

Abstract
Thymine glycol (Tg), one of the oxidized bases formed in DNA by reactive oxygen species, is repaired by the DNA glycosylases such as NEIL1, NTH1 and Endo III. In our recent studies, we showed that NEIL1’s catalytic efficiency and lesion specificity are regulated by an RNA editing adenosine deamination reaction. In this study, we synthesized oligodeoxynucleotides containing 2′-fluorothymidine glycol with either ribo or arabino configuration and investigated the binding of these modified DNAs with the unedited and edited forms of human NEIL1 along with E. coli Endo III. For the two forms of hNEIL1, binding affinities to FTg-containing DNA were similar indicating the editing effect is more subtle than to simply alter substrate affinity. While the NEIL1 binding to FTg-containing DNAs was largely insensitive to C5 and 2′ stereochemistry, a preference was observed for the FTg-G pair over the FTg-A pair. In addition, we found that optimal binding is observed with Endo III and duplex DNA with riboFTg (5S) paired with dG. The modified DNAs reported here will provide useful tools for further characterizing the interaction between DNA repair glycosylases and thymine glycol containing DNA.
Keywords: DNA repair, RNA editing, thymidine glycol, NEIL1, Endo III
Introduction
Oxidized base lesions arise in DNA at rates of thousands per day as a result of endogenous metabolic activity as well as from oxidative stress induced during inflammation, radiation or toxic agents.[1,2] The human protein NEIL1 plays a key role in the initiation of base excision repair of oxidized base lesions by catalyzing the cleavage of the N-glycosidic linkage.[3] NEIL1 is capable of removing a wide array of modified DNA bases including thymine glycol (Tg), 5-hydroxyC (5-OHC), 5-hydroxyU (5-OHU), dihydrothymine (DHT), dihydrouracil (DHU), the formamidopyrimidines (FapyG and FapyA), guanidinohydantoin (Gh), spiroiminodihydantoin (Sp) and psoralen photoadducts.[3–6] Our recent studies have shown that NEIL1’s catalytic efficiency and lesion specificity are regulated by an RNA editing adenosine deamination reaction that occurs at a specific codon in the NEIL1 pre-mRNA.[7] Deamination at C6 of adenosine (A) in RNA generates inosine (I) at the corresponding nucleotide position. Since inosine is decoded as guanosine during translation, this modification can lead to codon changes (recoding) and the introduction of amino acids into a gene product not encoded in the gene. In the case of NEIL1, deamination of adenosine within the AAA codon for K242 converts it to one for arginine (AIA, read as AGA). This reaction is catalyzed by the RNA editing enzyme ADAR1 that is induced by interferon and known to play a role in inflammation. Importantly, we observed large differences in glycosylase activity between K242 and R242 NEIL1 with DNA substrates containing Tg. Indeed, the unedited form of NEIL1 showed substantially enhanced glycosylase activity over edited NEIL1 with the Tg lesion (41-fold faster in duplex DNA when paired with A). Thymine glycol (Tg) is the most common pyrimidine base modification produced under oxidative stress and ionizing radiation.[2,8] Tg is also a substrate of the human DNA glycosylase NTH1 and, though not miscoding, its ability to strongly block DNA replication makes it a toxic lesion in cells. The bacterial protein Endo III is also capable of excising Tg from DNA.[9] An approach that has been exploited by several laboratories including ours, in studying BER glycosylases is the use of a 2′-F substitution on the damaged nucleotide in order to prevent (or stall) glycosidic bond hydrolysis.[10–14] Oligonucleotides containing 2′-FTg nucleotides will serve as useful tools for revealing the molecular details associated with damage recognition by NEIL1 and the effects of editing. Here we describe the synthesis of 2′-fluorothymidine glycol phosphoramidites with either ribo or arabino configuration at the 2′ position, their use in the generation of 2′-fluorothymidine glycol-containing DNAs and the binding of these modified DNAs with the unedited and edited forms of human NEIL1 and E. coli Endo III.
Results
Synthesis of 2′-fluorothymidine glycol phosphoramidite building blocks
The 2′-fluoronucleoside modification is well-known to slow the cleavage reaction of N-glycosylases.[10–14] These reactions occur with an intermediate 1′–4′ oxocarbenium ion and an electronegative fluorine atom at the adjacent 2′ position destabilizes the transition state to that intermediate.[15] Indeed, the 2′-ribofluorothymidine glycol 5R and 5S isomers have been incorporated into DNA and were shown to be resistant to the glycosylase activity of E. coli Endo III.[14] In addition, only with the 5S isomer containing duplexes was a DNA-protein complex observed in electrophoretic mobility shift assays (EMSA). Importantly, there have been no prior reports of the binding properties of NEIL1 with fluorinated damaged nucleotide-containing DNA.
We synthesized 2′-fluorothymidine glycol of both ribo and arabino configuration (Schemes 1–2). While the ribo-configured compound was known, the reported phosphoramidite bears tert-butyldimethylsilyl (TBDMS) protecting groups on the glycol hydroxyls necessitating an additional deprotection step for the modified DNA.[14] We wished to investigate the use of acetyl groups for this protection since they could be removed in the K2CO3/MeOH step standard to mild synthetic DNA deprotection. We also chose to prepare the arabino-configured isomer since this isomer is expected to adopt a sugar conformation more compatible with a B-form duplex and may more effectively mimic the natural DNA lesion.[16,17] The synthesis of 2′-ribofluorothymidine glycol (riboFTg) phosphoramidite 6 started with commercially available 5-methyluridine. Trityl-protected 2′-ribofluorothymidine 1 was synthesized according to the literature (Scheme S1),[18] and then oxidized with OsO4 to afford two isomers of thymidine glycol 2 (Scheme 1).[19] The ratio of isolation yields for these products was 3:5. The configuration of each isomer was determined from the 1H-NMR NOE data on the basis of a previous report.[14] For the major isomer obtained by the OsO4 oxidation, a relatively strong NOE was observed between H6 and H2′, whereas the minor one showed only a slight effect (Figure S1). From this result, the thymine glycols in the major and minor products were assigned as (5S,6R) (2b) and (5R,6S) (2a), respectively. We then protected both hydroxyl groups of the thymine glycol with acetyl groups.[20,21] The resulting compounds 3a, 3b were treated with trifluoroacetic acid to give the 5,6-O-acetyl-2′-ribofluorothymidine glycols 4a, 4b which were converted to the phosphoramidite building blocks 6a, 6b by standard procedures.
Scheme 1.

(a) see Scheme S1; (b) OsO4, pyridine, 31% (5R,6S) and 52% (5S,6R); (c) Ac2O, DMAP, pyridine, 3a: 97%, 3b: 97%; (d) TFA, CH2Cl2, 4a: 92%, 4b: 92%; (e) DMTrCl, pyridine, 5a: 83%, 5b: 88%; (f) amidite reagent, DIPEA, CH2Cl2, 6a: 86%, 6b: 81%.
Scheme 2.

(a) i) NH4OH, EtOH; ii) TrCl, pyridine, 100°C, 70% (2steps); (b) OsO4, pyridine, 92% (5S,6R/5R,6S = 10:1); (c) TBDMSCl, imidazole, DMF, 37°C, 88%; (d) TFA, CH2Cl2, 0°C, 72% (e) DMTrCl, pyridine, 80%; (f) amidite reagent, DIPEA, CH2Cl2, 80%.
For the synthesis of 2′-arabinofluorothymidine glycol (araFTg) phosphoramidite, we prepared benzoyl-protected 2′-arabinofluorothymidine 7 according to a literature report (Scheme 2).[22] The benzoyl groups were removed by ammonium hydroxide and the hydroxyls were protected with trityl groups. An osmium oxidation reaction was then performed to give two diastereomers in a ratio of 10:1 calculated from NMR integration values. Since it was difficult to isolate the minor product by normal column chromatography on silica gel, only the major product was protected with acetyl groups and carried forward. The phosphoramidite building block of acetyl-protected araFTg was synthesized by the same procedures as described above for riboFTg. However, unlike the case for riboFTg, the use of acetyl protecting groups for araFTg promoted an unwanted side reaction during the deprotection step leading to an oligonucleotide product with a mass different from that expected (see below). Therefore, we synthesized TBDMS-protected araFTg as an alternative. The mixture of diastereomers for thymidine glycol 9 was treated with TBDMS-Cl. The resulting compound 10 was allowed to react with trifluoroacetic acid to give the 5,6-O-TBDMS-2′-arabinofluorothymidine glycol 11. The two isomers were separated in this step by silica gel column chromatography, and the configuration of each isomer was determined from the 1H-NMR NOE data. The bulky TBDMS groups are expected to fix the glycosyl torsion angle in the syn conformation. For the minor isomer, a strong NOE was observed between H6 and H1′, whereas the major one did not show the effect (Figure S2). For the (5S,6R) isomer, the NOE between H6 and H1′ would not be expected because H6-equatorial is unfavorable due to the hindrance between the C6-axial O-TBDMS group and the 2′-fluorine. Therefore, the thymine glycols in the major and minor products were assigned as (5S,6R) (11a) and (5R,6S) (11b), respectively. Phosphoramidite building block 13 was then synthesized with 11a by normal DMTr protection and phosphitylation.
Synthesis of oligodeoxynucleotides containing FTg
With these three building blocks, 30 mer oligodeoxynucleotides (ODNs) were synthesized via automated solid phase DNA synthesis. The reaction time for the coupling of FTg was extended to 15 minutes. After the synthesis and removal of the 5′-terminal DMTr group on the synthesizer, the ODNs containing riboFTg were cleaved from the solid support and deprotected by a treatment with K2CO3/MeOH. The solvent was removed and the crude material was purified by ion exchange HPLC. A treatment with 30% aqueous ammonia instead of K2CO3/MeOH gave some decomposition. A single major peak and a significant minor peak (> 10% of major peak) were observed in the HPLC traces for the crude oligonucleotides (Figure S3). Since the masses observed by MALDI-TOFMS were the same, the two peaks were assigned as cis and trans FTg isomers. The trans isomer arises from epimerization at the C6 position, a reaction that has precedence for thymidine glycol lacking the 2′-F modification (Scheme 3).[23]
Scheme 3.

Epimerization reaction of thymidine glycol
The ODN containing acetyl-protected araFTg was also cleaved and deprotected by a treatment with K2CO3/MeOH, but the mass value of the major product was 14 amu higher than the desired compound. However, this side reaction did not occur with the ODN containing TBDMS-protected araFTg. This ODN was cleaved and deprotected by a treatment with 30% aqueous ammonia and the TBDMS was removed with triethylamine trihydrofluoride.[14] HPLC analysis on an ion exchange column gave a single major peak. Epimerization was not observed for this ODN under these conditions.
Estimation of the ratio of cis, trans epimers
For the thymidine glycol lacking the sugar modification, it has been reported that the cis isomer is preferred to the trans isomer. The ratio of cis/trans was 80/20 for 5S and 87/13 for 5R.[23] Also, the ratio of cis/trans was 70/30 for 5R thymidine glycol in a duplex containing the Tg-A base pair.[24] We investigated the effect on cis-trans epimerization by the introduction of fluorine at the 2′ position. The monomers of ribo and araFTg 5S isomers were synthesized from 2b or 9 by treatment with trifluoroacetic acid. After heating at 90 °C for 5 min and cooling to room temperature gradually, the monomers were analyzed by NMR (Figure S4). For the 5S riboFTg monomer, the ratio of cis/trans was 75/25. For the 5S araFTg monomer, the ratio was 90/10. Thus, the percentage of trans isomer of riboFTg increased slightly and that of araFTg decreased compared to the ratio of the unmodified thymidine glycol monomer. The ratio of cis/trans isomers for the FTg-containing ODNs was measured by HPLC analysis. The major ODN products containing 5R-riboFTg, 5S-riboFTg and 5S-araFTg were purified by HPLC to > 95% purity (Figure 1). After heating at 90 °C for 5 min and cooling to room temperature gradually, the ODNs were analyzed by HPLC again (Figure 1). For 5R-riboFTg, the percentage of the minor isomer after this treatment was 11%. For 5S-riboFTg, the percentage of minor isomer was 32% and that of 5S-araFTg was 7%. Since the ratios of major/minor products for the 5S-FTg-containing ODNs were similar to that observed by NMR for the monomers (e.g. ribo: 25% vs 32%, arabino: 7% vs 11%), the minor ODN products appear to be the trans isomers.
Figure 1.
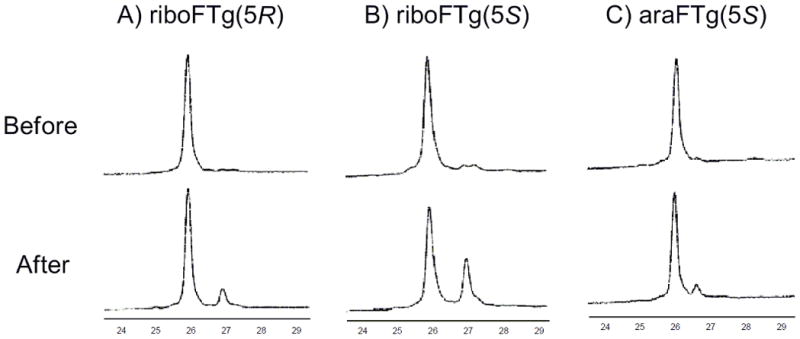
HPLC trace of before and after 90°C, 5 min treatment. The oligonucleotides containing (A) riboFTg(5R), (B) riboFTg(5S) or (C) araFTg(5S) were analyzed by HPLC using a Dionex DNAPac PA-100 ion exchange column. The analysis was achieved with buffer solutions consisting of 30% solvent B and 70% solvent A initially, with a gradient to 100% solvent B for 35min (solvent A consisted of 10% acetonitrile and 90% H2O, while solvent B consisted of 10% acetonitrile and 90% 1.5 M ammonium acetate (pH 7).
N-Glycosylase assays
We evaluated the ability of the 2′ fluorine modification of Tg to block the glycosylase activity of hNEIL1 and E. coli Endo III using a 30-bp duplex. After 32P-labeling, ODNs containing either the 5R or 5S isomers of riboFTg or the 5S isomer of araFTg were separately hybridized to the complementary strand. Tg would be expected to be found in base pairing context with A but may also be found in Tg-G base pair; the latter bp results from oxidation of 5-methylcytosine paired with G followed by hydrolytic deamination of the unstable 5-methylcytosine glycol.[25,26] Thus we evaluated the effect of having either A or G opposite the FTg in the duplex substrates. For comparison, a Tg-containing duplex was prepared and analyzed in side-by-side reactions. To ensure observance of strand scission at all abasic sites produced by the glycosylase reaction, the reactions were quenched with NaOH and heated at 90 °C for 5 min, rather than relying on the lyase activity of enzymes to provide strand scission. With the duplex containing Tg, edited and unedited hNEIL1 and Endo III mediated strand cleavage (Figure 2). On the other hand, the glycosylase reactions with the duplexes containing FTg were all inhibited and not influenced by the opposite base context (Figure S5). For the duplex bearing araFTg (5S), a trace amount of product was observed at longer reaction times with EndoIII. These results indicated that hNEIL1 and Endo III are unable to catalyze efficient thymine glycol base removal from the riboFTg- and araFTg-containing DNA duplexes. In addition, these results are consistent with a previous reported showing the resistance of riboFTg with DNA to base excision mediated by Endo III.[14]
Figure 2.
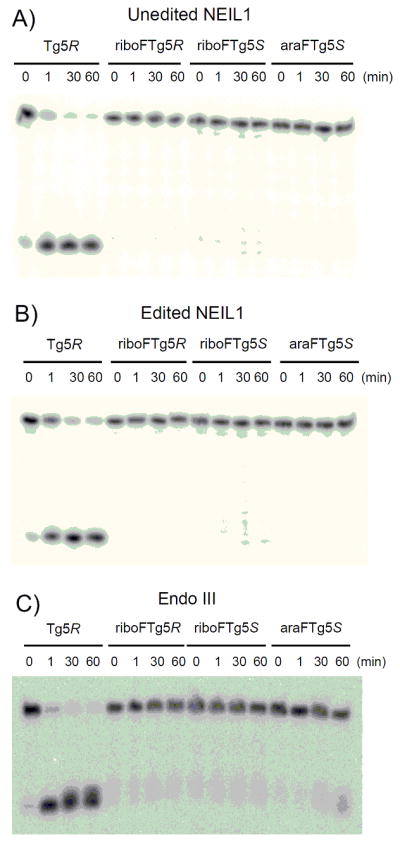
Storage phosphor autoradiogram of N-glycosylase assays with duplexs containing Tg(5R)/A, riboFTg(5R)/A, riboFTg(5S)/A and araFTg(5S)/A. Aliquots of 20 nM DNA and 200 nM enzyme (A: unedited NEIL1, B: edited NEIL1, C: Endo III) were incubated at 37°C for various times (0, 1, 30 and 60 min).
Binding affinities for NEIL1 and Endo III with DNA duplexes
Electrophoretic mobility shift assays (EMSA) were used to measure the relevant dissociation constants of the binding of the unedited and edited forms of human NEIL1 and E. coli Endo III to FTg-containing duplexes. These experiments involved titration of the enzyme into a solution 5′-32P-end-labeled DNA duplex and analysis on native PAGE to separate and detect both the free DNA duplex and protein-DNA complex (Figure 3, Figure 4). The same 30 bp DNA duplexes containing either the FTg-A or the FTg-G base pair that were analyzed for the glycosylase activity were used in these experiments. In addition, DNA duplexes containing either a T-A or a T-G base pair at the position corresponding to the FTg-X bp were analyzed to ascertain the lesion-independent DNA binding affinities for NEIL1 and EndoIII with this duplex sequence under these conditions.
Figure 3.
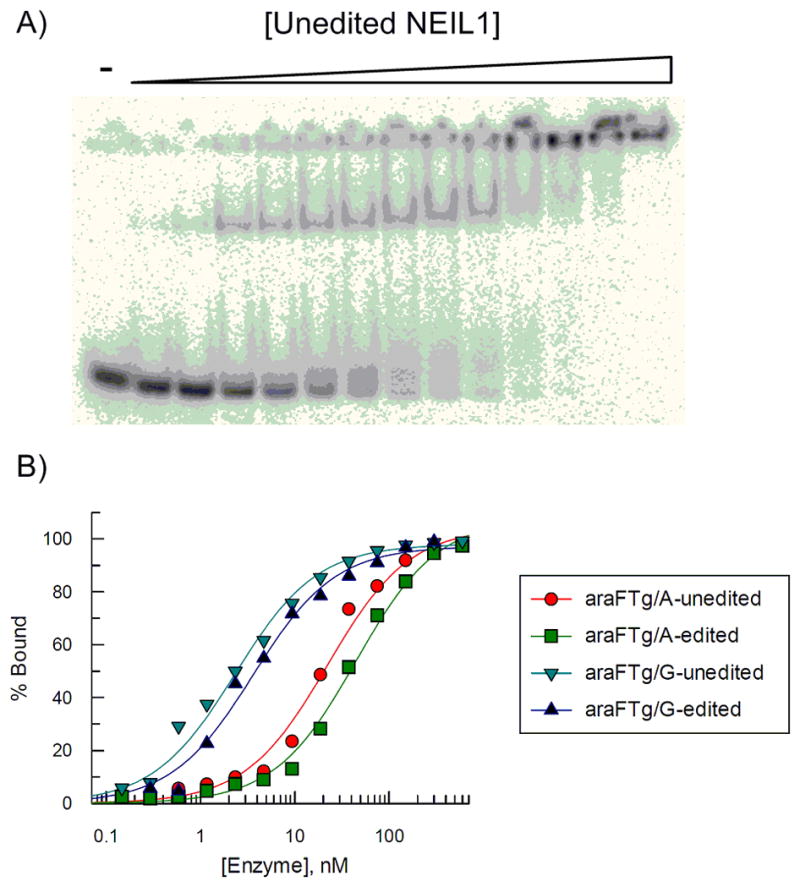
(A) Binding of unedited NEIL1 to duplexes containing araFTg(5S)/G. (B) Plot of percent of DNA-NEIL1 complex (●: araFTg(5S)/A-unedited NEIL1, ■: araFTg(5S)/A-edited NEIL1, ▼: araFTg(5S)/G-unedited NEIL1 ▲: araFTg(5S)/G-edited NEIL1). Aliquots of 10 pM DNA and NEIL1 (0.15–600 nM) were incubated at 25°C for 30 min.
Figure 4.

Binding of Endo III to duplexes containing A: riboFTg(5R)/G, B: riboFTg(5S)/G, C: T/G, D: araFTg(5S)/G. Aliquots of 10 pM DNA and Endo III (0.0011–150 nM) were incubated at 25°C for 30 min. Bracket indicates lesion-independent complexes, star indicates lesion-dependent complex.
Both the edited and unedited forms of NEIL1 bind the DNA duplexes containing FTg more tightly than DNA lacking any lesion (Table 1). The magnitude of this difference depends primarily on the identity of the opposite base with the largest difference observed between the riboFTg(5R)-G duplex compared to the T-G duplex (5.1-fold). A clear preference is observed for NEIL1 binding to FTg-G DNA compared to FTg-A DNA. This preference mirrors the enhanced rate of cleavage of Tg in duplexes paired with G compared to those with Tg-A pairs.[7,27] Surprisingly, NEIL1 binding affinity to the FTg duplexes is relatively insensitive to glycol or sugar stereochemistry. Binding with FTg (5R) was only slightly tighter than that with FTg (5S), with the ratio again corresponding with that observed for the glycosylase activity (5R/5S Tg cleavage rate = 1.5).[28] Importantly, we observed little difference in binding affinity to these DNAs between edited and unedited NEIL1 (Table 1). This stands in stark contrast to the effect editing has on NEIL1 kinetics where the unedited form (K242) showed a 41-fold higher single turnover rate constant for Tg cleavage when it was paired with A and 30-fold higher when paired with G.[7] Moreover, by measuring the single turnover rate constant as a function of enzyme concentration, we were able to determine the apparent dissociation constant (Kd) for natural Tg(5R)-G DNA with edited NEIL1 (Figure S6). The Kd value measured by this technique (2.7 nM) was similar to Kd value measured by EMSA for FTg (5R)-G DNA binding to edited NEIL1 (Kd = 1.5 nM). These results indicate that 2′-fluorination of the Tg lesion does not substantially perturb binding affinity for edited NEIL1.
Table 1.
Dissociation constants (Kd, nM) of unedited NEIL1 and edited NEIL1 for 30 bp DNA duplexes[a]
| Central base pair | Unedited NEIL1 | Edited NEIL1 |
|---|---|---|
|
| ||
| riboFTg(5R)-A | 12±3 | 15±4 |
| riboFTg(5S)-A | 18±1 | 22±6 |
| araFTg(5S)-A | 21±3 | 37±8 |
| T-A | 42±2 | 40±7 |
| riboFTg(5R)-G | 1.4±0.2 | 1.5±0.2 |
| riboFTg(5S)-G | 2.4±0.3 | 3±1 |
| araFTg(5S)-G | 2.4±0.4 | 2.9±0.7 |
| T-G | 7 ± 2 | 7 ±3 |
All measurements were performed at 25°C with 10 pM DNA and various amounts of enzyme. Errors reported in dissociation constants are standard deviation of the average of at least three trials. The concentrations of enzyme are active enzyme concentrations.
A similar EMSA series was performed using Endo III to provide for a comparison to NEIL1. Of note, distinctly different preferences for removal of the two Tg diastereomers (5R-Tg/5S-Tg) for NEIL1 (1.5) and Endo III (0.4) have been reported.[28] Consistent with the diastereoselectivity in the cleavage reaction, Endo III had a very high affinity for FTg (5S) in duplex and much lower affinity for the FTg (5R) duplex. This observation is similar to that previously reported for Endo III binding to FTg-containing DNA.[14] Multiple complex bands were observed (Figure 4), and the binding titration curves were biphasic and best fit with a two-site binding isotherm yielding two distinct Kd values (Table 2). Given the presence of the slower moving, low affinity complexes in the titration with the T-G and T-A control DNAs, these most likely arise from the lesion-independent binding of EndoIII to this duplex and involve more than one equivalent of protein. The dissociation constants for lesion-independent binding of Endo III under these conditions were in the 8–10 nM range (Table 2). We noted that the fraction of DNA bound in the lesion-specific complex differed among the duplexes tested. This fraction depended on the 2′ configuration and opposite base with the riboFTg(5S):G duplex exhibiting the largest fraction (73 ± 2%) with the extremely tight binding (Kd < 10 pM) (Table 2). On the other hand, the binding with 5R-isomer was similar to that observed with lesion-free DNA (Kd = 7.4 nM). As a result, a remarkable > 700-fold diastereoselectivity was observed in the binding of 5S/5R isomers. A high affinity complex was also observed with the araFTg(5S):G duplex indicating that Endo III is less sensitive to the sugar 2′ configuration in the FTg analog paired with G. However, with the duplex containing the araFTg(5S):A pair, <10% of the DNA bound in the lesion-specific complex whereas the riboFTg(5S):A duplex showed 51 ± 3% binding in the complex with high affinity (Table 2, Figure S7). Therefore, Endo III appears to prefer the 2′-F in the ribo configuration, at least when the fluorinated lesion is paired with A. We also noted the difference in the fraction bound in the lesion-specific complex for the riboFTg(5S):G duplex (73 ± 2%) vs the riboFTg(5S):A duplex (51 ± 3%). While the apparent dissociation constants for these two complexes are < 10 pM and too low to accurately measure under our assay conditions, the difference in fraction bound in the lesion-specific complex is noteworthy. This indicates a higher affinity for the FTg analog in a pair with G than A, since the lesion-specific complex with the G-containing duplex is saturated to a greater extent at Endo III concentrations where nonspecific binding is low and does not interfere with its quantification. This difference is even larger for duplexes containing araFTg(5S), FTg:G (38 ± 1%); FTg:A (< 10%) (Table 2, Figure S7).
Table 2.
Dissociation constants (Kd, nM) of Endo III for 30 bp DNA duplexes[a]
| Central base pair | Endo III |
|---|---|
|
| |
| riboFTg(5R)-A | 7 ± 1 |
| riboFTg(5S)-A | < 0.01 (51±3%), 4.7±1 (47±2%)[b] |
| araFTg(5S)-A | 6 ± 1 (>90%)[c] |
| T-A | 10±2 |
| riboFTg(5R)-G | 7.4±0.9 |
| riboFTg(5S)-G | < 0.01 (73±2%), 4.7±0.1 (27±1%)[b] |
| araFTg(5S)-G | ~ 0.01 (38±1%), 10±2 (61±3%)[b] |
| T-G | 8.3±0.9 |
All measurements were performed at 25°C with 10 pM DNA and various amounts of enzyme. Errors reported in dissociation constants are standard deviation of the average of at least three trials. The concentrations of enzyme are active enzyme concentrations.
The data fit best using a two-site binding isotherm that provides two Kd values with relative capacities indicated in closed parentheses.
DNA-enzyme complex of less than 10% with tighter Kd was observed.
Discussion
In this study we sought to generate reagents that would be useful in defining further the lesion recognition process for DNA repair enzymes capable of removing Tg from DNA with a particular emphasis on human NEIL1. We had previously shown that the N-glycosylase reaction of hNEIL1 with DNA containing Tg is regulated by an RNA editing event that changes the identity of an amino acid residue in the lesion-recognition loop of the protein (K/R242). However, the basis for the change in enzymatic activity arising from the conservative arginine for lysine replacement is not currently known. Introduction of a 2′-F group into the sugar of the lesion has proven useful for trapping DNA repair glycosylases in their corresponding lesion-recognition complexes.[10–14] Thus, we envisioned trapped FTg DNA complexes of NEIL1, in the edited and unedited forms, to be useful for defining differences that result in the altered glycosylase rate constants observed. Such information would serve to extend our understanding of how DNA repair glycosylases are regulated and the impact of RNA editing on the proteome.
Given the different stereoisomers of Tg known to exist and the two possible configurations at the 2′ position when fluorine is substituted for hydrogen in 2′-deoxyribose (ribo vs arabino), it was important to evaluate the different stereoisomers of FTg for their effect on repair protein recognition. This had been addressed qualitatively in a previous literature report of E. coli Endo III, where binding studies with DNA containing either 5R or 5S riboFTg showed the 5S isomer is preferred.[14] This stereochemical preference could not be assumed for NEIL1, particularly given the known preference for 5R Tg over 5S Tg in the NEIL1 glycosylase reaction.[7,26] In addition, no guiding information was available for the preferred configuration for the 2′-F, since the araFTg had not been incorporated into DNA prior to this study.
In the synthesis of ribo-FTg phosphoramidite building blocks, the OsO4 oxidation of trityl-protected riboF-thymidine afforded two isomers of thymidine glycol 2 in a ratio of 3:5. The major isomer was (5S,6R)-thymine glycol. Interestingly, when 5′-DMTr-3′-Bz-protected riboF-thymidine was used for this oxidation, the major isomer was (5R,6S)-thymine glycol, and the ratio was 3:1.[14] This stereoselectivity is most likely a function of the 3′ protecting group. Since trityl is bulkier than benzoyl, the approach of the OsO4 molecule from the pro-5R,6S face would be more effectively blocked. Also, the ratio was 1:9 when trityl-protected araF-thymidine was used. This change in stereoselectivity could be attributed to changes in the sugar pucker induced by the fluorine substitution and possibly to the hindrance of the fluorine itself.
Thymidine glycol exists as two diastereomeric pairs of epimers, the 5R cis, trans pair (5R, 6S; 5R, 6R) and the 5S cis, trans pair (5S, 6R; 5S, 6S).[23] Although our synthesis generated only cis (5S, 6R) riboFTg, (5R, 6S) riboFTg and (5S, 6R) araFTg isomers, we observed a small amount of product in the crude DNA samples that we assigned to the trans isomer in each case based on their masses, the known epimerization reaction and the ratio of epimers determined by NMR for the corresponding fluorinated mononucleosides. For the riboFTg(5S)-containing ODN, heating the purified sample to 90 °C and cooling to room temperature lead to the formation of this putative trans product in 32% yield (Figure 1). However, araFTg(5S)-containing DNA was more resistant to the epimerization reaction (7%). This may prove to be an advantage of using the arabino isomer in applications that require stability under a variety of conditions or over long periods of time, such as during crystallization trials. However, the TBDMS protecting group must be used for the glycol hydroxyls with the araFTg isomer, since the use of acetyl protecting groups promoted an unwanted side reaction during the DNA deprotection with K2CO3/MeOH.
Interestingly, NEIL1 binding to the fluorinated DNAs appears to be largely insensitive to the stereochemical differences evaluated in this study. For instance, the dissociation constants for unedited NEIL1 binding to DNA containing riboFTg(5R), riboFTg(5S) or araFTg(5S) in a base pair with A are all within a factor of two (Kd = 12 nM, 18 nM and 21 nM, respectively) (Table 1). The similarity is also seen when these fluorinated lesions are paired with G (Kd = 1.4 nM, 2.4 nM and 2.4 nM, respectively) (Table 1). This is in contrast to the clear preference Endo III shows for the 5S configuration, both in this work and in a previously published qualitative study.[14] Here, we measured a difference in affinity of Endo III for DNA containing riboFTg(5S) and riboFTg(5R) when paired with G of > 700-fold (Table 2). The origin of the different stereoselectivity for these two repair glycosylases is unknown at this time since neither a structure of Endo III bound to Tg-containing DNA nor a structure of hNEIL bound to Tg-containing DNA has been reported. However, a catalytically inactive mutant of the NEIL1 ortholog from mimivirus (Mv Nei1) in complex with Tg-containing DNA has recently been structurally characterized.[29] In this case, only a single direct H-bonding contact was observed between the protein and the thymine glycol base (Y221 backbone NH to Tg O4). The lack of extensive interaction with the Tg base may explain the insensitivity to the C5 configuration for the related hNEIL1 observed here.
NEIL1 binding affinity for the FTg-containing DNAs was also essentially independent of editing. In all the cases tested, unedited (K242) and edited (R242) NEIL1 bound the different target DNAs with dissociation constants that differed by less than a factor of two. For instance, the DNA duplex with riboFTg(5R) paired with A bound unedited NEIL1 with a Kd = 12 nM and edited NEIL1 with a Kd = 15 nM. This result was surprising given that Tg is cleaved from these DNAs much more efficiently by unedited NEIL1.[7] The results indicate the editing reaction generates NEIL1 isoforms with very similar affinities for lesion-containing DNA and the editing effect is more subtle than to simply alter substrate affinity. It may be the case that the change in the lesion recognition loop structure arising from editing serves to reorient the reactive nucleotide in the active site in a manner that effects the chemical steps of the glycosylase reaction but that does not change the overall substrate binding affinity. Further studies will be necessary to test this idea. The F-Tg phosphoramidites described here will be helpful in this regard to generate F-Tg DNAs for crystallography with unedited and edited hNEIL1.
We also investigated the effect of the opposite base on NEIL1 binding to FTg DNAs. NEIL1 binding to DNAs containing the FTg-G base pair was tighter than that with the FTg-A base pair on the whole (~ 8-fold). The NEIL1 binding affinity with DNA containing a simple T-G mismatch also was higher than with a T-A base pair (5.8-fold). It has been reported that the T base of a T-G mismatch pair is removed by excess NEIL1,[30] explaining why NEIL1 might bind the mismatch more tightly. Also, Mv Nei1 makes two H-bonds to the Hoogsteen face of the estranged G in its complex with DNA containing a 5-hydroxyU:G pair.[29] A similar interaction with the estranged G with human NEIL1 could explain the observed binding preference for FTg:G compared to FTg:A.
Interestingly, the minimal interaction between Mv Nei and the Tg base led the authors of that study to suggest that lesion recognition primarily occurs prior to extrusion from the helix (i.e. base flipping) and occupation of the active site.[29] Our fluorinated Tg phosphoramidites should prove useful in probing the base-flipping step for the NEIL1 reaction, such as in the manner previously described by Stivers and colleagues for uracil DNA glycosylase (UDG).[31] They evaluated changes in 2-aminopurine (2-AP) fluorescence induced by UDG in duplexes with 2′-fluorouridine adjacent to the 2-AP. The fluorine modification stalls the UDG reaction at the glycosylase step such that the base extruded intermediate can be studied in the absence of further processing. A similar study with FTg/2-AP-containing DNA and NEIL1 should shed additional light on the base-flipping step for NEIL and the role of active site amino acids, including K/R 242, on this step.
Whereas NEIL1 binding affinity appeared to be largely unaffected by changes in C5 and 2′ stereochemistry, Endo III binding to FTg-containing DNA was more sensitive to these changes. Our results confirm that of an earlier study showing Endo III’s pronounced preference for binding to the 5S isomer.[14] In addition, we observed a binding preference for the riboFTg (5S) analog compared to the araFTg (5S) analog when paired with A. Endo III also appears to bind more tightly to DNA bearing the FTg lesion paired with G than paired with A (73 ± 2% lesion specific complex observed vs 51 ± 3%) (Table 2, Figure S7). Together these studies indicate that optimal binding is observed with EndoIII and duplex DNA with the riboFTg (5S) paired with dG. Attempts to crystallize EndoIII with FTg-containing DNAs should benefit from focusing on this combination.
Interestingly, two types of binding interactions were observed with Endo III and DNAs containing either riboFTg (5S) or araFTg (5S). At low concentrations of Endo III, lesion-specific, high affinity complexes are observed with dissociation constants estimated in the low pM range. At higher protein concentrations, slower moving bands are apparent in the gel. The slower mobility of these complexes suggest they are made up of more than one equivalent of Endo III, consistent with behavior that we have observed previously with the related glycosylase MutY.[32] Since similar complexes are observed at the higher protein concentrations and DNA lacking any lesion, this type of complex corresponds to nonspecific or lesion-independent binding. For DNA containing weak Endo III ligands (e.g. riboFTg (5R) or T), only the nonspecific binding is observed (Figure 4).
Conclusion
We have synthesized 2′-ribo and 2′-arabinofluorothymidine glycol-containing DNAs and evaluated their interaction with the DNA repair glycosylases NEIL1 and Endo III. As we expected, the 2′-fluorinated analogues were resistant to cleavage by both NEIL1 and Endo III. For the unedited and edited forms of hNEIL1, binding affinities to FTg-containing DNA were similar. These results were surprising given the significant difference in glycosylase activity the two forms of NEIL1 demonstrate with DNA containing the unmodified Tg lesion, indicating the editing effect is more subtle than to simply alter substrate affinity. Also, while the NEIL1 binding to FTg-containing DNAs was largely insensitive to C5 and 2′ stereochemistry, a preference was observed for the FTg-G pair over FTg-A. Furthermore, we found that optimal binding is observed with Endo III and duplex DNA with riboFTg (5S) paired with G. The modified DNAs reported here will provide useful tools for further characterizing the interaction between DNA repair glycosylases and thymine glycol containing DNA.
Experimental Section
All reagents were purchased from commercial sources (Sigma/Aldrich or Fischer Scientific) and were used without further purification unless otherwise stated. Reactions were carried out under an atmosphere of dry argon. 1H, 13C, and 31P nuclear magnetic resonance spectra were recorded with Varian VNMRS 600, Varian Mercury 300 spectrometers. High-resolution ESI mass spectra were obtained at the University of California, Davis Mass spectrometry facility, on an Orbitrap FTMS instrument. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra of oligonucleotides were measured in the positive ion mode, using 3-hydroxypicolinic acid or 6-aza-2-thiothymine as a matrix.
3′,5′-Di-O-trityl-5,6-dihydroxy-2′-ribofluorothymidine (2)
Compound 1 (1.47 g, 1.97 mmol) and OsO4 (0.50 g, 2.0 mmol) were dissolved in pyridine (5 mL), and the mixture was stirred at room temperature for 3 h. Sodium hydrogen sulfite (1.8 g) dissolved in a mixture of water (30 mL) and pyridine (20 mL) was added to the reaction mixture and the mixture was stirred for another 14 h. The product was extracted with CH2Cl2 (100 mL×2), and the organic phases were dried over Na2SO4. After evaporation, the pyridine was removed by coevaporation with toluene, and the residue was purified by column chromatography on a silica gel with hexane/EtOAc/MeOH (70:25:5) to give 5R,6S-isomer 2a (479 mg, 31%) and 5S,6R-isomer 2b (808 mg, 52%) as white foams, respectively. 5R,6S-isomer (2a): 1H NMR (CDCl3, 600 MHz, TMS): δ (ppm) 7.72 (s, 1H), 7.38-7.34 (m, 6H), 7.33-7.17 (m, 24H), 6.16 (dd, J = 16, 4.2 Hz, 1H), 5.05 (s, 1H), 4.63 (ddd, J = 51, 5.4, 4.2 Hz, 1H), 4.23 (ddd, J = 11, 5.4, 4.2 Hz, 1H), 3.64-3.62 (m, 1H), 3.52 (s, 1H), 3.29 (d, J = 11 Hz, 1H), 2.84-2.82 (m, 1H), 2.82 (s, 1H), 1.31 (s, 3H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 173.1, 150.1, 143.9, 143.5, 129.1, 128.9, 128.1, 128.0, 127.5, 127.4, 90.6 (d, J = 192 Hz), 87.8, 87.6, 86.7 (d, J = 33 Hz), 81.3, 78.0, 71.9, 71.8 (d, J = 14 Hz), 63.1, 22.8. ESIHRMS (m/z): calcd for C48H43FN2O7 (M+Na)+ 801.2946, obsd 801.2954. 5S,6R-Isomer (2b): 1H NMR (CDCl3, 600 MHz, TMS): δ (ppm) 7.57 (s, 1H), 7.36-7.34 (m, 6H), 7.31-7.22 (m, 24H), 6.33 (dd, J = 13, 6.0 Hz, 1H), 4.89 (s, 1H), 4.55 (ddd, J = 53, 6.0, 5.4 Hz, 1H), 4.13-4.10 (m, 1H), 3.50-3.48 (m, 1H), 3.31 (d, J = 11 Hz, 1H), 3.28 (s, 1H), 3.19 (s, 1H), 2.82 (dd, J = 11, 3.0 Hz, 1H), 1.41 (s, 3H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 172.6, 151.1, 143.7, 143.1, 128.8, 128.7, 128.1, 127.5, 127.4, 89.3 (d, J = 196 Hz), 88.2, 87.7, 86.3 (d, J = 32 Hz), 81.5, 80.2, 72.2 (d, J = 14 Hz), 72.0, 62.6, 22.0. ESIHRMS (m/z): calcd for C48H43FN2O7 (M+Na)+ 801.2946, obsd 801.2951.
3′,5′-Di-O-trityl-(5R,6S)-5,6-di-O-acetyl-2′-ribofluorothymidine (3a)
Compound 2 (282 mg, 0.362 mmol) was coevaporated with anhydrous pyridine, and the dried residue was dissolved again in anhydrous pyridine. Acetic anhydride (0.685 mL, 7.24 mmol) and 4-dimethylaminopyridine (8.8 mg, 0.072 mmol) were added into the solution under argon atmosphere, and the mixture was stirred at room temperature for 16 h. The reaction mixture was dried under reduced pressure, and the dried residue was redissolved in EtOAc (100 mL). The organic phase was washed with water (100 mL×2) and brine (100 mL), dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on a silica gel with CH2Cl2/ EtOAc (29:1→19:1) to give 3a (302 mg, 97%) as a white foam. 1H NMR (CDCl3, 600 MHz, TMS): δ (ppm) 7.36 (d, J = 7.2 Hz, 6H), 7.33 (d, J = 7.2 Hz, 6H), 7.27-7.13 (m, 18H), 6.64 (s, 1H), 5.56 (dd, J = 22, 2.4 Hz, 1H), 4.17 (ddd, J = 15, 7.2, 5.4 Hz, 1H), 4.07-4.05 (m, 1H), 4.01 (ddd, J = 53, 5.4, 2.4 Hz, 1H), 3.06 (dd, J = 10, 1.8 Hz, 1H), 2.87 (dd, J = 10, 7.8 Hz, 1H), 2.07 (s, 3H), 1.90 (s, 3H), 1.76 (s, 3H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 169.0, 168.5, 166.9, 149.6, 143.9, 143.7, 129.1, 128.8, 128.3, 128.2, 127.8, 127.7, 127.2, 126.9, 92.0 (d, J = 41 Hz), 91.2 (d, J = 189 Hz), 87.4, 86.6, 81.6, 79.8, 71.8 (d, J = 15 Hz), 64.0, 21.4, 20.6. ESIHRMS (m/z): calcd for C52H47FN2O9 (M+Na)+ 885.3158, obsd 885.3168.
3′,5′-Di-O-trityl-(5S,6R)-5,6-di-O-acetyl-2′-ribofluorothymidine (3b)
A white foam (285 mg, 97%). 1H NMR (CDCl3, 600 MHz, TMS): δ (ppm) 7.38 (d, J = 8.4 Hz, 6H), 7.34 (d, J = 8.4 Hz, 6H), 7.29-7.15 (m, 18H), 6.41 (s, 1H), 5.65 (d, J = 22 Hz, 1H), 4.04-4.00 (m, 2H), 3.78 (ddd, J = 53, 3.0, 3.0 Hz, 1H), 3.34 (d, J = 10 Hz, 1H), 2.84 (dd, J = 10, 6.0 Hz, 1H), 2.08 (s, 3H), 1.84 (s, 3H), 1.77 (s, 3H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 168.9, 168.8, 166.8, 149.2, 143.9, 143.7, 129.1, 128.8, 128.7, 127.8, 127.7, 127.2, 127.0, 91.7 (d, J = 36 Hz), 91.3 (d, J = 189 Hz), 87.4, 87.0, 80.8, 79.8, 72.0 (d, J = 15 Hz), 64.2, 21.4, 20.5, 20.1. ESIHRMS (m/z): calcd for C52H47FN2O9 (M+Na)+ 885.3158, obsd 885.3163.
(5R,6S)-5,6-Di-O-acetyl-2′-ribofluorothymidine (4a)
To a solution of compound 3a (86 mg, 0.10 mmol) in anhydrous CH2Cl2 (2 mL) was added trifluoroacetic acid (0.11 mL, 1.5 mmol) under argon atmosphere, and the mixture was stirred at room temperature for 20 min. The reaction mixture was diluted with dimethyl ether (15 mL), and extracted with water (8 mL×2). The aqueous solutions were combined and evaporated under reduced pressure. The residue was coevaporated with a mixture of toluene and acetonitrile (1:1) twice to give 4a (35 mg, 92%) as a white solid. 1H NMR (CD3OD, 600 MHz): δ (ppm) 6.77 (s, 1H), 5.78 (dd, J = 20, 3.6 Hz, 1H), 4.96 (ddd, J = 54, 4.8, 3.6 Hz, 1H), 4.19 (ddd, J = 16, 7.2, 4.8 Hz, 1H), 3.88-3.86 (m, 1H), 3.82 (dd, J = 13, 3.0 Hz, 1H), 3.70 (dd, J = 13, 5.4 Hz, 1H), 2.09 (s, 3H), 2.05 (s, 3H), 1.82 (s, 3H). 13C NMR (CD3OD, 150 MHz): δ (ppm) 170.9, 170.5, 169.6, 152.1, 93.5 (d, J = 185 Hz), 90.4 (d, J = 35 Hz), 84.3, 79.7, 78.6, 70.7 (d, J = 16 Hz), 63.0, 21.3, 20.8, 20.6.
(5S,6R)-5,6-Di-O-acetyl-2′-ribofluorothymidine (4b)
A white solid (101mg, 92%). 1H NMR (CD3OD, 600 MHz): δ (ppm) 6.76 (s, 1H), 5.72 (dd, J = 20, 3.0 Hz, 1H), 5.05 (ddd, J = 54, 4.8, 3.0 Hz, 1H), 4.26 (ddd, J = 17, 7.2, 4.8 Hz, 1H), 3.88-3.86 (m, 1H), 3.78 (dd, J = 12, 2.4 Hz, 1H), 3.62 (dd, J = 12, 4.8 Hz, 1H), 2.06 (s, 3H), 2.05 (s, 3H), 1.84 (s, 3H). 13C NMR (CD3OD, 150 MHz): δ (ppm) 170.0, 169.5, 168.4, 151.1, 93.0 (d, J = 185 Hz), 89.9 (d, J = 35 Hz), 83.2, 79.2, 77.3, 69.0 (d, J = 17 Hz), 61.5, 20.1, 19.4.
5′-O-(4,4′-Dimethoxytrityl)-(5R,6S)-5,6-di-O-acetyl-2′-ribofluorothymidine (5a)
Compound 4a (70 mg, 0.19 mmol) was coevaporated with anhydrous acetonitrile and pyridine/toluene (1:1), and the dried residue was dissolved in anhydrous pyridine (0.7 mL). 4,4′-Dimethoxytrityl chloride (125 mg, 0.369 mmol) was added into the solution under argon atmosphere, and the mixture was stirred at room temperature for 1 h. The reaction mixture was diluted with EtOAc (30 mL), and washed with water (20 mL) and brine (20 mL). The organic phase was dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on a silica gel with hexane/EtOAc (2:1→3:2) to give 5a (105 mg, 83%) as a pale yellow foam. 1H NMR (CD2Cl2, 600 MHz): δ (ppm) 7.83 (s, 1H), 7.45 (d, J = 7.8 Hz, 2H), 7.34 (d, J = 8.4 Hz, 4H), 7.27 (t, J = 7.8 Hz, 2H), 7.20 (t, J = 7.8 Hz, 1H), 6.82 (d, J = 8.4 Hz, 4H), 6.75 (s, 1H), 5.62 (dd, J = 25, 2.4 Hz, 1H), 5.06 (ddd, J = 55, 4.8, 2.4 Hz, 1H), 4.38-4.31 (m, 1H), 3.90 (dt, J = 9.6, 4.8 Hz, 1H), 3.77 (s, 6H), 3.31 (d, J = 4.8 Hz, 2H), 2.13-2.10 (m, 1H), 2.07 (s, 3H), 2.04 (s, 3H), 1.84 (s, 3H). 13C NMR (CD2Cl2, 150 MHz): δ (ppm) 169.6, 169.1, 167.2, 159.0, 150.3, 145.4, 136.3, 130.5, 128.5, 128.2, 127.1, 113.5, 93.2 (d, J = 182 Hz), 91.8 (d, J = 37 Hz), 86.5, 82.0, 80.0, 77.3, 70.8 (d, J = 17 Hz), 63.8, 55.6, 21.6, 21.0, 20.8. ESIHRMS (m/z): calcd for C35H37FN2O11 (M+Na)+ 703.2273, obsd 703.2277.
5′-O-(4,4′-Dimethoxytrityl)-(5S,6R)-5,6-di-O-acetyl-2′-ribofluorothymidine (5b)
A pale yellow foam (90 mg, 88%). 1H NMR (CD2Cl2, 600 MHz): δ (ppm) 7.77 (s, 1H), 7.44 (d, J = 7.8 Hz, 2H), 7.32 (d, J = 7.2 Hz, 4H), 7.27 (t, J = 7.8 Hz, 2H), 7.21 (t, J = 7.8 Hz, 1H), 6.81 (d, J = 7.2 Hz, 4H), 6.66 (s, 1H), 5.57 (d, J = 25 Hz, 1H), 5.24 (ddd, J = 55, 4.8, 1.2 Hz, 1H), 4.48-4.40 (m, 1H), 3.89-3.85 (m, 1H), 3.77 (s, 6H), 3.33 (dd, J = 10, 3.0 Hz, 1H), 3.23 (dd, J = 10, 5.4 Hz, 1H), 2.11 (s, 3H), 2.03 (s, 3H), 1.90 (s, 3H). 13C NMR (CD2Cl2, 150 MHz): δ (ppm) 170.0, 169.7, 167.2, 159.0, 149.7, 145.3, 136.4, 136.3, 130.5, 130.4, 128.5, 128.2, 127.2, 113.4, 93.8 (d, J = 180 Hz), 93.6 (d, J = 37 Hz), 86.5, 81.9, 81.4, 77.3, 70.3 (d, J = 17 Hz), 63.7, 55.6, 21.7, 20.9, 20.1. ESIHRMS (m/z): calcd for C35H37FN2O11 (M+Na)+ 703.2273, obsd 703.2276.
5′-O-(4,4′-Dimethoxytrityl)-3′-O-[2-(cyanoethoxy)(N,N-diisopropylamino)phosphino]-(5R,6S)-5,6-di-O-acetyl-2′-ribofluorothymidine (6a)
Compound 5a (112 mg, 0.165 mmol) was coevaporated with anhydrous acetonitrile twice and the dried residue was dissolved in anhydrous CH2Cl2 (1.1 mL). To the solution were added N,N-diisopropylethylamine (170 μL, 0.98 mmol) and (2-cyanoethyl)-N,N-diisopropylchlorophosphoramidite (95 μL, 0.43 mmol) at 0°C, and the mixture was stirred at 0°C for 1 h. The reaction mixture was quenched with MeOH (0.2 mL) and 2% NaHCO3 (5 mL), and extracted with EtOAc (10 mL×2). The organic phases were washed with brine (10 mL), dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on a silica gel with hexane/ EtOAc (2:1) to give 6a (125 mg, 86%) as a white foam. 31P NMR (CD2Cl2, 121 MHz): δ (ppm) 151.8 (d, J = 7.6 Hz), 151.4 (d, J = 13 Hz). ESIHRMS (m/z): calcd for C44H54FN4O12P (M+H)+ 881.3533, obsd 881.3542.
5′-O-(4,4′-Dimethoxytrityl)-3′-O-[2-(cyanoethoxy)(N,N-diisopropylamino)phosphino]-(5S,6R)-5,6-di-O-acetyl-2′-ribofluorothymidine (6b)
A white foam (105 mg, 81%). 31P NMR (CD2Cl2, 121 MHz): δ (ppm) 151.6 (d, J = 7.7 Hz), 150.9 (d, J = 13 Hz). ESIHRMS (m/z): calcd for C44H54FN4O12P (M+H)+ 881.3533, obsd 881.3547.
3′,5′-Di-O-trityl-2′-arabinofluorothymidine (8)
NH4OHaq (30%, 70 mL) was added into a suspension of compound 7 (2.80 g, 5.98 mmol) in EtOH (70 mL), and the mixture was stirred at room temperature for 48 h. The reaction mixture was evaporated under reduced pressure, and the residue was used for next reaction without purification. The residue was coevaporated with anhydrous acetonitrile and pyridine, and then dissolved in anhydrous pyridine (16 mL). To the solution was added tritylchloride (4.17 g, 15.0 mmol) at room temperature, and the mixture was heated to 100°C. After 18h, the reaction mixture was cooled to room temperature, and evaporated under reduced pressure. The residue was purified by column chromatography on a silica gel with CH2Cl2/AcOEt (19:1) to give 8 (3.11 g, 70%) as a pale yellow foam. 1H NMR (CDCl3, 600 MHz, TMS): δ (ppm) 8.43 (s, 1H), 7.37-7.34 (m, 12H), 7.29-7.21 (m, 18H), 6.18 (dd, J = 23, 2.4 Hz, 1H), 4.44-4.43 (m, 1H), 4.26 (dd, J = 17, 3.0 Hz, 1H), 3.77 (dd, J = 50, 2.4 Hz, 1H), 3.30 (dd, J = 10, 2.4 Hz, 1H), 3.23 (dd, J = 10, 5.4 Hz, 1H), 1.67 (s, 3H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 163.5, 150.1, 143.7, 143.2, 137.4, 128.8, 128.3, 128.0, 127.9, 127.3, 110.0,93.7 (d, J = 192 Hz), 88.7, 87.0, 84.0 (d, J = 21 Hz), 84.0, 77.9 (d, J = 27 Hz), 63.6, 12.4. ESIHRMS (m/z): calcd for C48H41FN2O5 (M+H)+ 745.3073, obsd 745.3083.
3′,5′-Di-O-trityl-5,6-dihydroxy-2′-arabinofluorothymidine (9)
Compound 8 (732 mg, 0.983 mmol) and OsO4 (250 mg, 0.983 mmol) were dissolved in pyridine (2.5 mL), and the mixture was stirred at room temperature for 3 h. Sodium hydrogen sulfite (0.9 g) dissolved in a mixture of water (15 mL) and pyridine (10 mL) was added to the reaction mixture and the mixture was stirred for another 14 h. The product was extracted with CH2Cl2 (50 mL×2), and the organic phases were dried over Na2SO4. After evaporation, the pyridine was removed by coevaporation with toluene, and the residue was purified by column chromatography on a silica gel with hexane/EtOAc/MeOH (70:25:5) to give 9 (705 mg, 92%, 5R,6S-isomer/5S,6R-isomer=1:10 from NMR) as a white solid. 5S,6R-Isomer: 1H NMR (CDCl3, 600 MHz, TMS): δ (ppm) 7.68 (s, 1H), 7.37-7.34 (m, 12H), 7.30-7.21 (m, 18H), 6.19 (dd, J = 26, 3.0 Hz, 1H), 4.88 (s, 1H), 4.42-4.40 (m, 1H), 4.11 (dd, J = 16, 1.8 Hz, 1H), 3.78 (dd, J = 51, 3.0 Hz, 1H), 3.48 (d, J = 2.4 Hz, 1H), 3.22 (dd, J = 9.0, 3.0 Hz, 1H), 3.19 (s, 1H), 3.10 (dd, J = 10, 9.0 Hz, 1H), 1.27 (s, 3H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 173.1, 151.1, 143.5, 143.3, 128.8, 128.6, 128.4, 128.1, 127.9, 127.4, 94.7 (d, J = 190 Hz), 88.8, 87.0, 84.7 (d, J = 15 Hz), 83.4, 81.3 (d, J = 7.2 Hz), 77.5, 72.2, 63.3, 22.2. ESIHRMS (m/z): calcd for C48H43FN2O7 (M+Na)+ 801.2946, obsd 801.2948.
5,6-Bis-O-(tert-butyldimethylsilyl)-2′-arabinofluorothymidine (11)
To a solution of compound 9 (920 mg, 1.18 mmol) in anhydrous DMF were added imidazole (803 mg, 11.8 mmol) and tert-butyldimethylchlorosilane (889 mg, 5.90 mmol), and the mixture was stirred at 37 °C for 40 h. The reaction mixture was diluted with ether (100 mL) and washed with saturated aqueous NH4Cl (100 mL). The aqueous phase was extracted with ether (100 mL×2), and the organic phases were dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel with hexane/EtOAc (7:1→2:1) to give 10 (1.05 g, 88%) as a white foam. To a solution of compound 10 (160 mg, 0.159 mmol) in anhydrous CH2Cl2 (3 mL) was added trifluoroacetic acid (355 μL, 4.78 mmol) under argon atmosphere at 0 °C, and the mixture was stirred for 10 min. The reaction mixture was quenched with saturated aqueous NaHCO3 (20 mL) and extracted with CH2Cl2 (15 mL×2). The organic phases were dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel with CH2Cl2/EtOAc (4:1→2:1) to give 11 (60 mg, 72%) as a white foam. 5S,6R-Isomer (11a): 1H NMR (CDCl3, 600 MHz, TMS): δ (ppm) 7.93 (s, 1H), 5.63 (dd, J = 20, 3.0 Hz, 1H), 5.13 (ddd, J = 51, 3.0, 1.2 Hz, 1H), 5.00 (s, 1H), 4.43 (d, J = 19 Hz, 1H), 3.97 (dd, J = 9.0, 4.8 Hz, 1H), 3.92-3.84 (m, 2H), 3.35 (s, 1H), 2.23 (s, 1H), 1.45 (s, 3H), 0.89 (s, 9H), 0.83 (s, 9H), 0.26 (s, 3H), 0.21 (s, 3H), 0.11 (s, 3H), 0.10 (s, 3H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 173.5, 151.8, 95.9 (d, J = 191 Hz), 85.0 (d, J = 17 Hz), 84.1, 82.6, 77.5, 75.1 (d, J = 27 Hz), 62.2, 26.4, 25.9, 23.7, 18.7, 18.3, −1.86, −2.27, −4.24, −4.89. ESIHRMS (m/z): calcd for C22H43FN2O7Si2 (M+H)+ 523.2666, obsd 523.2665. 5R,6S-Isomer (11b): 1H NMR (CDCl3, 600 MHz, TMS): δ (ppm) 8.14 (s, 1H), 5.25 (dd, J = 9.6, 6.6 Hz, 1H), 5.16 (ddd, J = 53, 6.6, 5.4 Hz, 1H), 4.94 (d, J = 22 Hz, 1H), 4.57 (s, 1H), 3.93-3.91 (m, 2H), 3.81 (d, J = 7.2 Hz, 1H), 3.80-3.78 (m, 1H), 3.55 (d, J = 7.2 Hz, 1H), 1.46 (s, 3H), 0.88 (s, 9H), 0.84 (s, 9H), 0.26 (s, 3H), 0.20 (s, 3H), 0.16 (s, 3H), 0.12 (s, 3H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 173.0, 152.6, 97.4 (d, J = 197 Hz), 89.4, 87.8, 80.9 (d, J = 9.2 Hz), 77.6, 73.3 (d, J = 24 Hz), 61.2, 26.5, 25.8, 23.6, 18.7, 18.2, −2.02, −2.41, −4.05, −4.70. ESIHRMS (m/z): calcd for C22H43FN2O7Si2 (M+H)+ 523.2666, obsd 523.2662.
5′-O-(4,4′-Dimethoxytrityl)-(5S,6R)-5,6-bis-O-(tert-butyldimethylsilyl)-2′-arabinofluorothymidine (12)
Compound 11a (53 mg, 0.10 mmol) was coevaporated with anhydrous acetonitrile and pyridine/toluene (1:1), and the dried residue was dissolved in anhydrous pyridine (0.6 mL). 4,4′-Dimethoxytrityl chloride (69 mg, 0.204 mmol) was added into the solution under argon atmosphere, and the mixture was stirred at room temperature for 1 h. The reaction mixture was diluted with EtOAc (30 mL), and washed with water (20 mL) and brine (20 mL). The organic phase was dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on a silica gel with hexane/ EtOAc (4:1) to give 12 (76 mg, 80%) as a white foam. 1H NMR (CD2Cl2, 600 MHz): δ (ppm) 7.58 (s, 1H), 7.46 (d, J = 7.8 Hz, 2H), 7.33 (d, J = 9.0 Hz, 4H), 7.30 (t, J = 7.8 Hz, 2H), 7.23 (t, J = 7.8 Hz, 1H), 6.84 (d, J = 9.0 Hz, 4H), 5.50 (dd, J = 21, 2.4 Hz, 1H), 5.24 (ddd, J = 52, 2.4, 1.2 Hz, 1H), 5.01 (s, 1H), 4.37 (dt, J = 19, 3.6 Hz, 1H), 3.94 (dd, J = 9.0, 5.4 Hz, 1H), 3.78 (s, 6H), 3.39-3.36 (m, 2H), 2.43 (d, 4.2 Hz, 1H), 1.40 (s, 3H), 0.87 (s, 9H), 0.83 (s, 9H), 0.24 (s, 3H), 0.21 (s, 3H), 0.07 (s, 3H), 0.05 (s, 3H). 13C NMR (CD2Cl2, 150 MHz): δ (ppm) 173.4, 159.1, 151.5, 145.1, 136.2, 136.1, 130.4, 130.3, 128.4, 128.3, 127.2, 113.5, 96.3 (d, J = 190 Hz), 86.8, 85.8 (d, J = 18 Hz), 83.6, 82.4, 77.8, 76.4 (d, J = 26 Hz), 63.2, 55.6, 26.5, 26.0, 23.9, 18.9, 18.4, −1.93, −2.27, −4.23, −4.81. ESIHRMS (m/z): calcd for C43H61FN2O9Si2 (M+Na)+ 847.3792, obsd 847.3796.
5′-O-(4,4′-Dimethoxytrityl)-3′-O-[2-(cyanoethoxy)(N,N-diisopropylamino)phosphino]-(5S,6R)-5,6-bis-O-(tert-butyldimethylsilyl)-2′-arabinofluorothymidine (13)
Compound 12 (65 mg, 0.079 mmol) was coevaporated with anhydrous acetonitrile twice and the dried residue was dissolved in anhydrous CH2Cl2 (0.7 mL). To the solution were added N,N-diisopropylethylamine (85 μL, 0.49 mmol) and (2-cyanoethyl)-N,N-diisopropylchlorophosphoramidite (44 μL, 0.20 mmol) at 0 °C, and the mixture was stirred at 0 °C for 1 h. The reaction mixture was quenched with MeOH (0.2 mL) and saturated aqueous NaHCO3 (5 mL), and extracted with EtOAc (10 mL×2). The organic phases were washed with brine (10 mL), dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel with hexane/ EtOAc (6:1) to give 13 (65 mg, 80%) as a white foam. 31P NMR (CD2Cl2, 121 MHz): δ (ppm) 151.9, 151.6. ESIHRMS (m/z): calcd for C52H78FN4O10PSi2 (M+H)+ 1025.5051, obsd 1025.5068.
Synthesis of oligodeoxynucleotides
Oligodeoxynucleotides were synthesized on an ABI 394 synthesizer (DNA/Peptide Core Facility, University of Utah) using nucleoside phosphoramidites for ultramild DNA synthesis (Glen Research) on a 0.2 or 1.0 μmol scale with coupling times of 15 min for the coupling of FTg. 4,4′-Dimethoxytrityl (DMTr) group of the 5′ end was removed on the synthesizer. The 30-nucleotide (nt) sequence that was synthesized is as follows: d(5′-TGTTCATCATGGGTCXTCGGTATATCCCAT-3′), in which X = Tg, riboFTg5R, riboFTg5S or araFTg5S and the complementary strand d(3′-ACAAGTAGTACCCAGYAGCCATATAGGGTA-5′), in which Y = A or G.
Deprotection and purification of oligodeoxynucleotides containing acetyl-protected riboFTg
The oligonucleotides containing acetyl-protected riboFTg were treated with 0.05M potassium carbonate in methanol at room temperature for 4 h. The solvent was concentrated to dryness under reduced pressure. The residues were dissolved in 0.1 M triethylammonium acetate (pH 7.0), and the oligonucleotides were analyzed and purified by HPLC using a Dionex DNAPac PA-100 ion exchange column. The purification was achieved with buffer solutions consisting of 30% solvent B and 70% solvent A initially, with a gradient to 100% solvent B for 35min (solvent A consisted of 10% acetonitrile and 90% H2O, while solvent B consisted of 10% acetonitrile and 90% 1.5 M ammonium acetate (pH 7); flow rate of 3 mL/min). After lyophilization, the purified oligonucleotides were desalted with Sep-pac column. The correct composition of the 30-nt oligonucleotide containing riboFTg(5R) was confirmed by MALDI-TOF/MS: calcd: 9202.4 [M+H]+; obsd: 9203.3, riboFTg(5S): calcd: 9202.4 [M+H]+; obsd: 9203.6.
Deprotection and purification of oligodeoxynucleotides containing TBDMS-protected araFTg
The oligonucleotides containing TBDMS-protected riboFTg were treated with 30% aqueous ammonia (2 mL) at room temperature for 2 h. The resulting ammonia solutions were concentrated to dryness under reduced pressure. The residue was dissolved in triethylamine trihydrofluoride (500 μL), and the mixtures were kept at 40 °C overnight. After desalting on a NAP-10 column (GE Healthcare), the oligonucleotides were analyzed and purified by the above conditions. The correct composition for 30 nt DNA containing araFTg(5S) was confirmed by MALDI-TOF/MS: calcd: 9202.4 [M+H]+; obsd: 9203.0,
32P-labeling of oligonucleotide
Oligonucleotides containing Tg, riboFTg5R, riboFTg5S or araFTg5S were radiolabeled on the 5′ end using [γ-32P]ATP by T4 polynucleotide kinase at 37°C. Excess [γ-32P]ATP was removed using a Pharmacia Microspin G-50 spin column, according to the manufacturer’s protocol. The labeled DNA was then annealed to the complement (added at 20% excess) by heating to 90 °C for 5 min and allowing to cool overnight in annealing buffer (20 mM Tris-HCl pH 7.6, 10 mM EDTA, and 150 mM NaCl).
Enzyme purification
Unedited (K242) and edited (R242) NEIL1 were purified as described previously.[5] Active site concentration was determined using a 30-bp duplex containing central spiroimindihydantoin (Sp1) lesion base-paired to G and concentrations listed through-out are active site, rather than total protein, concentrations.[5] Endonuclease (Endo) III was purified using a pET24 nth vector (provided by Dr. R.P. Cunningham, SUNY, Albany) using BL21DE3 cells. The purification was as described previously with minor modifications.[33] A binding assay using an abasic site analog (tetrahydrofuran) containing 30 bp duplex was used to estimate the “binding” competent concentration of Endo III. This concentration was used through-out the manuscript.
Glycosylase Assays
The glycosylase activity of NEIL1 and Endo III was evaluated with single-turnover experiments. Briefly, 20 nM of substrate DNA was incubated at 37°C with 200 nM of active enzyme in assay buffer containing 20 mM Tris-HCl pH 7.6, 10 mM EDTA, 0.1 mg/mL BSA and 150 mM (for NEIL1) or 100 mM (for Endo III) NaCl, and aliquots were removed from the reaction mixture at various time points and quenched with NaOH. The products were analyzed by denaturing PAGE.
Measurement of dissociation constant (Kd)
Electrophoresis mobility shift assays were performed using 32P-labeled duplexes containing 5R-riboFTg, 5S-riboFTg or 5S-araFTg. Reactions contained 10 pM duplex, 20 mM Tris–HCl pH 7.6, 150 mM (for NEIL1) or 100 mM (for Endo III) NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 0.1 mg/ml BSA and varying amounts of enzyme. Enzyme solutions of varying concentrations were freshly prepared by diluting aliquots of enzyme at 4°C with dilution buffer containing 20 mM Tris–HCl pH 7.6, 10 mM EDTA and 20% glycerol. The samples containing DNA and enzyme were incubated at 25 °C for 25–35 min. Then, the samples were electrophoresed at 4°C on a 6% non-denaturing polyacrylamide gel with 0.5×TBE buffer at 250 V for 15–25 min followed by 150 V for 90–120 min. The gel was dried and exposed to a Molecular Dynamics phosphorimager screen for at least 18 h. Dissociation constants were determined by fitting the data (percent bound substrate versus log[enzyme]) to the equation for one-site or two-site ligand binding using Grafit 5.0 software. Kd values were determined from four to six separate experiments.
Supplementary Material
Acknowledgments
P.A.B and S.S.D acknowledge the National Institutes of Health (USA) for financial support in the form of grants R01GM061115 and R01CA090689. We are also grateful for the Research Fellowship from the Japan Society for the Promotion of Science (JSPS) for Young Scientists (K.O.). We thank Dr. R. P. Cunningham (SUNY, Albany) for providing the pET24 endo III expression plasmid, and Ms. Agnes Hahn and Dr. Nikolas Chmiel for purification of Endo III used in this work.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.((Please delete if not appropriate))
Contributor Information
Sheila S. David, Email: david@chem.ucdavis.edu.
Peter A. Beal, Email: beal@chem.ucdavis.edu.
References
- 1.Neeley WL, Essigmann JM. Chem Res Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 2.Geacintov NE, Broyde S. The chemical biology of DNA damage. Wiley-VCH; 2010. [Google Scholar]
- 3.David SS, O’Shea VL, Kundu S. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaruga P, Birincioglu M, Rosenquist TA, Dizdaroglu M. Biochemistry. 2004;43:15909–15914. doi: 10.1021/bi048162l. [DOI] [PubMed] [Google Scholar]
- 5.Krishnamurthy N, Zhao X, Burrows CJ, David SS. Biochemistry. 2008;47:7137–7146. doi: 10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bandaru V, Sunkara S, Wallace SS, Bond JP. DNA Repair. 2002;1:517–529. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 7.Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. Proc Natl Acad Sci USA. 2010;107:20715–20719. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace SS. Free Radic Biol. 2002;33:1–14. doi: 10.1016/s0891-5849(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 9.Dizdaroglu M, Laval J, Boiteux S. Biochemistry. 1993;32:12105–12111. doi: 10.1021/bi00096a022. [DOI] [PubMed] [Google Scholar]
- 10.Schärer OD, Kawate T, Gallinari P, Jiricny J, Verdine GL. Proc Nat Acad Sci USA. 1997;94:4878–4883. doi: 10.1073/pnas.94.10.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schärer OD, Verdine GL. J Am Chem Soc. 1995;117:10781–10782. [Google Scholar]
- 12.Stivers JT, Jiang YL. Chem Rev. 2003;103:2729–2760. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 13.Chepanoske CL, Porello SL, Fujiwara T, Sugiyama H, David SS. Nucleic Acids Res. 1999;27:3197–3204. doi: 10.1093/nar/27.15.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doi Y, Katafuchi A, Fujiwara Y, Hitomi K, Tainer JA, Ide H, Iwai S. Nucleic Acids Res. 2006;34:1540–1551. doi: 10.1093/nar/gkl061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marquez VE, Tseng CK-H, Mitsuya H, Aoki S, Kelley JA, Ford H, Jr, Roth JS, Broder S, Johns DG, Friscoll JS. J Med Chem. 1990;33:978–985. doi: 10.1021/jm00165a015. [DOI] [PubMed] [Google Scholar]
- 16.Bowman BR, Lee S, Wang S, Verdine GL. Structure. 2008;16:1166–1174. doi: 10.1016/j.str.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger I, Tereshko V, Ikeda H, Marquez VE, Egli M. Nucleic Acids Res. 1988;26:2473–2480. doi: 10.1093/nar/26.10.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun W, Wilson J, Kumar P, Knaus E, Wiebe L. Current Radiopharmaceuticals. 2009;2:75–82. [Google Scholar]
- 19.Iwai S. Angew Chem Int Ed. 2000;39:3874–3876. doi: 10.1002/1521-3773(20001103)39:21<3874::AID-ANIE3874>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Wang Y. Chem Res Toxicol. 2006;19:837–843. doi: 10.1021/tx060032l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumoto N, Toga T, Hayashi R, Sugasawa K, Katayanagi K, Ide H, Kuraoka I, Iwai S. Nucleic Acids Res. 2010;38:e101. doi: 10.1093/nar/gkq022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elzagheid MI, Viazovkina E, Damha MJ. Current Protocols in Nucleic Acid Chemistry. 2002;(Unit 1.7) doi: 10.1002/0471142700.nc0107s10. [DOI] [PubMed] [Google Scholar]
- 23.Lustig MJ, Cadet J, Boorstein RJ, Teebor GW. Nucleic Acids Res. 1992;20:4839–4845. doi: 10.1093/nar/20.18.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown KL, Adams T, Jasti VP, Basu AK, Stone MP. J Am Chem Soc. 2008;130:11701–11710. doi: 10.1021/ja8016544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuo S, Boorstein RJ, Teebor GW. Nucleic Acids Res. 1995;23:3239–3243. doi: 10.1093/nar/23.16.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfeifer GP. Mutat Res. 2000;450:155–166. doi: 10.1016/s0027-5107(00)00022-1. [DOI] [PubMed] [Google Scholar]
- 27.Ocampo-Hafalla MT, Altamirano A, Basu AK, Chan MK, Ocampo JE, Cummings A, Jr, Boorstein RJ, Cunningham RP, Teebor GW. DNA Repair. 2006;5:444–454. doi: 10.1016/j.dnarep.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 28.Katafuchi A, Nakano T, Masaoka A, Terato H, Iwai S, Hanaoka F, Ide H. J Biol Chem. 2004;279:14464–14471. doi: 10.1074/jbc.M400393200. [DOI] [PubMed] [Google Scholar]
- 29.Imamura K, Averill A, Wallace SS, Doublié S. J Biol Chem. 2012;287:4288–4298. doi: 10.1074/jbc.M111.315309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takao M, Kanno S, Kobayashi K, Zhang QM, Yonei S, van der Horst GT, Yasui A. J Biol Chem. 2002;277:42205–42213. doi: 10.1074/jbc.M206884200. [DOI] [PubMed] [Google Scholar]
- 31.Stivers JT, Pankiewicz KW, Watanabe KA. Biochemistry. 1999;38:952–963. doi: 10.1021/bi9818669. [DOI] [PubMed] [Google Scholar]
- 32.Porello SL, Williams SD, Kuhn H, Micheals ML, David SS. J Am Chem Soc. 1996;118:10684–10692. [Google Scholar]
- 33.Asahara H, Wistort PM, Bank JF, Bakerian RH, Cunningham RP. Biochemistry. 1989;28:4444–4449. doi: 10.1021/bi00436a048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.