0 | 0 | 0 | 30 |
| 21 | S | 6 | 0 | 0 | 0 | 0 | 0 | 6 | 1 | 3 | 0 | 6 | 1 | 0 | 12 | 12 | 37 | 16 | 0 | 0 | 0 | 0 | 26 |
| 39 | T | 6 | 0 | 0 | 12 | 0 | 0 | 0 | 1 | 19 | 0 | 0 | 0 | 12 | 0 | 0 | 13 | 19 | 18 | 0 | 0 | 0 | 39 |
| 43 | S | 0 | 0 | 1 | 21 | 0 | 49 | 0 | 7 | 0 | 0 | 0 | 1 | 7 | 0 | 0 | 14 | 0 | 0 | 0 | 0 | 0 | 62 |
| 58 | T | 0 | 0 | 0 | 12 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 56 | 18 | 0 | 0 | 0 | 65 |
| 60 | S | 0 | 0 | 8 | 11 | 0 | 0 | 1 | 0 | 6 | 0 | 0 | 1 | 0 | 5 | 6 | 25 | 37 | 0 | 0 | 0 | 0 | 75 |
| 73 | Y | 1 | 0 | 0 | 8 | 7 | 0 | 0 | 15 | 0 | 0 | 1 | 0 | 0 | 8 | 0 | 9 | 1 | 14 | 0 | 37 | 0 | 26 |
| 89 | S | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 19 | 1 | 0 | 0 | 0 | 0 | 7 | 12 | 19 | 14 | 23 | 0 | 0 | 0 | 42 |
| Mid-hydrophilic (5) | |||||||||||||||||||||||
| 16 | T | 0 | 0 | 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 19 | 0 | 6 | 1 | 9 | 46 | 6 | 0 | 0 | 0 | 63 |
| 40 | G | 0 | 0 | 0 | 19 | 0 | 19 | 6 | 0 | 6 | 0 | 0 | 28 | 0 | 6 | 7 | 3 | 1 | 6 | 0 | 0 | 0 | 86 |
| 49 | T | 6 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | 12 | 0 | 18 | 0 | 1 | 18 | 6 | 26 | 6 | 0 | 0 | 0 | 48 |
| 69 | T | 1 | 0 | 0 | 12 | 0 | 0 | 2 | 2 | 6 | 6 | 0 | 19 | 0 | 0 | 0 | 0 | 25 | 25 | 0 | 3 | 0 | 40 |
| 92 | Y | 14 | 0 | 0 | 0 | 13 | 0 | 0 | 6 | 6 | 0 | 0 | 0 | 0 | 39 | 1 | 0 | 0 | 14 | 0 | 7 | 0 | 36 |
| Hydrophobic (10) | |||||||||||||||||||||||
| 1 | V | 27 | 0 | 1 | 7 | 0 | 13 | 0 | 0 | 8 | 0 | 0 | 15 | 8 | 6 | 1 | 6 | 3 | 7 | 0 | 0 | 0 | 59 |
| 4 | V | 24 | 0 | 0 | 0 | 0 | 7 | 0 | 1 | 7 | 0 | 0 | 0 | 28 | 0 | 7 | 14 | 0 | 12 | 0 | 0 | 0 | 22 |
| 11 | V | 1 | 0 | 6 | 12 | 0 | 0 | 0 | 6 | 0 | 6 | 1 | 0 | 0 | 0 | 0 | 8 | 47 | 12 | 0 | 0 | 0 | 37 |
| 12 | A | 22 | 0 | 25 | 24 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | 0 | 10 | 1 | 1 | 3 | 0 | 0 | 0 | 0 | 54 |
| 13 | A | 10 | 0 | 0 | 6 | 0 | 1 | 0 | 16 | 0 | 7 | 0 | 6 | 0 | 0 | 0 | 2 | 14 | 39 | 0 | 0 | 0 | 30 |
| 19 | L | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 6 | 18 | 0 | 0 | 6 | 0 | 6 | 8 | 36 | 16 | 0 | 0 | 0 | 42 |
| 45 | V | 1 | 0 | 0 | 19 | 0 | 0 | 6 | 0 | 0 | 0 | 6 | 0 | 26 | 6 | 0 | 14 | 3 | 13 | 6 | 0 | 0 | 51 |
| 48 | F | 6 | 0 | 12 | 6 | 13 | 0 | 0 | 6 | 6 | 12 | 0 | 0 | 12 | 1 | 19 | 0 | 0 | 0 | 0 | 6 | 0 | 32 |
| 66 | V | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 6 | 1 | 0 | 0 | 0 | 0 | 11 | 33 | 35 | 0 | 0 | 0 | 31 |
| 90 | I | 6 | 0 | 7 | 0 | 6 | 41 | 0 | 20 | 3 | 0 | 0 | 3 | 0 | 7 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 25 |
The amino acid frequencies from 155 type III domains of fibronectin in 10 species are shown for each site. SASA indicates the solvent accessible surface area of each amino acid relative to the same side chain in a random coil peptide. Sites are listed from N- to C-terminus within the indicated categories.
Twenty-eight sites remained in consideration for mutation. The amino acid frequencies at each position were calculated from the 155 type III domains of fibronectin from 10 species. Each site was evaluated for the potential to tolerate a mutation to a side chain of increased hydrophilicity, according to the hydrophobicity scale of Kyte and Doolittle (1982). The amide-containing side chains of asparagine and glutamine have the highest hydrophilicity of non-charged residues and were considered optimal mutants. Thus, N42, Q46 and N91 were conserved. Next in hydrophilicity, the hydroxyl-containing side chains of tyrosine, serine and threonine were also considered viable mutant alternatives. Tryptophan was not considered as a mutant because of its potential for non-specific binding (Birtalan et al., 2010). Ten hydroxyl sites were conserved because more hydrophilic asparagine and glutamine occurred with <15% frequency. The remaining 15 sites were experimentally tested for hydrophilic mutation.
Five mildly hydrophilic residues were mutated to more hydrophilic residues, using phylogenetic frequency to decide between asparagine and glutamine. Ten hydrophobic sites were mutated to the most frequent acceptable hydrophilic (N, Q, Y, S and T). One exception is that V1S was tested because serine enables more efficient cleavage of N-terminal methionine in the event of future genetic removal of the Ala-Ser dipeptide resulting from the NheI cloning site. The 15 mutants are presented in bold in Table I.
Hydrophilic mutation analysis
Mutations can have variable impacts on effective hydrophilicity depending on their location in the protein including solvent accessibility and surrounding amino acid environment. Each mutation was tested individually for its impact on hydrophilicity and protein yield. Each mutant was expressed at 200-ml scale under identical bacterial culture conditions, purified by metal affinity chromatography, and analyzed by RP-HPLC. Relative hydrophilicity was analyzed by retention time and relative yield was measured as the peak area. Thirteen of 15 mutants increase hydrophilicity including five that shift retention time by >0.5 min (Fig. 2). Eleven of 15 mutants decrease protein yield but only five by >50%. Eight mutants have both retention time shifts >0.1 min. and yields >50%. Of note, seven of these resulted from hydrophobic wild-type residues (of 10 total) and only one resulted from a mid-hydrophilic wild type (of five total). Alternate mutants were then tested for the three ‘failed’ hydrophobic sites as well as V45 given its borderline yield (51%). V45Q and F48Q improved yield but at the expense of hydrophilicity. I90T was essentially a silent mutant. Conversely, V66Q shifted retention time 0.3 min. and increased the yield to 199% of the parental clone. Thus, nine mutants (V1S, V4S, V11T, A12N, A13T, T16N, L19T, V45S and V66Q) provide at least 0.1 min. shift with at least 50% yield (Fig. 2).
Fig. 2.
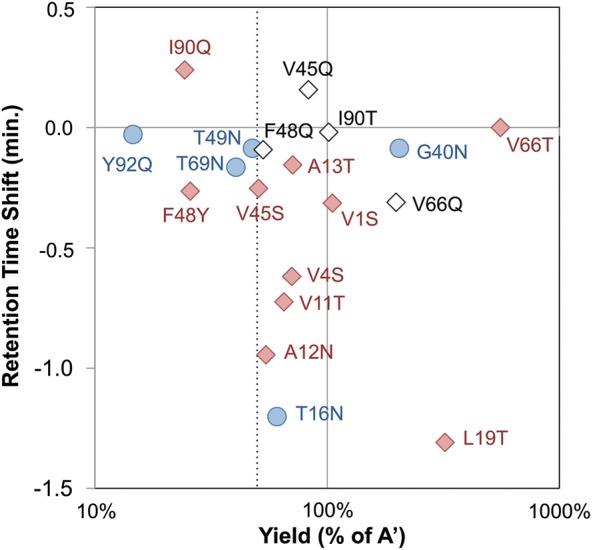
Single mutant analysis. Mutant clones were produced in BL21(DE3), purified and analyzed by RP-HPLC using a 10–90% acetonitrile gradient over 30 min. The yield and retention time shift, both relative to the parental clone, are indicated. Red diamonds indicate mutation from hydrophobic residue. Blue circle indicates mutation from mid-hydrophilic residue. Open diamond indicates mutation alternative to original attempt.
It was then tested whether combination of these mutations yields a substantially more hydrophilic protein. Three sites, V11, A12 and L19, are proximal in the wild-type structure (Fig. 3A); thus, their mutation may impact each other. The single, double and triple mutants were tested. The triple mutant provides the optimal hydrophilic shift while retaining high yield (Fig. 3B). Given the modest hydrophilic impacts of A13T (0.2-min shift) and V45S (0.3-min shift) and their reduced yields (71 and 51%, respectively), the benefit of these mutations was tested within the context of the combination mutant. The seven other beneficial mutations (V1S, V4S, V11T, A12N, T16N, L19T and V66Q) were combined. The resulting clone (7x mutant, Fig. 3B) had a retention time shift of −3.0 min and was produced in 49% yield relative to the parental clone. Inclusion of V45S improved the shift to −3.2 min and increased the yield to 73%. Further inclusion of A13T had a nominal improvement in hydrophilicity but reduced yield to 44%. Thus, only V45S was included in the final mutant A-phil: V1S, V4S, V11T, A12N, T16N, L19T, V45S and V66Q (Table II).
Fig. 3.

Multiple mutant analysis. (A) The NMR structure of the tenth type III domain of human fibronectin (1TTG (Main et al., 1992)) is presented in cartoon form with sites of beneficial mutation depicted in red. (B) Mutant clones were produced in BL21(DE3), purified, and analyzed by RP-HPLC using a 10–90% acetonitrile gradient over 30 min. The yield and retention time shift, both relative to the parental clone, are indicated for single (blue diamonds), double (green squares) and triple (red circle) mutants of V11T, A12N and L19T. The 7x mutant (V1S, V4S, V11T, A12N, T16N, L19T, V66Q) and derivatives are shown in black triangles.
Table II.
Protein sequences and RP-HPLC retention times
| Name | Sequence | tR (min) |
|---|---|---|
| A | ASVSDVPRDLEVVAATPTSLLISWFDYAVTYYRITYGETGGNSPVQEFTV PGWISTATISGLNPGVDYTITVYAVTDNSHWPFRSTPISINYRTGSHHHHHH | 21.2 |
| A-phil | ASSSDSPRDLEVTNATPNSLTISWFDYAVTYYRITYGETGGNSPSQEFTV PGWISTATISGLNPGQDYTITVYAVTDNSHWPFRSTPISINYRTGSHHHHHH | 18.0 |
| D | VSDVPRDLEVVAATPTSLLISWLHHRSDVRSYRITYGETGGNSPVQEFTV PGSRSLATISGLRPGVDYTITVYAVTWGSYCCSNPISINYRTHHHHHH | 17.8 |
| D-phil | ASSSDSPRDLEVTNATPNSLTISWLHHRSDVRSYRITYGETGGNSPSQEFTV PGSRSLATISGLRPGQDYTITVYAVTWGSYCCSNPISINYRTGSHHHHHH | 15.7 |
Mutations from the parental protein are boldfaced and underlined. tR denotes retention time for RP-HPLC on analytical C18 column with a gradient of 10–90% acetonitrile in water with 0.1% trifluoroacetic acid from 2 to 32 min.
To test the generality of these hydrophilic mutations and their influence on biodistribution, these eight mutations were applied to clone D′. It is noteworthy that clone D′ is already substantially more hydrophilic than clone A′ (3.4 min earlier elution) because of the composition of its engineered loops. Application of these mutations to D′ resulted in D-phil with 5.5 min earlier elution on RP-HPLC and a 334% yield (both relative to A′; 2.1 min and 74% relative to D′, Table II).
Small animal imaging: hydrophilicity
The hydrophilic mutants were tested by small animal PET imaging to determine the impact of hydrophilicity on protein biodistribution. Nu/nu mice were injected via the tail vein with 64Cu-fibronectin domain, and static PET images were acquired at 1 h post-injection. Both hydrophilic mutants increased renal signal relative to their parental proteins (Fig. 4A). In fact, the four clones collectively exhibit a strong correlation between hydrophilicity and renal signal (Pearson's correlation coefficient = 0.97) ranging from 29 ± 11 to 100 ± 22%ID/g. Conversely, hepatic signal demonstrated a strong negative correlation with hydrophilicity (Pearson's correlation coefficient = −0.92), ranging from 30 ± 7 to 3 ± 1%ID/g (Fig. 4B).
Fig. 4.

Impact of hydrophilicity on renal and hepatic signal. nu/nu mice were injected with 64Cu-fibronectin domain via the tail vein. Five-minute static scans were acquired. Signals in the (A) kidney and (B) liver were quantified with AsiProVM. Value and error bars represent mean and standard deviation (n = 3–5). A′ and A-phil (open squares), non-binding scrambled mutants (filled squares), and D′ and D-phil (diamonds).
As healthy liver can express relatively high levels of EGFR (Carver et al., 2002), it was desired to separate the effects of EGFR-targeting from other protein characteristics. Clone D′ has previously been demonstrated to possess only 2.5 ± 1.3 μM affinity for murine EGFR (Hackel et al., 2012). Clone A′ was analyzed for murine cross-reactivity and was found to bind with 13 ± 5 nM affinity (Supplemental Fig. S2). Thus, four amino acids in the FG loop of A′ were scrambled from PFRS to RSFP, which eliminated binding (Supplemental Fig. S2). Elimination of EGFR targeting reduced hepatic signal from 30 ± 7 to 16 ± 3%ID/g for A′ (P = 0.008) and from 16 ± 3 to 8 ± 1%ID/g for A′-phil (P = 0.01). Yet the remaining hepatic signal maintains a strong negative correlation with hydrophilicity (Pearson's correlation coefficient = −0.91, Fig. 4B). Protein scrambling did not have a statistically significant impact on renal signal (Fig. 4A).
Charge
We also sought to explore the impact of charge. Thus, we identified mutants to remove charge while retaining hydrophilicity. Single mutants were produced at 200-ml scale and were generally tolerated (Supplemental Table SI). E38S was not tolerated but the phylogenetically dominant E38P (70%, Table I) was produced in 90% yield relative to the parental clone. R33S, R33N and R33Q mutants were not produced in sufficient yield. Eight mutations (D3S, R6T, D7T, E9S, E38P, E47T, D67T, R93T) were combined in clone A′, and the resultant clone (A-22) was produced in 23% yield with only 0.2-min HPLC shift relative to A′ despite the removal of eight surface charges. As in the hydrophilicity study, to test generality, these mutations were applied to clone D′. As D′ is produced in substantially higher yield than A′, we were able to incorporate the R33S mutation to further remove charge, resulting in clone D-14 (19% yield and 0.4-min shift relative to D′).
The charge mutants were tested via PET imaging. Nu/nu mice were injected via the tail vein with 64Cu-fibronectin domain, and static PET images were acquired at 1 h post-injection. D-14 and A-22 do not yield significant reductions in renal uptake. D-14 is highly positively charged. One of the engineered positively charged residues, R54, exists in the DE loop, which is not always critical for binding (Hackel et al., 2008); in fact, mutation does not hinder binding affinity (Supplementary Fig. S2). Removal of this charge, along with removal of the positive charge at K63, yielded clone D-12, which demonstrated significant reduction in renal uptake (P = 0.007 vs. D-14, P = 0.013 vs. D′ at 1 h, Fig. 5A; P < 0.0001 vs. D′ at 1–4 h, Fig. 5C). To further explore the impact of these number and type of charges, a clone (D-32) was produced with five charges residues like D-14 but net negative charge. D-32 is clone D-12 with reversion of D3 and E9. This clone exhibits reduced renal uptake, though not to a statistically significant extent (Fig. 5A).
Fig. 5.

Impact of charge on renal signal. nu/nu mice were injected with 64Cu-fibronectin domain via the tail vein. Five-minute static scans were acquired at 1, 2, 4 and 24 h post-injection. Signals in the kidney were quantified with AsiProVM. Value and error bars represent mean and standard deviation (n = 3–5). (A) D′ and mutants at 1 h post-injection. (B) A′ and mutants at 1 h post-injection. Net charges are calculated from the protein sequence at pH 7.4 assuming independent, accessible amino acids. (C) D′ (filled squares) and D-12 (open diamonds) at 1, 2, 4 and 24 h post-injection. (D) A′ (filled squares) and A-42 (open diamonds) at 1, 2, 4 and 24 h post-injection.
Application of the eight tolerable charge mutations in clone A′ (to A-22) did not yield a change in renal uptake (Fig. 5B). We then explored if a more negative net charge would impact renal uptake as it did for D-14 compared with D-32. As the remaining charges in A-22 could not be mutated without impacting affinity or yield, acidic residues were re-instated. Reversion of two acidic residues, D3 and E9, to yield clone A-42 resulted in significant reduction in renal uptake (P = 0.007 vs. A′, P = 0.002 vs. A-22 at 1 h, Fig. 5B; P = 0.0005 vs. A′ at 1 to 4 h, Fig. 5D).
Discussion
Engineered proteins are useful in a multitude of applications including molecular imaging and therapy. Although these proteins can be engineered for high molecular specificity with high affinity, physiological mechanisms can result in off-target delivery. Non-specific localization can hinder the diagnostic value of molecular imaging, create dose limitations for nuclear imaging, and cause substantial side effects in therapeutic applications. Two common organs for such unintended accumulation are the kidneys and liver. As many of the engineered proteins scaffolds fall below the ∼50 kDa glomerular filtration limit, renal uptake is especially problematic. Yet the factors impacting such off-target delivery are poorly understood and strategies to modulate biodistribution would be valuable for molecular imaging and drug delivery.
As we tested the generalizability of the fibronectin domain scaffold for PET tracers, we discovered that clones A′ and D′ have vastly different renal and hepatic signals despite 79% sequence identity (Fig. 1). One hypothesis for this discrepancy is their differing hydrophobicity. Thus, we systematically quantified kidney and liver signal in mice by PET imaging with 64Cu-labeled fibronectin domains with a range of hydrophilicities.
The selection criteria (>20% SASA, non-structural, Kyte-Doolittle hydrophobicity >−0.7, mutation to phylogenetically prevalent residue) to identify the primary mutants was relatively effective as 7 of 10 identified sites yielded improved hydrophilicity upon mutation with at least 50% yield. Two exceptions, F48Y and I90Q, have low yield, which could have been predicted by infrequent phylogenetic occurrence of hydrophilic residues at these positions. Mutant analysis reveals a strong correlation (Pearson's correlation coefficient = 0.91) between the logarithm of relative yield and phylogenetic frequency of amide or hydroxyl residues with the exception of V11T (Fig. 6). It is noteworthy, though, that the outlying reduced yield of V11T in A′ (65%) is not observed when V11T is applied to the A12N mutant (168% yield) or the L19T mutant (100% yield). The other ineffective mutant, V66T, has high yield but lacks an increase in hydrophilicity. Mutation to an inherently more hydrophilic residue, V66Q, provides a 0.3-min hydrophilic shift with good yield (199%). Only one of five mid-hydrophilic sites provided a significantly useful mutation, as the other four did not yield a substantial increase in hydrophilicity. Yet T16N did provide a 1.2-min shift thereby justifying screening mid-hydrophilics experimentally. It is worth noting that four of the five mid-hydrophilic sites fit the yield:phylogenetic frequency correlation, which could help to guide mutant selection. Overall, the current approach enables rational identification of hydrophilic mutants with strong accuracy. These correlations can also be used to guide combinatorial library design and selection of lead clones. Taken in tandem with the strong correlations of renal and hepatic uptake with hydrophobicity, we demonstrate that designed mutation can be used to tailor biodistribution. The opposite impacts of hydrophilicity on renal and hepatic uptake do not facilitate dual reduction of these off-target sites, but rather enable modulation of the more clinically or scientifically impactful site. This decision is dependent on multiple factors including physiological localization of the molecular target and application-dependent impact of off-target delivery (e.g. radiation dosimetry or therapeutic index). For example, for molecular imaging applications in the abdomen it would likely be best to minimize hepatic uptake while maximizing renal clearance.
Fig. 6.

Relation of yield to phylogenetic frequency of hydrophilic residues. The logarithm of the yield of a mutant relative to the yield of A′ is plotted on the ordinate. The phylogenetic frequency of hydrophilic amide or hydroxyl residues (N, Q, S, T or Y) at the site of that mutation is plotted on the abscissa. The best-fit linear correlation is shown for 9 of the 10 hydrophobic sites mutated to their most phylogenetically frequent hydrophilic residue (closed diamonds); the outlier, V11T, is also shown (open diamond). Four secondary mutants whose primary mutant had low yield or low hydrophilic shift are shown (open triangles). Five mutants from mid-hydrophilic side chains to amides are shown with their phylogenetically amide frequency only (cross).
Applicability to alternative protein scaffolds must be evaluated and is the subject of future studies. The strong negative correlation between kidney uptake and hydrophobicity is in agreement with results for 99mTc-labeled cyclic RGD peptide variants (Kunstler et al., 2010) and two metronidazole derivatives (Giglio et al., 2011) but directly contrary to the inverse trend observed for albumin derivatives (Ono et al., 2002) and the lack of a trend observed for variants of octreotide peptide (Schottelius et al., 2002), Fab fragments (Ono et al., 2002), and cyclin-dependent kinase 4 inhibitors (Koehler et al., 2010) (Supplementary Fig. S1). The strong positive correlation between liver uptake and hydrophobicity is in agreement with octreotide (Schottelius et al., 2002), 18F-labeled cyclic RGD peptides (Glaser et al., 2008), and two small molecule organics (Giglio et al., 2011) but directly contrary to the negative correlation for albumin derivatives (Ono et al., 2002), the variance of Fab fragments (Ono et al., 2002) and the lack of a trend for 99mTc-labeled cyclic RGD peptides (Kunstler et al., 2010). In both cases, the contrary results could result from other unquantified molecular variables. Future studies on the physiological mechanisms responsible for organ retention may enlighten these discrepancies.
Previous reports have implicated charge in renal uptake. Thus, we also explored the ability to modulate renal uptake through mutagenic removal of charged amino acids. Eight mutations were tolerated as inferred by their phylogenetic frequency. Note that mutants of R33 were not expressed well, which is perhaps predictable by the phylogenetic lack of hydrophilic side chains (only 6% Q and no occurrence of N, S or T). Likewise, E38 only exhibits 12% S, and E38S is expressed at <10% of the parental clone. Conversely, the phylogenetically frequent (70%) E38P is well expressed (90% relative to parental). Despite the removal of eight and nine charged residues from A′ and D′, respectively, the combination mutants A-22 and D-14 did not exhibit modified renal uptake. Reduction in the net positive charge of both mutants, to A-42 and D-12, decreased kidney retention by 36% in each case. Thus, charge modification, via removal of charged residues and consideration of net charge, can be implemented to alleviate renal retention of engineered protein scaffolds. The impact of both net and total charge will be further elucidated through study of more mutants. Future studies will also investigate the impact of charge residue location and test the generalizability to other protein scaffolds.
In addition to engineering binding paratopes to provide molecular specificity, protein hydrophilicity and charge can be engineered to modulate in vivo biodistribution. Effective mutation can be guided by structural and phylogenetic data.
Supplementary data
Funding
This work was supported by funding from the Canary Foundation (to S.S.G.); the National Cancer Institute at the National Institutes of Health (CA119367, CA136465, CA083636 to S.S.G.); and the American Cancer Society (postdoctoral fellowship to B.J.H.).
Supplementary Material
Footnotes
Edited by Dario Neri
References
- Birtalan S., Fisher R.D., Sidhu S.S. Mol. Biosyst. 2010;6:1186–1194. doi: 10.1039/b927393j. [DOI] [PubMed] [Google Scholar]
- Carver R.S., Stevenson M.C., Scheving L.A., Russell W.E. Gastroenterology. 2002;123:2017–2027. doi: 10.1053/gast.2002.37060. [DOI] [PubMed] [Google Scholar]
- Dickinson C.D., Veerapandian B., Dai X.P., Hamlin R.C., Xuong N.H., Ruoslahti E., Ely K.R. J. Mol. Biol. 1994;236:1079–1092. doi: 10.1016/0022-2836(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz R., Braun W. J. Comput. Chem. 1998;19:319–333. [Google Scholar]
- Gainkam L.O.T., Huang L., Caveliers V., et al. J. Nucl. Med. 2008;49:788–795. doi: 10.2967/jnumed.107.048538. [DOI] [PubMed] [Google Scholar]
- García Garayoa E., Schweinsberg C., Maes V., Brans L., Bläuenstein P., Tourwe D.A., Schibli R., Schubiger P.A. Bioconjug. Chem. 2008;19:2409–2416. doi: 10.1021/bc800262m. [DOI] [PubMed] [Google Scholar]
- Giglio J., Fernández S., Rey A., Cerecetto H. Bioorg. Med. Chem. Lett. 2011;21:394–397. doi: 10.1016/j.bmcl.2010.10.130. [DOI] [PubMed] [Google Scholar]
- Gilbreth R.N., Esaki K., Koide A., Sidhu S.S., Koide S. J. Mol. Biol. 2008;381:407–418. doi: 10.1016/j.jmb.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser M., Morrison M., Solbakken M., Arukwe J., Karlsen H., Wiggen U., Champion S., Kindberg G.M., Cuthbertson A. Bioconjug. Chem. 2008;19:951–957. doi: 10.1021/bc700472w. [DOI] [PubMed] [Google Scholar]
- Hackel B.J., Ackerman M.E., Howland S.W., Wittrup K.D. J. Mol. Biol. 2010;401:84–96. doi: 10.1016/j.jmb.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel B.J., Kapila A., Wittrup K.D. J. Mol. Biol. 2008;381:1238–1252. doi: 10.1016/j.jmb.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel B.J., Kimura R.H., Gambhir S.S. Radiology. 2012;263:179–188. doi: 10.1148/radiol.12111504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Koide A., Makabe K., Koide S. Proc. Natl. Acad. Sci. U S A. 2008a;105:6578–6583. doi: 10.1073/pnas.0801097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Gainkam L.O.T., Caveliers V., Vanhove C., Keyaerts M., De Baetselier P., Bossuyt A., Revets H., Lahoutte T. Mol. ImagingBiol. 2008b;10:167–175. doi: 10.1007/s11307-008-0133-8. [DOI] [PubMed] [Google Scholar]
- Huang J., Makabe K., Biancalana M., Koide A., Koide S. J. Mol. Biol. 2009;392:1221–1231. doi: 10.1016/j.jmb.2009.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L., Kimura R.H., Miao Z., et al. J. Nucl. Med. 2010;51:251–258. doi: 10.2967/jnumed.109.069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R.H., Teed R., Hackel B.J., Pysz M.A., Chuang C.Z., Sathirachinda A., Willmann J.K., Gambhir S.S. Clin. Cancer Res. 2012;18:839–849. doi: 10.1158/1078-0432.CCR-11-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler L., Graf F., Bergmann R., Steinbach J., Pietzsch J., Wuest F. Eur J Med Chem. 2010;45:727–737. doi: 10.1016/j.ejmech.2009.11.020. [DOI] [PubMed] [Google Scholar]
- Koide A., Abbatiello S., Rothgery L., Koide S. Proc. Natl. Acad. Sci. U S A. 2002;99:1253–1258. doi: 10.1073/pnas.032665299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A., Gilbreth R.N., Esaki K., Tereshko V., Koide S. Proc. Natl. Acad. Sci. U S A. 2007;104:6632–6637. doi: 10.1073/pnas.0700149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunstler J.-U., Seidel G., Bergmann R., et al. Eur. J. Med. Chem. 2010;45:3645–3655. doi: 10.1016/j.ejmech.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Kyte J., Doolittle R.F. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lipovsek D. Protein Eng. Des Sel. 2011;24:3–9. doi: 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main A.L., Harvey T.S., Baron M., Boyd J., Campbell I.D. Cell. 1992;71:671–678. doi: 10.1016/0092-8674(92)90600-h. [DOI] [PubMed] [Google Scholar]
- Miao Z., Levi J., Cheng Z. Amino acids. 2010;41:1037–1047. doi: 10.1007/s00726-010-0503-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M., Arano Y., Mukai T., Saga T., Fujioka Y., Ogawa K., Kawashima H., Konishi J., Saji H. Bioconjug. Chem. 2002;13:491–501. doi: 10.1021/bc010043k. [DOI] [PubMed] [Google Scholar]
- Pimm M.V., Gribben S.J., Bogdan K., Hudecz F. J. Control. Release. 1995;37:161–172. [Google Scholar]
- Ren G., Zhang R., Liu Z., Webster J.M., Miao Z., Gambhir S.S., Syud F.A., Cheng Z. J. Nucl. Med. 2009;50:1492–1499. doi: 10.2967/jnumed.109.064287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schottelius M., Wester H.-J., Reubi J.C., Senekowitsch-Schmidtke R., Schwaiger M. Bioconjug. Chem. 2002;13:1021–1030. doi: 10.1021/bc0200069. [DOI] [PubMed] [Google Scholar]
- Tolmachev V., Friedman M., Sandström M., Eriksson T.L.J., Rosik D., Hodik M., Ståhl S., Frejd F.Y., Orlova A. J. Nucl. Med. 2009;50:274–283. doi: 10.2967/jnumed.108.055525. [DOI] [PubMed] [Google Scholar]
- Tran T., Engfeldt T., Orlova A., Sandström M., Feldwisch J., Abrahmsén L., Wennborg A., Tolmachev V., Karlström A.E. Bioconjug. Chem. 2007;18:1956–1964. doi: 10.1021/bc7002617. [DOI] [PubMed] [Google Scholar]
- Tran T.A., Ekblad T., Orlova A., Sandström M., Feldwisch J., Wennborg A., Abrahmsén L., Tolmachev V., Eriksson Karlström A. Bioconjug. Chem. 2008;19:2568–2576. doi: 10.1021/bc800244b. [DOI] [PubMed] [Google Scholar]
- Wojcik J., Hantschel O., Grebien F., et al. Nat. Struct. Mol. Biol. 2010;17:519–527. doi: 10.1038/nsmb.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahnd C., Kawe M., Stumpp M.T., et al. Cancer Res. 2010;70:1595–1605. doi: 10.1158/0008-5472.CAN-09-2724. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.