

Abstract
We have designed bispecific antibodies that bind one target (anti-Her3) in a bivalent IgG-like manner and contain one additional binding entity (anti-cMet) composed of one VH and one VL domain connected by a disulfide bond. The molecules are assembled by fusing a VH,Cys44 domain via flexible connector peptides to the C-terminus of one H-chain (heavy chain), and a VL,Cys100 to another H-chain. To ensure heterodimerization during expression in mammalian cells, we introduced complementary knobs-into-holes mutations into the different H-chains. The IgG-shaped trivalent molecules carry as third binding entity one disulfide-stabilized Fv (dsFv) without a linker between VH and VL. Tethering the VH and VL domains at the C-terminus of the CH3 domain decreases the on-rates of the dsFv to target antigens without affecting off-rates. Steric hindrance resolves upon removal of one side of the double connection by proteolysis: this improves flexibility and accessibility of the dsFv and fully restores antigen access and affinity. This technology has multiple applications: (i) in cases where single-chain linkers are not desired, dsFvs without linkers can be generated by addition of furin site(s) in the connector that are processed during expression within mammalian cells; (ii) highly active (toxic) entities which affect expression can be produced as inactive dsFvs and subsequently be activated (e.g. via PreScission cleavage) during purification; (iii) entities can be generated which are targeted by the unrestricted binding entity and can be activated by proteases in target tissues. For example, Her3-binding molecules containing linkers with recognition sequences for matrix metalloproteases or urokinase, whose inactivated cMet binding site is activated by proteolytic processing.
Keywords: antibody engineering, bispecific antibodies, disulfide stabilization, protease activation
Introduction
Bispecific antibodies have advantages for applications where dual targeting elicits better potency or specificity than molecules with single functionality. They can simultaneously address individual components of proliferation or inflammatory pathways or viral attachment processes. Therefore, such molecules may confer enhanced therapeutic activity in malignant, inflammatory or viral diseases (Lu et al., 2005; Muller and Kontermann, 2007, 2010; Kontermann, 2012). Furthermore, dual specificity of bispecific antibodies may lead to preferred enrichment at locations which display both targets. The latter may depend on antigen density as well as affinity of the components of bispecific antibodies. Many bispecifics contain fully functional binding entities and therefore will bind upon target exposure equivalent to conventional antibodies. Such molecules can bind to surfaces that only possess one of the desired targets. Moreover, high-affinity binding to abundant antigens on non-target cells may provide an undesired sink which interferes with efficacy of the second binding entity. One possibility to avoid depletion prior to encountering the target tissue would be a bispecific format that permits targeting with one specificity, while cloaking accessibility of the second specificity until the molecule has localized at the tissue of interest.
Many approaches to generate bispecific entities are based on a format that was first described by Morrison and colleagues: single-chain Fvs (scFvs) fused to whole antibodies or antibody fragments (Coloma and Morrison, 1997). Although this format works for selected scFvs, linker peptides between variable domains may not be desirable because it may restrict antigen access. Furthermore, peptide linkers between VH and VL are in some instances not sufficient to confer good stability to the scFv, a feature frequently observed for hybridoma-derived Fvs, which are not selected for stability without accompanying constant regions (e.g. by ‘surviving’ display technologies). In consequence, such scFvs and scFv fusion proteins, including bispecific antibodies have a high tendency to aggregate (Schanzer et al., 2011; Croasdale et al., 2012). One solution to this problem is to replace the linker of scFvs with interchain disulfide bonds between VH and VL, to generate disulfide-stabilized Fvs (dsFvs) (Glockshuber et al., 1990). Among different possibilities to stabilize VH–VL heterodimers by interchain disulfides, the connection between VH,cys44 and VL,cys100 turned out as a versatile position that works for most Fvs without affecting structure and interfering with antigen binding (Jung et al., 1994; Reiter et al., 1995, 1996). This stabilizing principle can be combined with peptide linkers and the resulting disulfide-stabilized single-chain Fvs can be applied to generate bispecific antibodies with good stability (Schanzer et al., 2011; Croasdale et al., 2012).
In contrast to these studies which applied scdsFvs as additional (and inherently functional) binding entities for bispecific antibodies, the approach that we describe here utilizes a dsFv without linker peptides between VH and VL as additional binding module to generate bispecifics that are activateable by proteolytic processing. Utilizing bacterial expression systems, it has been shown that linkerless dsFvs and fusion proteins can be produced with good yields within bacteria, as separate components in inclusion bodies which can be assembled in refolding reactions (Brinkmann et al., 1992; Buchner et al., 1992; Reiter et al., 1996; Schmiedl et al., 2000; Kleinschmidt et al., 2003). In mammalian cells, however, co-expression and assembly of the components of dsFvs without linker is ineffective. This is probably due to inefficient association of the separate dsFv modules (without help of binding protein (BIP), 36) to their partner within the secretion pathway.
The bispecific antibody format that we describe here enables in mammalian cells the production of antibody derivatives containing dsFvs without any linker. The format was subsequently adapted to generate entities with freely accessible binding activity towards one target and another ‘restricted’ specificity. Compared with bivalent IgG arms, this second binding activity is reduced by monovalency and—most important—in its ‘restricted’ form severely compromised by steric hindrance. Activation by specific proteases releases this restriction and thus renders the second binding entity fully functional.
Materials and methods
Generation of expression plasmids
Methods for manipulation of DNA are described in Sambrook and Russell (2001) and molecular biological reagents were used according to the manufacturer's instructions.
Sequences of variable regions of Her3 and cMet binding antibodies were previously described (Brinkmann et al., 2012a,b). The Vector NTI Advance suite version 9.0 was used for sequence data management and processing. Gene segments flanked by endonuclease cleavage sites were prepared by automated gene synthesis (Geneart AG, Regensburg, Germany), integrated into plasmids which were subsequently subjected to DNA sequencing (SequiServe, Vaterstetten, Germany and Geneart AG, Regensburg, Germany). The Her3-heavy chain fusion gene 1 containing ‘knobs-into-holes’ S354C and T366W within CH3 and an additional C-terminal cMET VH region carrying a G44C mutation linked by a (G4S)6 peptide connector was prepared by gene synthesis with flanking BamHI and XbaI restriction sites. A similar BamHI and XbaI module was synthesized encoding the complementary Her3-heavy chain fusion gene 2 containing ‘knobs-into-holes’ Y349C, T366S, L368A and Y407V mutations with a C-terminal cMET VL region carrying a Q100C mutation linked by a (G4S)6 peptide connector. In the same manner, BamHI and XbaI modules were generated that harbored different protease cleavage sites in the (G4S)6 peptide connector, or encoded unmodified heavy and light chains of the Her3 antibody or the VL of the cMET antibody carrying a Q100C. All constructs were designed with a 5′-end DNA sequence coding for a leader peptide. The expression vectors for transient expression in mammalian cells contained in addition to prokaryotic and eukaryotic replication origins (ColE1, oriP) and selection markers (amp, hygromycin) the CMV promoter and a polyA sequence.
Transient expression of immunoglobulin variants in HEK cells
Expression plasmids were co-transfected into human embryonic kidney suspension cells utilizing transfection reagents according to the manufacturer's instructions. The ratios for co-transfection were ‘Knobs-into-holes’ heavy chains 1 and 2 and light chain plasmid DNA in a 1 : 1 : 2 molar ratio. Cell culture supernatants were harvested 7 days after transfection (centrifugation at 14 000 g for 45 min followed by 0.22 µm filtration, storage at −20°C).
Purification of bispecific antibodies
Bispecific antibodies were purified from cell culture supernatants by affinity chromatography on Protein A-Sepharose™ (GE Healthcare, Sweden) and Superdex200 size exclusion chromatography. The sterile filtered cell culture supernatants were applied on a HiTrap ProteinA HP (5 ml) column equilibrated with phosphate-buffered saline (PBS) buffer (10 mM Na2HPO4, 1 mM KH2PO4, 137 mM NaCl and 2.7 mM KCl, pH 7.4). Non-bound proteins were removed by washing with equilibration buffer and desired recombinant protein was recovered from the column with 0.1 M citrate buffer, pH 2.8. The fractions were neutralized with 1 M Tris, pH 8.5, pooled, concentrated (Amicon Device 30 K, Millipore), and loaded on a Superdex200 HiLoad 120 ml 16/60 gel filtration column (GE Healthcare) using 20 mM Histidine, 140 mM NaCl, pH 6 as running buffer. Fractions containing purified bispecific antibodies with less than 5% high molecular weight aggregates were pooled and stored as 1.0 mg/ml aliquots at −80°C.
Protein characterization by biochemical methods and mass spectrometry
Protein concentrations were calculated by measuring OD280, using the molar extinction coefficient based on the amino acid sequence. Purity and molecular weight was evaluated by SDS-PAGE using NuPAGE®, 4–20% Tris-Glycine gels (Invitrogen, USA) followed by Coomassie staining. The integrity of the bispecific antibodies was analyzed by NanoElectrospray Q-TOF mass spectrometry after removal of N-glycans by enzymatic treatment with Peptide-N-Glycosidase F (Roche Molecular Biochemicals). The aggregate content of protein preparations was determined by high-performance SEC (Superdex 200, GE Healthcare). For stability analysis, concentrations of 1 mg/ml of purified proteins were incubated at 4 and 40°C for 7 days and then evaluated by high-performance SEC.
Surface plasmon analysis
Experiments were performed on a Biacore3000 with HBS-P as running and dilution buffer (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), using the BIAevaluation software for data processing. For HER3 binding kinetics, goat anti-human IgG (Jackson ImmunoResearch Europe Ltd) was immobilized to a CM5 Sensor Chip at around 500 RU using the amine coupling kit (GE Healthcare). After capturing, different concentrations of HER3 were passed through the flow cell with a flow rate of 30 µl/min. Kinetic rate constants were obtained by curve fitting according to a 1 : 1 Langmuir binding model. For cMet binding kinetics, a cMet-Fc fusion protein was coupled to a CM5 Sensor Chip using standard conditions at around 500 RU. Different concentrations of antibody were passed over the surface with a 30 µl/min flow rate and the derived curves were fitted to a 1 : 1 Langmuir binding model. Bulk refractive index differences were subtracted automatically by using a blank control flow cell.
Proteolytic processing
The conditions for protease treatment in vitro (protease concentrations, incubation temperature and time) were chosen as to achieve complete processing of the precursor molecules without afflicting further ‘damage’ to the generated products (as assessed by mass spectrometry, see Supplementary data S1). For proteolytic cleavage of the bispecific antibody derivatives, recombinant PreScission protease (GE Healthcare), recombinant active human MMP2 (Calbiochem) or recombinant human u-plasminogen activator (uPA/urokinase, R&D Systems) was used. PreScission is a recombinant protease which specifically cleaves at one defined position in its recognition sequence (Walker et al., 1994). For processing with PreScission, 0.3 mg bispecific antibody derivative containing a PreScission-cleavage site was incubated with 6 units PreScission protease overnight at 4°C in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl and 1 mM EDTA. For MMP2 cleavage, 1 mg bispecific antibody derivative containing a MMP2/9-cleavage site was incubated with 1.2 µg MMP2 overnight at 37°C in PBS. For proteolytic processing with uPA, 0.8 mg bispecific antibody derivative containing an uPA-cleavage site was incubated with 1.5 µg uPA for 48 h at 37°C in 50 mM Tris, pH 8.5 containing 0.01% Tween. The antibody derivatives were subsequently purified by Protein A affinity chromatography and dialyzed against PBS.
Fluorescence-activated cell sorting analysis
For c-Met binding analyses we used A549 cells (cMet positive/Her3 low) and for Her3 binding T47D cells (cMet negative/Her3 positive) were used. 3 × 105 cells per well of a 96-well-plate were incubated with 6.86 nM of the indicated processed or non-processed bispecific antibody in PBS for 1 h on ice, thereafter washed with PBS. Cy5 labeled anti-human antibodies [Jackson Immunoresearch 709176-1490; Cy5 F(ab′)2 Donkey anti-human IgG (H + L)] were added for 1 h on ice to a final concentration of 6.86 nM to detect the bound (bisecific) antibodies. After another washing step with PBS cells were analyzed with the FACS canto II (BD Biosciences).
Immunoblot analysis of Her3 phosphorylation
MCF7 cells maintained in RPMI1640 (Gibco) supplemented with 10% fetal calf serum (FCS) were seeded in full medium and upon reaching 90% confluency after 48 h, further starved for 24 h in RPMI containing 0.5% FCS. Cell were incubated for 1 h with indicated compounds, followed by addition of heregulin (R&D) to a final concentration of 500 ng/ml for another 10 min. Cells were harvested, whole-cell lysates prepared and equal amounts of total protein lysate were subjected to reducing SDS-PAGE followed by immunoblot analysis. For detection, a Y1289 phospho-specific Her3/ErbB3 antibody (Cell Signaling) and a β-actin antibody (Abcam) were used.
Enzyme-linked immunosorbent assay-based quantitation of phosphorylated AKT
A549 cells were propagated in RPMI1640 (Gibco) supplemented with 10% FCS, seeded and incubated for 48 h in medium containing 0.5% FCS. Prior to addition of compounds, medium was replaced for 1 h with RPMI1640 containing 0.2% bovine serum albumin. Ten minutes post addition of antibody derivatives hepatocyte growth factor (HGF) (R&D) was added to a final concentration of 50 ng/ml. After a total of 20 min cells were harvested, lyzed and protein content was determined by the BCA method. Quantitation of phosphorylated AKT was performed with an enzyme-linked immunosorbent assay detecting phosphorylated S473 as well as total AKT (ActiveMotif). For quantitation, the ratio of phosphorylated to total AKT was determined.
Results
Trivalent bispecific antibodies with unrestricted bivalent binding to target 1 and restricted binding to target 2
Our objective was the generation of stable bispecific antibody derivatives that contain, as second binding entity, linkerless dsFvs instead of scFv modules. In our first attempt, we applied the dsFv technology to molecule types that have Fvs connected to the C-termini of IgG heavy-chains. This format was adapted from ‘Morrison-type’ molecules (Coloma and Morrison, 1997) which contain scFvs fused to the C-termini of heavy-chains, with the exception that we generated derivatives that carry only one additional Fv as second binding moiety (Fig. 1A). We removed the linker peptides of the scFv and introduced as replacement interchain disulfides between VH,Cys44 and VL,Cys100 (Jung et al., 1994; Reiter et al., 1995, 1996). The VH,Cys44 of the dsFv remained fused to the CH3 domain as in the original scFv bispecific, but due to removal of the linker peptide, the corresponding VL,Cys100 module was expressed as separate entity. It was previously shown that dsFvs can assemble from separately expressed modules by bacterial inclusion body refolding or periplasmic secretion (Brinkmann et al., 1992; Buchner et al., 1992; Reiter et al., 1996; Schmiedl et al., 2000; Kleinschmidt et al., 2003). However, in contrast to bacterial systems where co-expression of separate entities enables formation of linkerless dsFvs, assembly of dsFv-bipecifics was inefficient in our secretory HEK expression system. Size exclusion chromatography (SEC) indicated a large amount of aggregates to be present in the cell culture supernatants. Only marginal amounts of bispecific antibody-sized protein were recovered after purification by affinity chromatography on Protein A and SEC. The purified material was instable and had a very high tendency to aggregate. A bottleneck for production of dsFvs without linker attachment in mammalian secretion systems may be ineffective assembly of VH and VL domains without the help of chaperons: dsFv components do not contain constant regions that are recognized by BIP (Feige et al., 2009). To overcome this limitation, we connected one component of the dsFv via a connector peptide to the C-terminus of one heavy chain (H-chain), and the corresponding other component to the C-terminus of the second H-chain via another peptide. The resulting proteins are shown in Fig. 1A and the connector peptide sequences are listed in Fig. 1B. The rationale for this approach was that the effective dimerization of H-chains facilitates heterodimerization of dsFv components. To reduce non-productive assembly of molecules containing two VH or two VL modules, complementary knobs-into-holes mutations were engineered into the H-chain of the IgG. These mutations were first devised by Paul Carter and colleagues (Ridgway et al., 1996; Merchant et al., 1998) to force heterodimerization of different H-chains and consist of a T366W (knob) mutation in one H-chain chain and T366S, L368A and Y407V (hole) mutations in the corresponding second H-chain. Our design for generation of dsFv-containing bispecifics had the ‘knob’ on the CH3 domain that was fused to VH,Cys44 and the complementary ‘hole’ on the H-chain that carried VL,Cys100. An additional disulfide bond was engineered into the heavy chain carrying the ‘knob’ mutation (S354C) and also using the mutation Y349C into the heavy chain carrying the ‘hole’ mutation (Ridgway et al., 1996; Merchant et al., 1998). The combination of C-terminal attachment of dsFv components with the knobs-into-holes technology enabled us to generate molecules that have two fully accessible IgG-type binding arms, and one single additional linkerless dsFv at the C-terminus of the H-chain (Fig. 1). The dsFv does not contain a peptide linker that is otherwise present in scFvs. However, both of its components are tethered to CH3. This simultaneous attachment of VH and VL at their N-termini to bulky CH3 domains does not affect the structure of the Fv. Effective antigen binding of such dual-tethered molecules has previously been observed for selected examples (Wu et al., 2007; Brinkmann et al., 2012a,b). In many other cases, however, this format restricts the accessibility towards the antigen because the CDR region points upwards towards the CH3 domain. In addition, tethering at two connection points adds additional constraints. The Fv has a limited degree of rotation when positioned close to the CH3, due to which antigens need to squeeze between CH3 and Fv. This affects accessibility to antigen and reduces affinity, which we indeed observed for the double-connected dsFv moiety of the bispecific antibody (surface plasmon resonance (SPR) data in Table I and Figs. 1C and 3C). Consistent with limited antigen accessibility due to steric hindrance, affinity determination revealed a significantly reduced on-rate for the double-tethered dsFv. Nevertheless, structural integrity of the Fv appears to be intact because once the antigen is bound, the off-rate is the same as that of the parental antibody. The affinity values for binding of the IgG-like accessible arms of the bispecific antibody (which expectedly have full affinity), as well as for the additional double-tethered dsFv, are listed in Table I. The term ‘restricted binding mode’ is used to describe dsFv modules with reduced on-rates due to double tethering of the dsFv components.
Fig. 1.
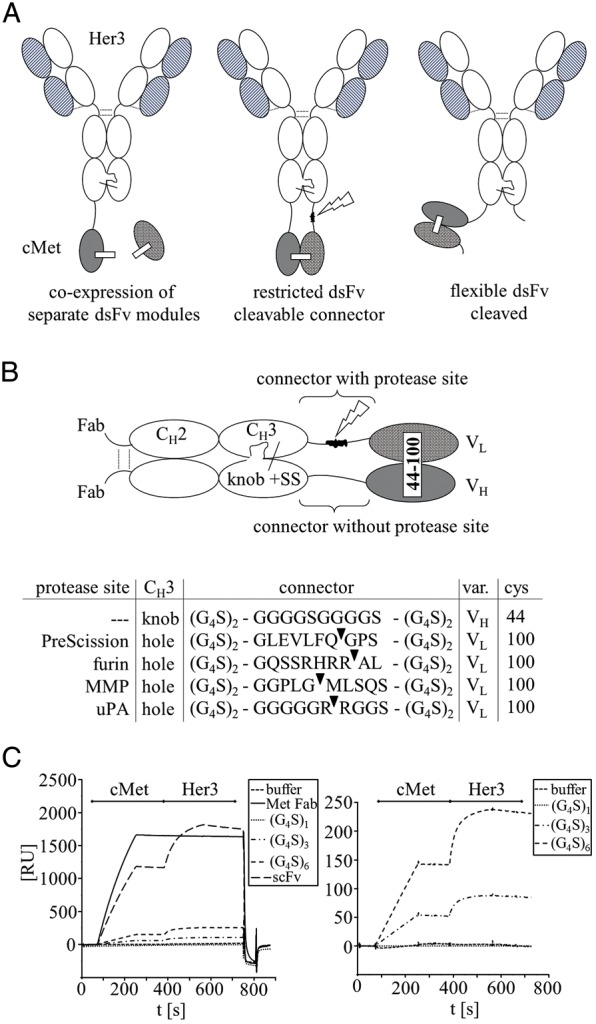
Composition of trivalent bispecific antibody derivatives. (A) Schematic presentation of bispecific antibodies (BsAb) containing dsFvs. (B) Magnification of Fc part with detailed composition of connector-peptides comprising recognition sequences for proteolytic processing during or after expression. (C) SPR sensorgrams with binding kinetics of BsAb containing uncleavable connectors of different length. Sequential binding to cMet and Her3 extracellular domain (cMet, Her3) is shown. Right panel presents a magnification of BsAb to visualize differences.
Table I.
Restricted and unleashed binding of protease-activated bispecific antibodies
| Molecule | Processing | Her3 binding KD (M) | cMet binding Kon (M) | cMet binding Koff (M) | cMet binding KD (M) |
|---|---|---|---|---|---|
| Her3 IgG | n.a. | 2.4 × 10−9 | n.a. | n.a. | n.a. |
| cMet Fab | n.a. | n.a. | 6.9 × 105 | 1.6 × 10−4 | 2.3 × 10−9 |
| PreScission Connector | −(purified precursor) | 2.4 × 10−9 | 5.3 × 102 | 2.0 × 10−4 | 3.7 × 10−7 |
| PreScission Connector | + recomb. PreScission | 2.1 × 10−9 | 2.4 × 104 | 2.0 × 10−4 | 8.4 × 10−9 |
| MMP Connector | −purified precursor | 1.9 × 10−9 | 1.5 × 102 | 3.7 × 10−4 | 2.5 × 10−6 |
| MMP Connector | + recombinant MMP | 1.9 × 10−9 | 2.1 × 104 | 1.5 × 10−4 | 7.1 × 10−9 |
| uPA Connector | −purified precursor | 2.3 × 10−9 | 9.7 × 103 | 1.9 × 10−4 | 2.0 × 10−8 |
| uPA Connector | + recombinant uPA | 2.2 × 10−9 | 4.8 × 104 | 1.6 × 10−4 | 3.3 × 10−9 |
| Furin Connector | processing in expression | 2.1 × 10−9 | 3.1 × 104 | 1.9 × 10−4 | 6.1 × 10−9 |
| Furin Connector 2* | processing in expression | 2.1 × 10−9 | 1.7 × 104 | 3.3 × 10−4 | 1.9 × 10−8 |
| Format: 1st CH3-connector uncleavable; second CH3-connector cleavable | processing verified by SDS-PAGE & Mass Spec | Her3 binding not affected because modifications are on C-termini of CH3 | Reduced on-rate in unprocessed form | Off-rate unchanged | Reduced binding in unprocessed form; processing restores binding to cMet |
*Reducing the lengths of the furin connector by 3 G4S motifs generates Furin connector 2 with the sequence GGGGS-GQSSRHRRAL (the original furin connector sequence is shown in Fig. 1B).
Fig. 3.

Proteolytic activation of the restricted dsFv moieties. Coomassie blue-stained reducing SDS-PAGE of BsAb derivatives containing restricted dsFvs before and after proteolytic processing. (A) Protein-A elution profile of PreScission cleaved purified protein (ct.). (B) Proteins that contain furin cleavage sites are activated already during expression (cf. Fig. 2). Proteins containing MMP or uPA cleavage sites are generated as restricted molecules (cf. Fig. 2) and become activated upon exposure to MMP or uPA. (C) Exemplary SPR sensorgrams of unprocessed and processed BsAb with PreScission cleavage site. Shown are titration series with cMet extracellular domain as ligand.
Expression and purification of stable trivalent antibody derivatives containing linkerless dsFv
Transient expression was applied for production of secreted bispecific antibody derivatives. Plasmids encoding L-chains (light chains) and modified H-chains were co-transfected into HEK suspension cells. Culture supernatants containing secreted antibody derivatives were harvested after one week. These supernatants could be frozen and stored at −20°C before purification without affecting yields. The bispecific antibodies were purified from supernatants by Protein A-affinity chromatography and SEC in the same manner as conventional IgGs which proves that they were fully competent to bind Protein A (Metz et al., 2011). Expression yields within cell culture supernatants were lower than transiently expressed unmodified antibodies but still within a reasonable range. After completion of all purification steps, yields ranging between 4 and 20 mg/l of homogeneous protein were obtained (4–20 mg/l for PreScission, 4 mg/l for Furin, 6–15 mg/l for uPA (urokinase plasminogen activator) and 10–20 mg/l for the MMP (matrix metalloproteinase) connector containing variants). Despite having no peptide linker between VH and VL of the additional dsFv moiety, stability analyses revealed no indication for Fv disintegration or aggregation. The proteins were stable and freeze–thaw cycles were well tolerated. Size, homogeneity and composition of trivalent bispecific antibody derivatives and their components under reducing and non-reducing conditions are shown in Fig. 2A and B, respectively.
Fig. 2.

Expression and purification of trivalent BsAb containing dsFv. (A) Coomassie blue-stained reducing SDS-PAGE of protein preparations after Protein-A and size exclusion (SEC) purification (H = cleaved; H1 = H + VH; H2 = H + VL). Note that the disulfide-connected VL domains are visible as separate bands in molecules that are expressed without second or with furin connector. BsAb which contain connectors that are recognized by the proteases Prescission, MMP or uPA are produced with extended H-chains. (B) Exemplary SEC profile of the trivalent bispecific dsFv-containing antibody with a MMP2/9 site in its connector demonstrates pure monomeric compositions free of aggregates.
Connector peptides for specific activation of the restricted binding site by proteolytic processing
Double tethering of dsFv components to CH3-domains restricts antigen access and thereby reduces the functionality of the dsFv. Rotation of Fvs around one connector peptide would most likely facilitate access to antigen, but the fusion of a dsFv at two connection points does not permit a large degree of flexibility and hinders rotation. To re-activate the inactivated binding functionality of such restricted dsFvs moieties, we introduced specific protease recognition sites into one of the two connector peptides (shown in Fig. 1B). Our objective for that was to utilize proteolytic cleavage for the release of only one of the two connections. Upon proteolytic processing, the dsFv is still covalently linked to the IgG backbone of the bispecific antibody by its remaining intact connector. But in contrast to double connection, attachment at just one flexible connection point improves flexibility and allows rotation, and hence increases access for antigen binding. Figure 1B shows different connector sequences which we applied to enable processing by proteases. The non-cleavable connector is composed of six G4S repeats. Such G4S repeats of different lengths are frequently applied for generation fusion proteins composed of different domains (Muller and Kontermann, 2007, 2010; Kontermann, 2012; Metz et al., 2011). For proteolytic processing, we introduced specific recognition sequences into the central region of this connector: One connector contains a site that is cleaved by the PreScission protease. This protease can restore functionality of Fv modules that are expressed in restricted form. Processing with PreScission can be applied after purification. Other connector sequences that we introduced contain recognition sequences for furin, a protease that is present in endosomal and secretory compartments of mammalian cells, including in HEK cells. We chose furin sites to enable dsFv processing within the expression process. Bispecific entities carrying the restricted dsFv will encounter furin during secretion. Thereby, already processed and hence fully functional proteins should be secreted by the cells. Further connector sequences that we generated harbor recognition sequences for MMP2 and 9 or uPA. Many mammalian cells such as HEK cells that we use for recombinant expression do not express MMPs or uPA. Therefore, bispecific entities containing restricted dsFvs are expressed in these cells as inactive precursors, with the potential to become activated upon exposure to MMPs or uPA.
Bispecific antibodies containing a PreScission site are expressed in restricted form and can be activated in downstream processing
One application of bispecific antibody formats that contain restricted binding modules is to express them in restricted form and activate them post-production in downstream processing. As an example for this approach, we produced a Her3-cMet bispecific antibody carrying a restricted cMet dsFv module, and subsequently reactivated the dsFv activity by cleavage with the PreScission protease (see Materials and methods section for details). Figure 3A shows that after expression and purification from cell culture supernatants, bispecific Her3-cMet entities are obtained with the components of the dsFv connected to H-chains (left panel). Reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analyses show the unmodified L-chain of the Her3-binding entity, and in addition two protein bands of ∼65 kDa. These represent the two H-chains (50 kDa) to which additional connector peptides (2 kDa) and VH or VL domains (13 kDa) are fused at their C-termini. The affinity (prior to and after PreScission processing) of these bispecific molecules towards their fully accessible Her3 binding arms is the same as that of the parent antibody (Table I). In contrast, the affinity of the restricted dsFv moiety towards cMet is compromised. SPR analyses show a greatly reduced affinity compared to that of the Fab of the parent antibody (Table I). The reason for this reduced affinity lies in a reduced on-rate. Cleavage of the PreScission site within the connector between CH3 and VL resolves the restriction of the dsFv. This gives rise to molecules that have the dsFv still covalently attached to the IgG, but by only one connector. Reducing SDS-PAGE analyses prove that after cleavage one of the extended 65 kDa H-chains is converted to H-chain only (52 kDa) and to an additional VL domain of 13 kDa. These molecules are still held together by the Cys44–100 disulfide bond between VH and VL. Processing at the correct position and identity of the products of precursor conversion was confirmed by mass spectrometry (see Supplementary data S1 for experimental details and results). A comparison of affinities of restricted and processed forms of the bispecific antibodies are listed in Table I: as expected, processing at the dsFv moiety did not change the binding of Her3 antigen to the previously already fully accessible Fab arms. On the other hand, diminishing steric hindrance by cleaving one connector greatly improved the on-rate of the binding of the linkerless dsFv module to cMet. Affinities of the unleashed dsFv were fully restored to affinity levels of the parent antibody fragment.
Bispecific antibodies containing furin sites become effectively processed during expression and display full functionality of linkerless dsFv
Furin sites within connectors can be used for production of bispecific antibodies containing linkerless dsFvs with unrestricted functionality. Furin is present in endocytic and secretory vesicles, in the trans-Golgi network and in some cases on cell surfaces of many mammalian cells. Its recognition sites, frequently containing a RXK/RR motif, are present in a variety of secreted precursor proteins such as pro-transforming growth factor β1 or pro-von Willebrand Factor (Nakayama, 1997). We therefore selected this recognition sequence to generate connector sequences containing a furin recognition site (Fig. 1B). Because furin is present in the trans-Golgi network and in secretory vesicles, cleavage can occur within cells during the production. The expression yields of furin processed unrestricted dsFv molecules that we obtained was similar to those observed for restricted molecules. The dsFvs appear to become fully folded and assembled prior to encountering compartments with furin activity. Figure 2A (left panel) confirms that after expression and purification, processed molecules are obtained. Reducing PAGE shows (in addition to the standard Her3 L-chain) one larger H-chain of 65 kDa that carries the VH of the dsFv, and another H-chain that has been converted to unmodified H-chain size (52 kDa). The additional VL domain of 12 kDa is also detectable. Since the purification procedure involved Protein A and SEC (both of which would not recover unlinked VL domains), detection of these domains indicate the generation of fully processed functional bispecific antibodies. SEC further confirmed that all domains are held together by stable disulfides (schematically shown in Fig. 1A). Processing by furin at the correct position and identity of the products of precursor conversion was confirmed by mass spectrometry (Supplementary data S1). The furin recognition sites (Fig. 1) left upon cleavage two basic amino acids, RR or KR, at the carboxy-terminus of the processed H-chain. Our mass spectrometry analyses showed that these two amino acids become also quantitatively removed (by carboxypeptidases) during expression (Supplementary data S1). Because the processing via furin occurs during the expression process, the preparations obtained after purification are bispecific entities with fully active linkerless dsFvs. This was confirmed by SPR analyses (Table I). All binding entities of the bispecific antibody, those recognizing Her3 as well as the dsFv that binds cMet, have unrestricted binding capability. Their affinity to Her3 and cMet is comparable to that of unmodified antibodies (Her3) or Fab fragments (cMet), respectively.
Bispecific antibodies containing MMP2/9 or uPA sites are expressed and purified in restricted form and become activated upon exposure to cognate proteases
The introduction of sequences into the connector which are recognized by MMP2/9 or uPA provides the option to produce bispecific dsFv-containing entities whose second binding entity is inactive until it encounters these proteases. MMPs and uPA are expressed in some disease tissues, for example in the environment of tumors or inflamed tissues (Liotta et al., 1980; Stetler-Stevenson, 1994; Ruppert et al., 1997; van't Veer et al., 2002; Minn et al., 2005; Cudic and Fields, 2009; Bao et al., 2010; Mukhopadhyay et al., 2010; Scott and Taggart, 2010). The sequences GPLGMLSQ, GPLGLWAQ and GPLGIAGQ are substrates for MMP2 and MMP9 (Netzel-Arnett et al., 1991, 1993), and the peptide sequence GGGRR has been shown to be a substrate for uPA (Chung and Kratz, 2006). These cleavage sites were incorporated into connector sequences to generate dsFv-fusions which can become processed by MMPs or uPA (Fig. 1B). The HEK cells that we use for recombinant expression do not have significant levels of MMPs or uPA. Because of that, expression and purification of entities with such connectors resulted in restricted molecules with two extended H-chains. Final yields after affinity chromatography on Protein-A and SEC purification were between 6 and 10 mg/l, and the composition of purified molecules was virtually identical to the bispecific variants that contained the PreScission site (see above). SDS-PAGE revealed (in addition to the L-chain of the Her3-entity) the presence of two protein bands at the height of 65 kDa. These represent the H-chains (50 kDa) that carry additional connector peptides (2 kDa) and VH or VL domains (13 kDa) at their C-termini. Cleavage of the MMP2/9 or uPA site (see Materials and methods section for details) within the connector between CH3 and VL resolves the restriction of the dsFv and gives rise to unleashed dsFvs with full binding functionality. Figure 3 demonstrates that MMP-site-containing connectors are cleaved by MMP2- and uPA-site-containing connectors are cleaved upon exposure to uPA. The result of this processing is visible as conversion of one of the extended H-chains to its unmodified size (52 kDa), and appearance of an additional VL domain of 13 kDa. While cleaved, the molecules are stably held together by the VH–VL,Cys44–100 disulfide bond. A comparison of affinities of restricted and processed forms of the bispecific antibodies is listed in Table I: processing by MMP or uPA did not alter the binding to the already fully accessible antigen Her3. This indicates that MMP or uPA specifically cleaves its recognition sequence in the connector, but not other positions of the antibody (such as exposed CDRs). Processing at the correct position and identity of the products of precursor conversion by MMP or uPA was confirmed by mass spectrometry (see Supplementary data S1 for experimental details and results). Resolving steric hindrance by MMP or uPA processing completely restored functionality of the dsFv, in the same manner as shown above for cleavage by PreScission or furin and hence fully restored the affinity for cMet.
Bispecific antibodies with protease cleavage sites are functionally active in vitro
To compare activity of restricted versus activated bispecific antibodies, cellular binding was assessed either in T47D cells which express Her3 but no cMet or in A549 cells with high cMet expression and low Her3 levels. By flow cytometry, binding of the unprocessed and processed antibodies to cell surface receptors replicated the findings already obtained by SPR analysis. While Her3 binding was unaffected for all constructs, full cMet binding activity required release of the dsFv from one connector (Fig. 4 and Supplementary Fig. S2 for unprocessed and furin-processed molecules on MKN45 cells). In addition, the ability of processed and unprocessed antibodies to inhibit ligand-mediated Her3 receptor phosphorylation was evaluated in MCF-7, a cell line in which Her3 signaling can be activated by addition of heregulin (Fig. 5). As expected, all antibodies were able to suppress Her3 phosphorylation in a similar manner. Activation of the cMet pathway triggers downstream phosphorylation of AKT. This readout was used to monitor activity of the cMet dsFv. For this purpose, A549 cells were treated with HGF, the ligand of cMet, to specifically activate AKT by this pathway. Inhibition was measured relative to stimulated cells (Fig. 5C). Furin-activated dsFvs inhibited AKT phosphorylation, which further supports that functional activity is mediated by unleashing of the dsFv. As a control, bispecific antibody with an uncleaved PreScission site was compared to the respective in vitro cleaved construct. Only the cleaved construct was able to efficiently inhibit AKT phosphorylation which corresponded to the binding characteristics of the tethered or unleashed dsFv. Finally, MMP2- and uPA-cleavage-site-containing antibodies were cleaved in vitro with recombinant proteases and assessed similarly. The activity of these molecules was the same as those of furin-processed or PreScission-activated antibody derivatives, and the activity of PreScission and MMP processed molecules was significantly higher than that of their inactivated precursor molecules (Fig. 5C). Only the uPA connector containing precursor molecule (whose binding potency was reduced but to a lower degree in unprocessed form) inhibited AKT phosphorylation in the same manner as the processed mature form. This may indicate that a high level of inactivation is required to abolish activity of molecules that are enriched on cell surfaces in cellular assays.
Fig. 4.

Cellular binding of restricted and unrestricted trivalent BsAb. Binding of the bivalent unrestricted Her3-modules to Her3-expressing, cMet-negative T47D cells is shown on the left panels. Binding of the different restricted cMet-modules to Her3-low cMet-expressing A549 cells is shown on the right panels (gray fill = isotype control). Weak binding is observed for the restricted modules while protease-processing leads to full binding and accumulation on cells (solid line = uncleaved; dashed line = processed). Uncleaved Prescission connector BsAb was used as control for furin-connector containing BsAb.
Fig. 5.

Cellular activity of trivalent bispecific antibodies. (A) MCF7 cells were incubated with the respective antibodies (triangle: 0.1 and 1.0 µg/ml) and the Her3 ligand Heregulin. Whole-cell lysates were used to detect phosphorylated (p-Her3) and total Her3 (t-Her3). Note that the Her3 antibody induces internalization. Beta-actin was used as loading control. Reduction of phosphorylated Her3 levels is observed for all constructs and independent of previous protease treatment. (B) A549 cells were stimulated with the cMet ligand hepatocyte growth factor (HGF) and the indicated antibodies. Dose-dependent inhibition of cMet phosphorylation (p-Met) was observed. Total cMet (t-Met) and beta-actin bands were unchanged. (C) A549 cells were stimulated with HGF and treated with the indicated antibodies. The ratio of phosphorylated and total AKT was used as readout to determine efficacy of the restricted and unrestricted cMet binding moiety (triangle: 1.9, 7.5, 30 µg/ml BsAb). Consistent lack of activity was observed for unprocessed PreScission BsAb while BsAb with uncleaved MMP connector displayed activity in some instances.
Discussion
The assembly of linkerless dsFvs from separately expressed components has previously been shown to be successful in bacterial expression systems. For example, dsFvs and dsFv-fusion proteins can be assembled from separately purified inclusion bodies in refolding reactions. Similarly, co-secretion of separate domains and subsequent assembly to functional dsFvs is achieved in the periplasm of Escherichia coli without the need of a peptide linker connecting the domains (Brinkmann et al., 1992; Buchner et al., 1992; Reiter et al., 1996; Schmiedl et al., 2000; Kleinschmidt et al., 2003). In mammalian cells, however, co-expression and secretion of separate components of dsFvs has proven to be very inefficient. Compared with expression of IgG or IgG-scFv fusion proteins, equivalent formats which harbor stabilizing disulfides but do not carry an additional linker peptide between variable domains are produced with very low yields or cannot be made at all. The reason for that is most likely inefficient assembly of the non-linked VH–VL heterodimer in the secretion pathway of mammalian cells. Like VH and VL domains, whole IgGs are expressed as separate proteins and need to assemble in the secretory pathway. Chain assembly of IgGs is aided by chaperons (BIP) present in secretory vesicles (Knarr et al., 2002). These chaperons bind to the constant regions of antibodies (Feige et al., 2009). The small VH and VL domains of dsFvs do not carry constant regions and therefore may not be able to effectively recruit chaperons. This suggests that inefficient heterodimer assembly from separately expressed domains could be the bottleneck that restricts efficient production of linkerless dsFvs in mammalian cells. To test this hypothesis and to overcome this bottleneck, we chose to use the strong heterodimerization tendency of knobs-into-holes modified IgG-heavy chains to bring together the isolate VH and VL domains. The observation of good expression yields of correctly folded stable molecules containing linkerless dsFvs indicated (i) that heterodimerization is a bottleneck in VH–VL assembly of the dsFv moiety, and (ii) that the dimerization force of IgG H-chains can replace Ckappa-CH1 (chaperon)-mediated heterodimerization. Applying double tethering to CH3 domains (Fig. 1) of dsFv components permitted the production of dsFv-containing bispecific antibodies within the similar yield range as that of other single-chain fusion proteins (Glockshuber et al., 1990; Metz et al., 2011). Tethering both domains of the dsFv at their amino termini to the IgG restricts antigen access to the binding region of the dsFv, especially for large antigens such as receptors. Similar observations of steric hindrance and reduced on-rates have been described for some bispecific antibodies with the dual-variable-domain (DVD) format (Wu et al., 2007). It appears that the restriction of antigen binding that we observe in our format is determined not only by dual tethering, but that it depends to some degree also on the sequence of the connecting peptides. One example for that are molecules that harbor the uPA site, which has still some reasonable binding activity (and as consequence biological activity) even in unprocessed form. It appears that remaining activity depends more on sequence composition than on linker length since the uPA linker has the same length, but a different composition than the other linkers. We have furthermore generated and analyzed GlySer linkers of different length (Fig. 1C), but with those we detected only some influence of linker lengths on antigen binding. Regardless of which linker we applied, full antigen access was not achieved for this format and this antigen. Thus, the critical factor that determines antigen access may be steric orientation rather than distance from the CH3 domains, or a combination of both.
The restriction of steric hindrance and reduced on-rates can be fully resolved within our molecules by proteolytic processing of one of the two connector peptides. This ‘unleashing’ fully restores the affinity of the dsFv because it now has full flexibility and rotational freedom around the remaining single flexible connector. In fact, freedom to bind antigen may be even greater for such dsFv molecules than for similar scFv entities which still contain a single-chain linker and hence may have some constraints at N- and C-termini of variable domains.
For the generation of bispecific antibody derivatives with full accessibility to both antigens, restriction of the dsFv can be resolved either by proteolysis after expression (e.g. by PreScission cleavage) or during protein production (e.g. with furin). In the latter case, a furin site is introduced into one connector. Because furin is present in the trans-Golgi network and in secretory vesicles, molecules become processed during production. Where and to what degree does furin cleavage occur within the secretion process of bispecific antibodies? Considering that we observed co-expression/secretion of separate dsFv components to be very inefficient and with low yields, efficient early furin cleavage before dsFv assembly is highly unlikely, as early processing would generate similar low yields. Thus, we assume that processing occurs in compartments or on the cell surface after the dsFvs have assembled as (restricted) precursor molecules. Mass spectrometry data indicated complete processing of proteins that were produced in HEK cells with endogenous levels of furin (Supplementary data S1). Thus, furin cleavage does not seem to be a bottleneck during expression and processing in laboratory scale. The conclusion that furin cleavage does not provide any limitation to expression is further supported by the observation that we found no variance in processing for two different constructs that contained distinct furin sites. The production of our bispecific molecules appears to follow the classical path of secreted pre-pro-proteins: Secretion with processing of the signal sequences, folding and assembly to a precursor protein (inactive dsFv), followed by furin activation to the mature active protein that is released from the cell. In addition to the production of dual-activity harboring bispecific antibodies, entities with one inactivated binding moiety can be produced. One could envision bispecific molecules produced as inactivated precursor molecules that become activated upon exposure to uPA or MMPs. Thus, it may be possible to apply such or similar entities for antibody-mediated targeting followed by specific activation on target tissues.
Supplementary data
Funding
Funding to pay the Open Access publication charges for this article was provided by Roche Pharma Research and Early Development (pRED).
Supplementary Material
Acknowledgements
We thank Rosa-Maria Busl-Schuller, Klaus Mayer, Michael Antony, Christoffer von Schwerin, Sandra Praßl, Silke Schneid-Müller, Cornelia Wagner and Martina Wagner for excellent technical assistance.
Footnotes
Edited by James Huston
References
- Bao W., Fu H.J., Jia L.T., et al. Arch. Biochem. Biophys. 2010;499:49–55. doi: 10.1016/j.abb.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Brinkmann U., Buchner J., Pastan I. Proc. Natl Acad. Sci. USA. 1992;89:3075–3079. doi: 10.1073/pnas.89.7.3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann U., Haas A.K., Metz S., Schanzer J. 2012a. (March 1) World Intellectual Property Organization Publication WO/2012/025530 (patentscope.wipo.int)
- Brinkmann U., Croasdale R., Metz S., Schanzer J., Sustmann C., Umana P. 2012b. (March 1) World Intellectual Property Organization Publication WO/2012/025525. (patentscope.wipo.int)
- Buchner J., Pastan I., Brinkmann U. Anal. Biochem. 1992;205:263–270. doi: 10.1016/0003-2697(92)90433-8. [DOI] [PubMed] [Google Scholar]
- Chung D.E., Kratz F. Bioorg. Med. Chem. Lett. 2006;16:5157–5163. doi: 10.1016/j.bmcl.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Coloma M.J., Morrison S.L. Nat. Biotechnol. 1997;15:159–163. doi: 10.1038/nbt0297-159. [DOI] [PubMed] [Google Scholar]
- Croasdale R., Wartha K., Schanzer J.M., et al. Arch. Biochem. Biophys. 2012;526:206–218. doi: 10.1016/j.abb.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Cudic M., Fields G.B. Curr. Protein Pept. Sci. 2009;10:297–307. doi: 10.2174/138920309788922207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige M.J., Groscurth S., Marcinowski M., Shimizu Y., Kessler H., Hendershot L.M., Buchner J. Mol. Cell. 2009;34:569–579. doi: 10.1016/j.molcel.2009.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glockshuber R., Malia M., Pfitzinger I., Pluckthun A. Biochemistry. 1990;29:1362–1367. doi: 10.1021/bi00458a002. [DOI] [PubMed] [Google Scholar]
- Jung S.H., Pastan I., Lee B. Proteins. 1994;19:35–47. doi: 10.1002/prot.340190106. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt M., Rudolph R., Lilie H. J. Mol. Biol. 2003;327:445–452. doi: 10.1016/s0022-2836(03)00141-4. [DOI] [PubMed] [Google Scholar]
- Knarr G.U., Kies U., Bell S.M., Mayer M., Buchner J. J. Mol. Biol. 2002;318:611–620. doi: 10.1016/S0022-2836(02)00166-3. [DOI] [PubMed] [Google Scholar]
- Kontermann R. MAbs. 2012;4:182–197. doi: 10.4161/mabs.4.2.19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta L.A., Tryggvason K., Garbisa S., et al. Nature. 1980;284:67–68. doi: 10.1038/284067a0. [DOI] [PubMed] [Google Scholar]
- Lu D., Zhang H., Koo H., et al. J. Biol. Chem. 2005;280:19665–19672. doi: 10.1074/jbc.M500815200. [DOI] [PubMed] [Google Scholar]
- Merchant A.M., Zhu Z., Yuan J.Q., et al. Nat. Biotechnol. 1998;16:677–681. doi: 10.1038/nbt0798-677. [DOI] [PubMed] [Google Scholar]
- Metz S., Haas A.K., Daub K., et al. Proc. Natl Acad. Sci. USA. 2011;108:8194–8199. doi: 10.1073/pnas.1018565108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn A.J., Minn A.J., Gupta G.P., et al. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Sypek J., Tavendale R., et al. J. Allergy Clin. Immunol. 2010;126:70–76. doi: 10.1016/j.jaci.2010.03.027. [DOI] [PubMed] [Google Scholar]
- Muller D., Kontermann R.E. Curr. Opin. Mol. Ther. 2007;9:319–326. [PubMed] [Google Scholar]
- Muller D., Kontermann R.E. BioDrugs. 2010;24:89–98. doi: 10.2165/11530960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Nakayama K. Biochem. J. 1997;327:625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzel-Arnett S., Fields G.B., Birkedal-Hansen G.B., Van Wart H.E. J. Biol. Chem. 1991;266:6747–6755. [PubMed] [Google Scholar]
- Netzel-Arnett S., Sang Q.X., Moore W.G., et al. Biochemistry. 1993;32:6427–6432. doi: 10.1021/bi00076a016. [DOI] [PubMed] [Google Scholar]
- Reiter Y., Brinkmann U., Jung S.H., Pastan I., Lee B. Protein Eng. 1995;8:1323–1331. doi: 10.1093/protein/8.12.1323. [DOI] [PubMed] [Google Scholar]
- Reiter Y., Brinkmann U., Lee B., Pastan I. Nat. Biotechnol. 1996;14:1239–1245. doi: 10.1038/nbt1096-1239. [DOI] [PubMed] [Google Scholar]
- Ridgway J.B., Presta L.G., Carter P. Protein Eng. 1996;9:617–621. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- Ruppert C., Ehrenforth S., Scharrer I., Halberstadt E. Cancer Detect. Prev. 1997;21:452–459. [PubMed] [Google Scholar]
- Sambrook J., Russell D.W. Molecular Cloning: A Laboratory Manual. 3rd edn. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Schanzer J., Jekle A., et al. Antimicrob. Agents Chemother. 2011;55:2369–2378. doi: 10.1128/AAC.00215-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedl A., Breitling F., Dubel S. Protein Eng. 2000;13:725–734. doi: 10.1093/protein/13.10.725. [DOI] [PubMed] [Google Scholar]
- Scott C.J., Taggart C.C. Biochimie. 2010;92:1681–1688. doi: 10.1016/j.biochi.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Stetler-Stevenson W.G. Invasion Metastasis. 1994;14:259–268. [PubMed] [Google Scholar]
- van 't Veer L.J., Dai H., van de Vijver M.J., et al. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Walker P.A., Leong L.E.C., Ng P.W.P., Tan S.H., Waller S., Murphy D., Porter A.G. Biotechnol. 1994;12:601–605. doi: 10.1038/nbt0694-601. [DOI] [PubMed] [Google Scholar]
- Wu C., Ying H., Grinnell C., et al. Nat. Biotechnol. 2007;25:1290–1297. doi: 10.1038/nbt1345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.