

Abstract
The transcription factor interferon regulatory factor 5 (IRF5) has been identified as a human systemic lupus erythematosus (SLE) susceptibility gene by numerous joint linkage and genome-wide association studies. Although IRF5 expression is significantly elevated in primary blood cells of SLE patients, it is not yet known how IRF5 contributes to SLE pathogenesis. Recent data from mouse models of lupus indicate a critical role for IRF5 in the production of pathogenic autoantibodies and the expression of Th2 cytokines and type I IFN. In the current study, we examined the mechanism(s) by which loss of Irf5 protects mice from pristane-induced lupus at early time points of disease development. We demonstrate that Irf5 is required for Ly6C(hi) monocyte trafficking to the peritoneal cavity (PC), which is believed to be one of the initial key events leading to lupus pathogenesis in this model. Chemotaxis assays using peritoneal lavage from pristane-injected Irf5+/+ and Irf5−/− littermates support an intrinsic defect in Irf5−/− monocytes. We found the expression of chemokine receptors CXCR4 and CCR2 to be dysregulated on Irf5−/− monocytes and less responsive to their respective ligands, CXCL12 and CCL2. Bone marrow reconstitution experiments further supported an intrinsic defect in Irf5−/− monocytes since Irf5+/+ monocytes were preferentially recruited to the PC in response to pristane. Together, these findings demonstrate an intrinsic role for IRF5 in the response of monocytes to pristane, and their recruitment to the primary site of inflammation that is thought to trigger lupus onset in this experimental model of SLE.
Introduction
Systemic Lupus Erythematosus (SLE) is a complex autoimmune disorder characterized by multiple immunologic abnormalities that lead to a break in self-tolerance and the production of autoantibodies (1, 2). Patients display elevated type I IFN in their serum and IFN gene signature in their blood cells that correlates with disease activity and severity (3). At this point, nearly all key elements of the immune system have been implicated in the pathogenesis of SLE (1). Monocytes/macrophages have been increasingly recognized to play a dynamic role in the initiation and perpetuation of SLE given their hallmark functions in phagocytosis, antigen presentation and cytokine production (4). Increased influx of monocytes to sites of inflammation and aberrant expression of cytokines and surface activation markers on monocytes have been documented in SLE patients (4, 5). Recent studies in the pristane-induced model of murine lupus revealed that monocytes play a key pathogenic role by acting as a major producer of type I IFNs (6, 7).
The transcription factor interferon (IFN) regulatory factor 5 (IRF5) has been identified as an SLE susceptibility gene in numerous large-scale genetic association studies (8–13). IRF5 controls multiple inflammatory and immune responses through its regulation of type I IFN expression and IFN stimulated genes (ISGs) (14–17). IRF5 is also a key mediator of MyD88-dependent Toll-like receptor (TLR) signaling, thus a critical factor controlling the expression of proinflammatory cytokines (18). We have recently demonstrated that mice lacking Irf5 are protected from pristane-induced lupus in part due to significant alterations in cytokine expression, including elevated Th2 cytokines and significant weakening of the type I IFN gene signature (19). In support of this, overexpression of IRF5 was reported to induce M1 polarization (proinflammatory) in human macrophages and promote the Th1/Th17 response, while knockdown of IRF5 induced the M2 anti-inflammatory phenotype (20). In human SLE, we have shown that IRF5 expression is significantly elevated in primary peripheral blood mononuclear cells (PBMC) of SLE patients, as compared to healthy donors, and upregulation specifically associated with the IRF5 SLE risk haplotype in SLE monocytes (21). Subsequently, we demonstrated that IRF5 was constitutively activated (nuclear-localized) in SLE monocytes, and not other immune cell populations examined from SLE patients, further supporting a pathogenic role for IRF5 in SLE monocytes (22).
Given that our current findings in human SLE point towards a pathogenic role for IRF5 in SLE monocytes (21, 22), combined with the fact that monocytes are one of the key pathogenic triggers in pristane-induced lupus (6, 7, 23), we sought to use this model to examine further the functional role(s) of IRF5 in the early stages of disease development that require monocyte trafficking to the PC. We show that mice lacking Irf5 are significantly impaired in their chronic recruitment of Ly6C(hi) monocytes to the PC due to intrinsic defects in their chemokine receptor expression.
Materials and Methods
Mice
Irf5−/− mice backcrossed eight generations to C57BL6 were obtained from Dr. Ian Rifkin (Boston University School of Medicine) by approval from T. Taniguchi (University of Tokyo, Tokyo, Japan) and T. Mak (University of Toronto, Toronto, Ontario, Canada) (18). Wild-type C57BL/6 were purchased from The Jackson Laboratory (Bar Harbor, ME). After obtaining the Irf5−/− mice, they were back-crossed two additional times to parental C57Bl/6 in order to obtain heterozygotes for intercrossing to obtain a new cohort of Irf5+/+ and Irf5−/− littermates, by standard breeding techniques. Littermate Irf5+/+ mice were used as controls. The Dock2 mutation was analyzed by PCR genotyping of purified RNA from PBMC of Irf5+/+ and Irf5−/− littermates as described (24). All Irf5+/+ and Irf5−/− littermates used in this study lacked the Dock2 mutation. CD45.1 congenic mice B6.SJL-Ptprca Pepcb/BoyJ were purchased from The Jackson Laboratory (Bar Harbor, ME). Six to eight-week-old mice received a single intraperitoneal (i.p.) injection of 0.5 ml of pristane (Sigma-Aldrich), 1.5 ml 4% thioglycollate medium (Sigma-Aldrich) or PBS. Mice were sacrificed at indicated time-points post-injection. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Medicine and Dentistry of New Jersey, New Jersey Medical School.
Cell counting and flow cytometry
Cell counting was performed on a VI-Cell® (Beckman Coulter, Inc) or FACSCalibur flow cytometer (BD Biosciences) using CountBright™ Absolute Counting Beads (Invitrogen) according to manufacturer’s instruction. All antibodies were purchased from Biolegend except for anti-CXCR4-PE (clone 2B11; eBioscience) and anti-CCR2-PE (clone 475301; R&D Systems). The monocyte/macrophage population was gated on using anti-CD11b-FITC (clone M1/70) and anti-Ly6G-APC (clone 1A8) antibodies; monocyte subsets were distinguished from this population using anti-Ly6C-PE/CY7 (clone HK1.4) antibodies. To examine bone marrow chimeric mice, anti-CD45.1-PE (clone A20) and anti-CD45.2-PerCP/Cy5.5 (clone 104) antibodies were used. To examine monocyte maturation, anti-I-A-PE (clone M5/114.15.2) and anti-CD86-AF700 (clone GL-1) antibodies were used, and anti-IgG2b-PE Isotype Control (clone RTK4530) antibodies were used throughout the study. Surface staining was performed at 4° C for 20 min with an optimized amount of primary antibody or the appropriate isotype control. Intracellular staining was carried out in 0.5% saponin at 4° C for 30 min following surface staining. Samples were acquired on a FACSCalibur or LSR II (BD Biosciences) and data analyzed with FlowJo (Tree Star, Inc.) software.
Isolation of monocytes
Bone marrow cells collected from femurs and tibias were incubated with Biotin anti-mouse Ly6G Ab (clone 1A8; Biolegend) followed by incubation with Streptavidin microbeads (Miltenyi Biotec). Ly6G+ cells were depleted by magnetic separation with MS columns (Miltenyi Biotec). The negative fraction was incubated with CD11b microbeads (Miltenyi Biotec) and CD11b+ cells were isolated by magnetic separation. Alternatively, monocytes were isolated using EasySep® mouse monocyte enrichment kit (Stemcell Technologies). Briefly, bone marrow cells were incubated with antibody cocktail against non-monocytes followed by incubation with biotin selection cocktail and magnetic particles. Labeled cells were removed using EasySep magnet and monocytes were enriched. The two methods yielded a similar percentage of enriched monocytes per sample.
Chemotaxis Assay
Bone marrow cells were pooled from Irf5+/+ or Irf5−/− littermates and monocytes isolated using the EasySep® monocyte enrichment kit described above. 0.05 to 0.1 million cells were seeded in each of 6.6 mm transwells with 5 µm pores (Corning). Peritoneal lavage fluid or chemokine was added to the bottom well as the chemoattractant and RPMI-1640 plus 1% BSA medium was used as control. All chemokines were purchased from R&D Systems. Cultures were incubated under 5% CO2, 37° C for 3 hrs. Cells that migrated across the insert were trypsinized, collected and counted on a FACSCalibur (BD Biosciences).
Chemokine and chemokine receptor measurement
MILLIPLEX MAP multiplex mouse cytokine/chemokine kit (Millipore, Billerica, MA) was used to determine chemokine expression level according to the manufacturer's instruction. Samples were analyzed with a Luminex 100 Multi-Analyte Profiling System (Luminex Corp, Austin, TX). Cytokine/chemokine concentrations were determined by standard curve, which were generated using the mixed standard provided with the kit. For the analysis of chemokine receptor transcript expression, bone marrow monocytes were enriched, as described above, and RNA extracted using the RNeasy plus mini kit (Qiagen). Total RNA was converted to cDNA and the Mouse Chemokines and Receptors qPCR array (SABiosciences) was performed on the ABI 7300 Real-Time PCR system (Applied Biosystems) using RT2 qPCR master mixes (SABiosciences). Raw data were analyzed with SDS software (Applied Biosystems) and the web-based PCR array data analysis tool (SABiosciences).
Dual luciferase assay
HEK 293T cells were purchased from American Type Culture Collection (ATCC; Manassas, VA) and grown in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (Sigma) and 1 IU penicillin/1 µl/ml streptomycin (Mediatech, Hemdon, VA) at 37°C in a humidified incubator with 5% CO2/95% air. Cells were co-transfected with pGL3b-CCR2 or -CXCR4 promoter reporter plasmids and pCAGEN-mIRF5 or empty vector control plasmid and pRL using Lipofectamine® 2000 Reagent (Invitrogen) as previously described (14). Luciferase activity was measured 24 hr post-transfection and normalized to Renilla activity.
Generation of bone marrow chimeric mice
Bone marrow cells from CD45.1 Irf5+/+ and CD45.2 Irf5−/− C57BL/6 mice were harvested, treated with red blood cell lysis buffer (eBioscience), mixed at a 1:1 ratio and used as donor cells. Recipient Irf5+/+ CD45.2 C57BL/6 mice received 10 Gy of irradiation (split dose, 3 hrs apart) and were i.v. injected with 10 million donor cells 4 hrs after second irradiation. 6 weeks post-transplantation, each mouse was given 0.5 ml of pristane intraperitoneally and sacrificed 4 weeks later for PECs, spleen, blood and bone marrow. The monocyte/macrophage population was gated on using anti-Cd11b-FITC and anti-Ly6G-APC antibodies.
Phagocytosis assay
Splenic macrophages were isolated by positive selection with CD11b magnetic beads. Cells were seeded in a 96-well plate format and pre-incubated with LPS (100 ng/mL) or medium alone for 35 min (25). Cells were then incubated with 1 mg/mL FITC dextran for 40 min followed by 1 min incubation with 0.25 mg/mL trypan blue to quench extracellular signal. Phagocytosis was assessed by measuring fluorescein counts at absorbance 485/535.
Statistical analysis
Statistical analysis was done with the two-tailed Students t test or Mann-Whitney U test when variables were not normally distributed. Data are presented as mean ± SD (normal distribution) or mean ± SEM (non-normal distribution). p value <0.05 was considered significant. Statistical analyses were performed using Prism 4.0 (GraphPad Software, San Diego, CA).
Results
Pristane-induced Ly6C(hi) monocyte recruitment to the PC is impaired in Irf5−/− mice
Pristane (or 2,6,10,14-tetramethylpendadecane; TMPD) is a type of hydrocarbon oil that induces SLE in a variety of mouse strains (23). Diseased mice produce autoantibodies against small nuclear ribonucleoproteins (snRNPs) and dsDNA and develop immune complex-mediated glomerulonephritis (23). Irf5−/− mice are protected from pristane-induced lupus (19, 26). An early key event in this model is the recruitment of IFN-producing monocytes to the PC following i.p. injection of pristane (6, 7, 23). Type I IFN then stimulates the expression of inflammatory cytokines and chemokines leading to the sustained infiltration of leukocytes and formation of ectopic lymphoid tissues in the PC (6, 7, 27). There are two subsets of monocytes in murine blood; one subset expresses high levels of CX3CR1 and low levels of Ly6C and CCR2; the other subset expresses low levels of CX3CR1 and high levels of Ly6C and CCR2 (28, 29). The Ly6C(hi) subset is preferentially recruited by pristane and is the major producer of Type I IFN in this model (6, 7). In naïve C57BL/6 mice, the major components of peritoneal exudate cells (PECs) are CD11b+B220(dim) B1 cells and CD11b(hi)Ly6G−Ly6C− resident macrophages (7). Upon i.p. injection of pristane, however, CD11b+Ly6G+ neutrophils and CD11b+Ly6G− monocytes are recruited to the PC within 8 hrs (data not shown) and influx persists for months after (30) while B1 cells are dramatically reduced in the PC (7). Consistent with previous findings (6, 7), we found that the majority of monocytes elicited by pristane express high levels of Ly6C (Fig. 1A); we also found that there were less than 2% of B1 cells in the PC after pristane injection (Supplemental Fig. 1). We compared the numbers of peritoneal monocytes in Irf5+/+ (wt) and Irf5−/− littermates before pristane injection and 24, 48 hrs, 1, 2, 4 and 6 weeks after injection. No significant difference was observed at 24 and 48 hrs post-injection (data not shown). The earliest time point that showed a significant difference in the recruitment of monocytes/macrophages to the PC between littermates was at 2 weeks post-injection (Fig. 1B); this difference was even more striking at 4 and 6 weeks (Fig. 1C). Similar to total cell numbers in the PC, the frequency of CD11b+Ly6G− monocytes was also reduced in pristane-injected Irf5−/− mice (Supplemental Fig. 2A); the reduction in the Ly6C(hi) subset is not due to alterations in Ly6C expression between littermates (Supplemental Fig. 2B). A significant decrease in the number of Irf5−/− Ly6C(lo) cells in the PC was only observed at the later timepoints. Examination of IRF5 expression in sorted blood cells revealed dramatically higher levels in Ly6C(hi) monocytes as compared to Ly6C(lo) (Supplemental Fig. 3). In contrast to the pristane-induced chronic inflammation model, i.p. injection of thioglycollate (TG) induces acute inflammation with recruitment of monocytes/macrophages to the PC; recruitment generally peaks around 72 hrs post-injection and then declines (31). Upon examination of Ly6C(hi) monocyte recruitment to the PC in response to TG, we found no significant difference between littermates; notably, the recruitment of Ly6C(lo) cells was significantly decreased in Irf5−/− mice (Fig. 1D). In either model, the observed defect in monocyte migration to the PC was not due to differences in PC cellular apoptosis (data not shown). These data support a critical role for IRF5 in the recruitment of monocytes to the PC after induction of chronic or acute inflammation.
Figure 1. Ly6C(hi) monocyte recruitment to the PC after pristane-injection is impaired in Irf5−/− mice.
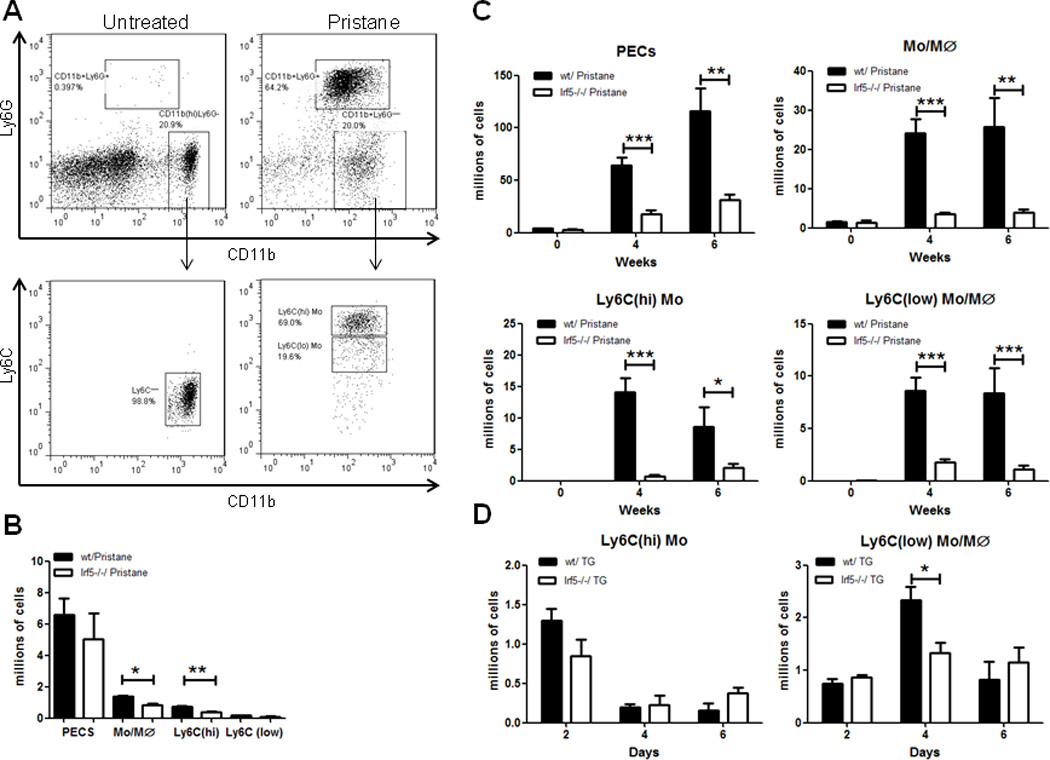
Irf5+/+ (wt) and Irf5−/− littermates were i.p. injected with 0.5 ml of PBS or pristane and PECs collected by peritoneal lavage with 3 ml PBS. (A) Gating strategy for the analysis of resident macrophages (CD11b(hi)Ly6G−), pristane-elicited monocytes/macrophages (CD11b+Ly6G−) in PECs and monocyte subsets (Ly6C(hi) and Ly6C(lo)) used for subsequent labeling experiments. Representative flow cytometry analysis of resident and pristane-elicited PECs in Irf5+/+ C57BL/6 mice. (B) Flow cytometry quantification of PECs, monocyte/macrophage (Mo/Mϕ), Ly6C(hi) and Ly6C(lo) monocyte subsets at 2 weeks post-pristane injection. (C) Same as in (B) except quantification was performed at 4 and 6 weeks post-injection and cells counted by Vi-Cell. (D) Flow cytometry quantification of Ly6C(hi) and Ly6C(lo) PECs after 1.5 ml i.p. injection of thioglycollate (TG). n = 4–7 mice per group, representative of two independent experiments. * p< 0.05; ** p< 0.01; *** p< 0.001 by unpaired Student t test.
Irf5−/− monocytes are defective in their response to peritoneal lavage of pristane-injected mice
Impaired Ly6C(hi) monocyte recruitment to the PC in pristane-injected Irf5−/− mice could be due to an intrinsic defect in Irf5−/− monocytes or an extrinsic defect in the signaling environment of the PC. To test these two possibilities, we performed chemotaxis assays on naïve monocytes from Irf5+/+ and Irf5−/− littermates using peritoneal lavage fluid from littermates 4 weeks post-pristane injection as the chemoattractant. Naïve monocytes were enriched from bone marrow cells by magnetic depletion of non-monocytes; a similar percent enrichment per sample and number of cells plated per experiment was achieved (Figs. 2A–B). Consistent with previous reports, enriched bone marrow monocytes are predominantly Ly6C(hi) (data not shown) (32, 33). We found that significantly fewer monocytes from Irf5−/− mice, as compared to Irf5+/+ littermates, could migrate towards peritoneal lavage fluid from pristane-injected Irf5+/+ mice (Fig. 2C), supporting an intrinsic defect in Irf5−/− monocytes. Little difference between the migration efficiency of Irf5+/+ and Irf5−/− monocytes to Irf5−/− lavage was observed.
Figure 2. Irf5−/− monocytes are defective in their migration towards peritoneal lavage fluid from pristane-injected Irf5+/+ mice.
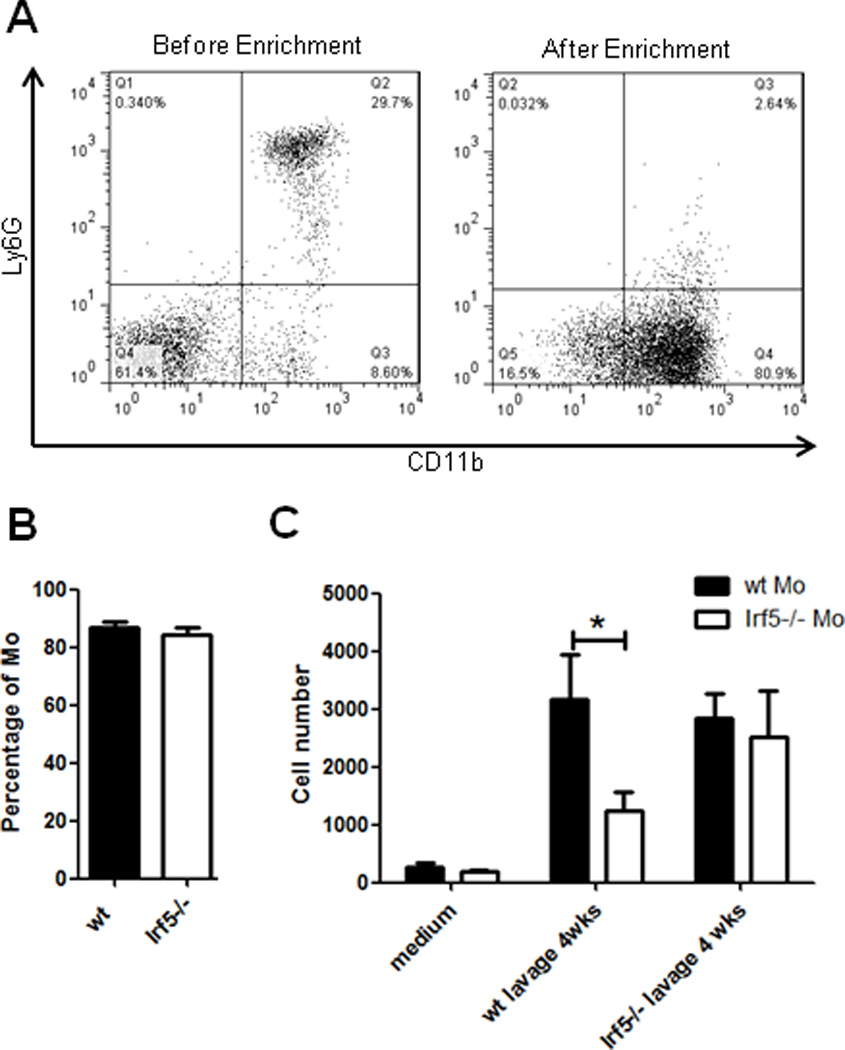
Monocytes were enriched from bone marrow cells of naïve Irf5+/+ (wt) and Irf5−/− littermates. (A) Representative flow cytometry analysis of CD11b+Ly6G− monocytes from Irf5+/+ C57BL/6 mice before and after enrichment. (B) Percentages of CD11b+Ly6G− monocytes were assessed by flow cytometry; similar percentages were obtained for each genotype after enrichment. (C) Peritoneal lavage fluid was collected from six littermates per genotype at 4 weeks post-pristane injection and used as chemoattractant in the chemotaxis assay. The number of Irf5+/+ and Irf5−/− monocytes that migrated to bottom wells in response to peritoneal lavage fluid were quantified on flow cytometry using counting beads. n = 10 mice per group, representative of three independent experiments. * p< 0.05 by Mann-Whitney U test.
Expression of a subset of chemokine receptors is dysregulated on Irf5−/− monocytes
Chemokine receptors are critical for monocyte trafficking under both steady-state and inflammatory conditions (34). CX3CR1 is required for Ly6C(lo) monocytes to migrate to the periphery to replenish tissue macrophages during homeostasis (35). CCR1, CCR2, CCR5, CCR8, CXCR2 and CXCR4 have been implicated in monocyte recruitment during infection and inflammation; they regulate monocyte egress from bone marrow, migration from the circulation to inflamed tissue, and/or homing to bone marrow (34). The observed decrease in the response of Irf5−/− monocytes to pristane could be due to defects in chemokine receptor expression, resulting in the inability of Irf5−/− monocytes to “sense” inflammatory signals. To test this, we isolated naïve monocytes from the bone marrow of Irf5+/+ and Irf5−/− littermates, that are predominantly Ly6C(hi), and examined the expression of chemokine receptors using a Mouse Chemokines and Receptors qPCR array (SABiosciences). Receptors that were upregulated or downregulated more than two-fold in Irf5−/− monocytes as compared to Irf5+/+ are listed, along with their respective ligands, in Table 1. We found a striking decrease in CXCR4 receptor expression on monocytes from Irf5−/− mice, as well as decreases in CXCR3, CCRL1, CCR5, and CCR2, and increases in CCR1 and CX3CR1 expression.
Table 1.
Chemokine receptors differentially expressed on naϊve bone marrow monocytes from Irf5−/− micea.
| Gene | Fold Changeb | Ligands |
|---|---|---|
| Ccr1 | 3.56 | RANTES |
| Cx3cr1 | 17.31 | CX3CL1 |
| Cxcr4 | −52.46 | CXCL12 |
| Cxcr3 | −8.19 | IP10 |
| Ccrl1 | −5.83 | CCL19, CCL21 |
| Ccr5 | −3.69 | RANTES, MIP1A/B |
| Ccr2 | −2.08 | CCL2, CCL7, CCL12 |
Bone marrow cells were pooled from n=4 Irf5+/+ and Irf5−/− littermates to isolate monocytes.
denotes the fold change as compared to naϊve Irf5+/+ monocytes; (+) upregulated or (−) downregulated.
Impaired CCR2 and CXCR4 protein expression and function on Irf5−/−monocytes
To determine whether there is a correlation between chemokine receptor transcript expression and protein expression on Irf5−/− monocytes, we collected blood and bone marrow from naïve and pristane-injected Irf5+/+ and Irf5−/− littermates and examined surface or intracellular staining of the chemokine receptors CXCR4, CXCR3, CCR2 and CCR5 on monocyte subsets. No significant difference in the expression of CCR5 or CXCR3 was observed on monocytes from the blood or bone marrow of naïve and pristane-injected littermates (data not shown). Instead, a significant decrease in CCR2 expression was found on naïve blood monocytes of Irf5−/− mice, as compared to Irf5+/+, and the decrease was observed in both Ly6C(hi) and Ly6C(lo) subsets (Fig. 3A&B). CCR2 expression is not altered by pristane treatment (7). Since nothing is known of CXCR4 expression in the pristane-induced model of lupus, we examined expression on monocytes from both naïve and pristane-injected mice. No defect in CXCR4 expression was observed on naïve blood or bone marrow monocytes of Irf5−/− mice (Fig. 3C and data not shown); however, a decrease on bone marrow monocytes of pristane-injected Irf5−/− mice was observed (Fig. 3D–F). Although the mean fluorescence intensity (MFI) of CXCR4 staining was not significantly reduced, the percentage of CXCR4 expressing cells was significantly decreased (Fig. 3D–F). Similar reductions were found on blood monocytes from pristane-injected Irf5−/− mice (data not shown).
Figure 3. Decreased expression of CCR2 and CXCR4 on Irf5−/− monocytes.
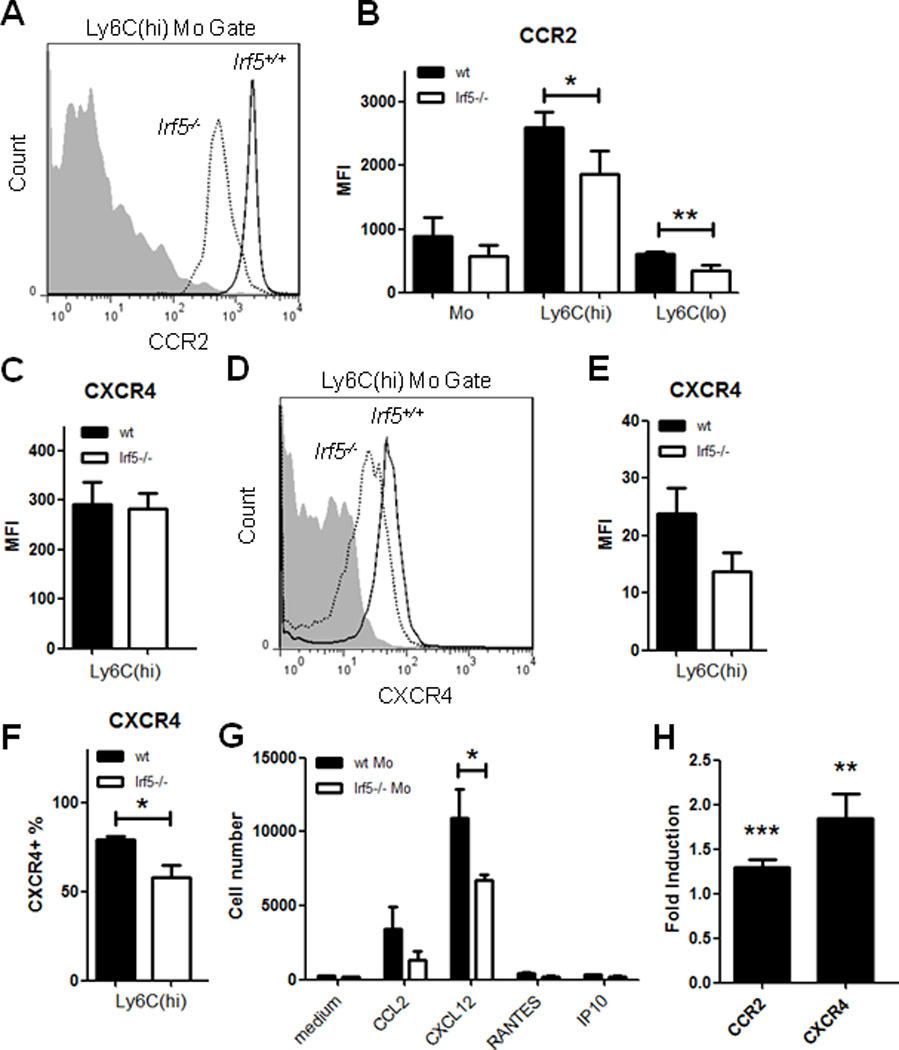
Blood and bone marrow cells were collected from naïve and pristane-injected Irf5+/+ (wt) and Irf5−/− littermates. Surface staining was performed with anti-CD11b, -Ly6G, -Ly6C and -CCR2 antibodies, and intracellular staining with anti-CXCR4 antibodies; receptor expression was analyzed by flow cytometry. (A) Representative histogram of CCR2 expression on naïve blood Ly6C(hi) monocytes from littermates and (B) quantification of the mean fluorescence intensity (MFI) on Ly6C(hi) and Ly6C(lo) monocytes; n = 10–11 mice per genotype. Gray area represents isotype control staining in (A and D). (C) Quantification of the MFI of CXCR4 on bone marrow monocytes from naïve mice. (D) Same as in (A) except CXCR4 expression was analyzed on bone marrow monocytes 4 weeks post-pristane injection. (E) Quantification of the MFI of CXCR4 and (F) percentage of CXCR4 expressing cells in Ly6C(hi) bone marrow monocytes from pristane-injected mice. n = 3 mice per genotype. (G) Irf5−/− monocytes are defective in their response to CCR2 and CXCR4 ligands. Enriched naïve bone marrow monocytes from Irf5+/+ and Irf5−/− littermates were seeded inside transwells and medium alone or supplemented with 50 ng/ml CCL2, CXCL12, RANTES or 300 ng/ml IP10 added to the other side as chemoattractants. Cells that migrated to bottom wells were quantified. n = 6 mice per genotype. Data are representative of two independent experiments. * p < 0.05; ** p < 0.01 by Mann-Whitney U test. (H) IRF5 regulates chemokine receptor promoter reporter activity. HEK 293T cells were transiently co-transfected with pGL3b-CCR2 or CXCR4 promoter reporters with pCAGEN-mIRF5 or empty vector and pRL. Luciferase activity was measured and normalized to Renilla activity. Data is presented as relative induction of luciferase activity by IRF5 over control empty vector. Data are representative of three independent experiments performed in duplicate. * p < 0.05; ** p < 0.01; *** p< 0.001 by unpaired Student t test.
In order to understand the functional consequence of impaired chemokine receptor expression on Irf5−/− monocytes, we examined the ability of Irf5+/+ and Irf5−/− monocytes to migrate towards specific receptor ligands using the chemotaxis assay. Naϊve monocytes from either Irf5+/+ or Irf5−/− littermates showed little response to RANTES and IP10 (Fig. 3G); however, a significant defect in the ability of Irf5−/− monocytes to migrate towards the CXCR4 ligand, CXCL12, was found. In addition, a consistent, yet non-significant, reduction in the migration ability of Irf5−/− monocytes towards the CCR2 ligand, CCL2, was observed (Fig. 3G). To begin to address how IRF5 might be regulating the expression of these two genes, we performed promoter reporter assays in HEK 293T cells and found a small yet significant transactivation of both reporters by IRF5 (Fig. 3H). Together, these data suggest that the observed defect in both in vivo and in vitro Irf5−/− monocyte recruitment is due to the impaired expression and function of CCR2 and CXCR4 receptors.
Expression of monocyte-related chemokines is not compromised in the PC of Irf5−/− mice
It has been shown that IRF5 expression promotes M1 polarization and Th1/Th17 cytokine expression while inhibiting M2-associated cytokines (19, 20, 36). Among the cytokines currently known to be regulated by IRF5, such as IFN-α, IL-6, IL-12, and TNF-α, some have been reported to be elevated in the serum of SLE patients (2, 18, 37–39). To this effect, we recently demonstrated that serum levels of the Th1 cytokine IL-6 was decreased, while Th2 cytokines IL-4, IL-5 and IL-10 were increased in pristane-injected Irf5−/− mice (19). In addition, we showed that CCL2 (MCP-1) expression was significantly reduced in Irf5−/− bone marrow cells (19). Although data in Figs. 1–3 support an intrinsic defect in Irf5−/− monocytes that results in impaired recruitment to the PC, we cannot exclude an extrinsic defect in serum or PC chemokine expression in Irf5−/− mice that may also contribute to impaired recruitment. To further examine this, we measured sera and PC chemokine levels using the MILLIPLEX MAP multiplex mouse cytokine/chemokine kit (Millipore) at 2 weeks post-pristane injection. Interestingly, we found no significant difference in the levels of monocyte-related chemokines (CCL2, IP10, RANTES, MIP1a and MIP1b) in the peritoneal lavage fluid between Irf5+/+ and Irf5−/− littermates; however, decreases were observed for some of these chemokines in the sera of Irf5−/− mice (Fig. 4). Although a significant defect in IP10 expression was observed in sera from Irf5−/− mice, it is currently not known whether IP10 mediates monocyte recruitment in the pristane model of murine lupus and neither Irf5+/+ or Irf5−/− monocytes responded to IP10 in vitro (Fig. 3G). Nonetheless, these data provide further support that impaired recruitment of Irf5−/− monocytes to the PC in response to pristane is primarily due to intrinsic defects in chemokine receptor expression and not the chemokines themselves.
Figure 4. Expression of monocyte-related chemokines is not impaired in the PC of Irf5−/− mice.
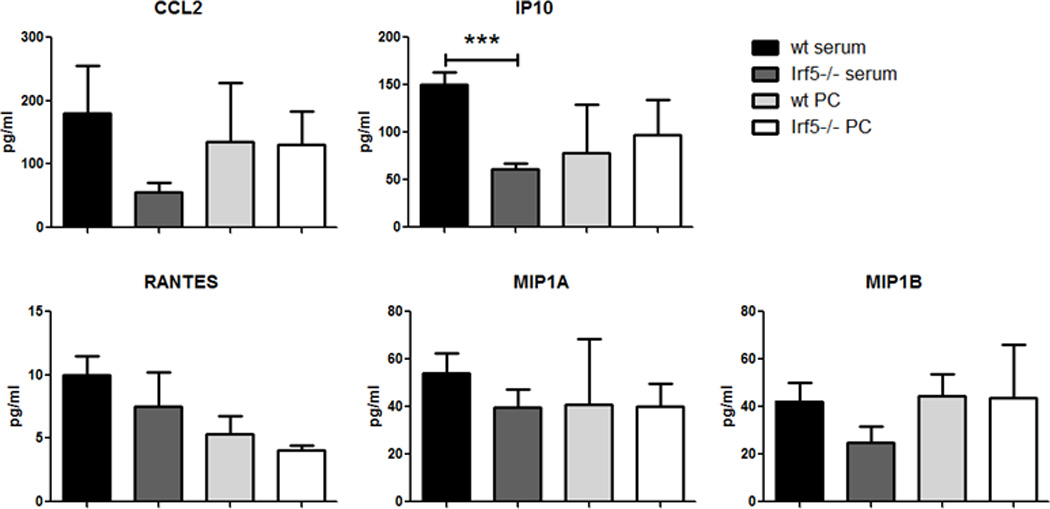
Serum and peritoneal lavage fluid (PC) from Irf5+/+ (wt) (n = 2–5) and Irf5−/− (n = 4–6) littermates were harvested at 2 weeks post-pristane injection. Chemokine levels were determined by beads-based immunoassay. *** p< 0.001 by unpaired Student t test.
Irf5+/+ monocytes/macrophages are preferentially recruited to the PC of pristane-injected chimeras
To unambiguously confirm the intrinsic defect in Irf5−/− monocytes, we generated bone marrow chimeras by injecting 1:1 mixed CD45.1 Irf5+/+ and CD45.2 Irf5−/− bone marrow cells to lethally irradiated CD45.2 Irf5+/+ mice (Fig. 5A). Six weeks after reconstitution, pristane was administered to the reconstituted mice, and 4 weeks later, the percentages of CD45.1 and CD45.2 monocytes/macrophages in the bone marrow, blood, PC and spleen were determined (Fig. 5B). Before injection of pristane, a slight enrichment of Irf5+/+ monocytes was observed in blood and this held true after pristane injection; however, a significant defect in the recruitment of Irf5−/− CD11b+Ly6G− bone marrow cells to the blood and PC of chimeras was observed as the ratio of Irf5+/+ (CD45.1) to Irf5−/− (CD45.2) monocytes went up from ~1 in bone marrow to ~1.8 in the blood and ~2.2 in the PC (Fig. 5C). Less than 5% of CD11b+Ly6G− cells from chimeras were CD11b+B220(dim) B1 cells (data not shown). These data support the impaired recruitment of Irf5−/− monocytes to the PC in response to pristane due to an intrinsic defect in Irf5−/− monocytes.
Figure 5. Impaired recruitment of Irf5−/− monocytes to the PC of pristane-injected bone marrow chimeras.
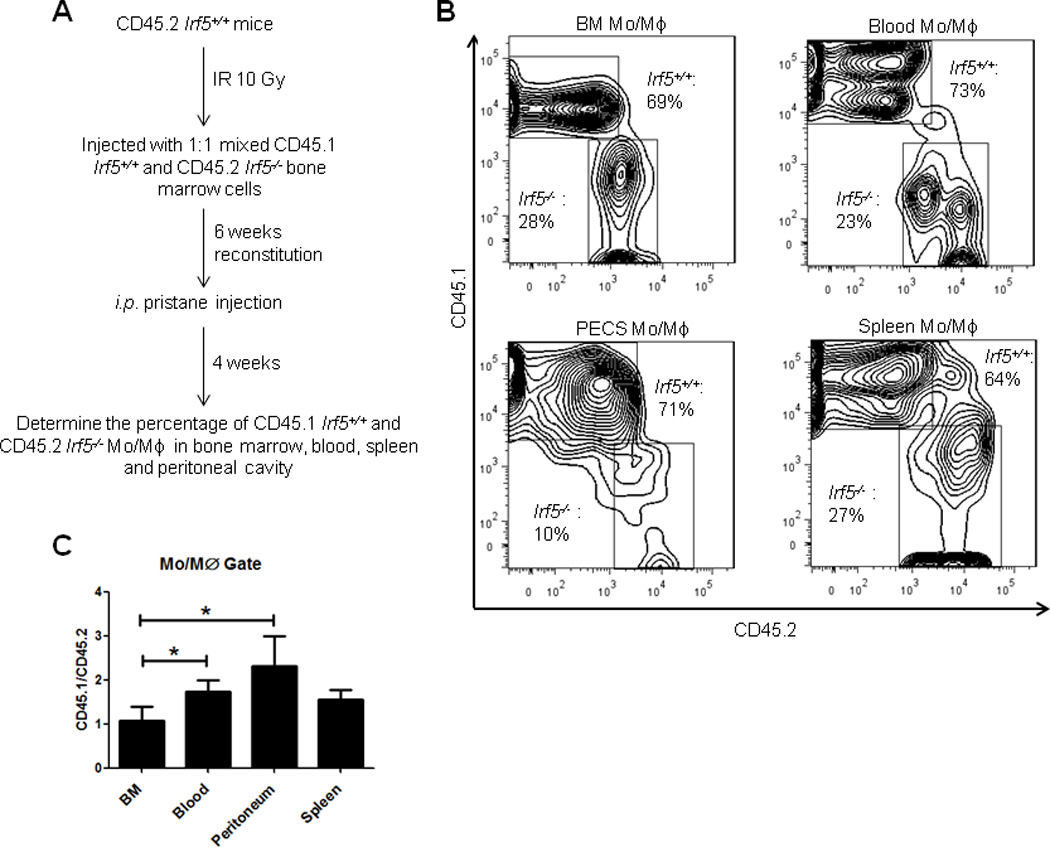
(A) Schematic of bone marrow reconstitution model. (B) Representative contour plots showing percentages of CD45.1 Irf5+/+ and CD45.2 Irf5−/− cells in the monocytes/macrophages (Mo/Mϕ) gate in different compartments. (C) Ratio of CD45.1 and CD45.2 Mo/Mϕ in the bone marrow (BM), blood, PC (PECs) and spleen of chimerical mice. n = 10 mice; representative of two independent experiments. * p< 0.05 by paired Student t test.
Enhanced phagocytic activity but not maturation of Irf5−/− monocytes/macrophages
In human SLE, impaired cell clearance and accumulation of apoptotic debris is thought to contribute to a break in self-tolerance leading to the production of pathogenic autoantibodies (40). Indeed, cells from SLE patients appear more susceptible to apoptosis than healthy donors, yet their macrophages are impaired in their ability to engulf apoptotic material (41, 42). In the pristane-induced model of murine lupus, it was found that the rapid turnover of recruited monocytes in the PC of pristane-injected wild-type mice was associated with a lack of differentiation into more phagocytic Ly6C(lo) monocytes/macrophages (7), which was consistent with previous observations that the uptake of carbon particles are substantially reduced after pristane injection (43). Furthermore, pristane induces apoptosis both in vivo and in vitro and is thought to be one of the critical first events in the pathogenesis of pristane-induced lupus that is similar to human SLE (44). First, we examined whether loss of Irf5 alters monocyte/macrophage differentiation. At 4 weeks post-pristane injection, we examined activation/maturation markers I-A (MHC II) and CD86 on PC monocytes from Irf5+/+ and Irf5−/− littermates and found no significant difference in expression (Fig. 6A). We next examined in vitro phagocytosis and found that macrophages from Irf5−/− mice had enhanced phagocytic ability in their response to LPS as compared to macrophages from Irf5+/+ littermates (Fig. 6B). These data support a potentially new role for IRF5 in phagocytosis that is not due to alterations in the activation/maturation of monocytes.
Figure 6. CD11b+ splenic macrophages from Irf5−/− mice display enhanced phagocytosis.
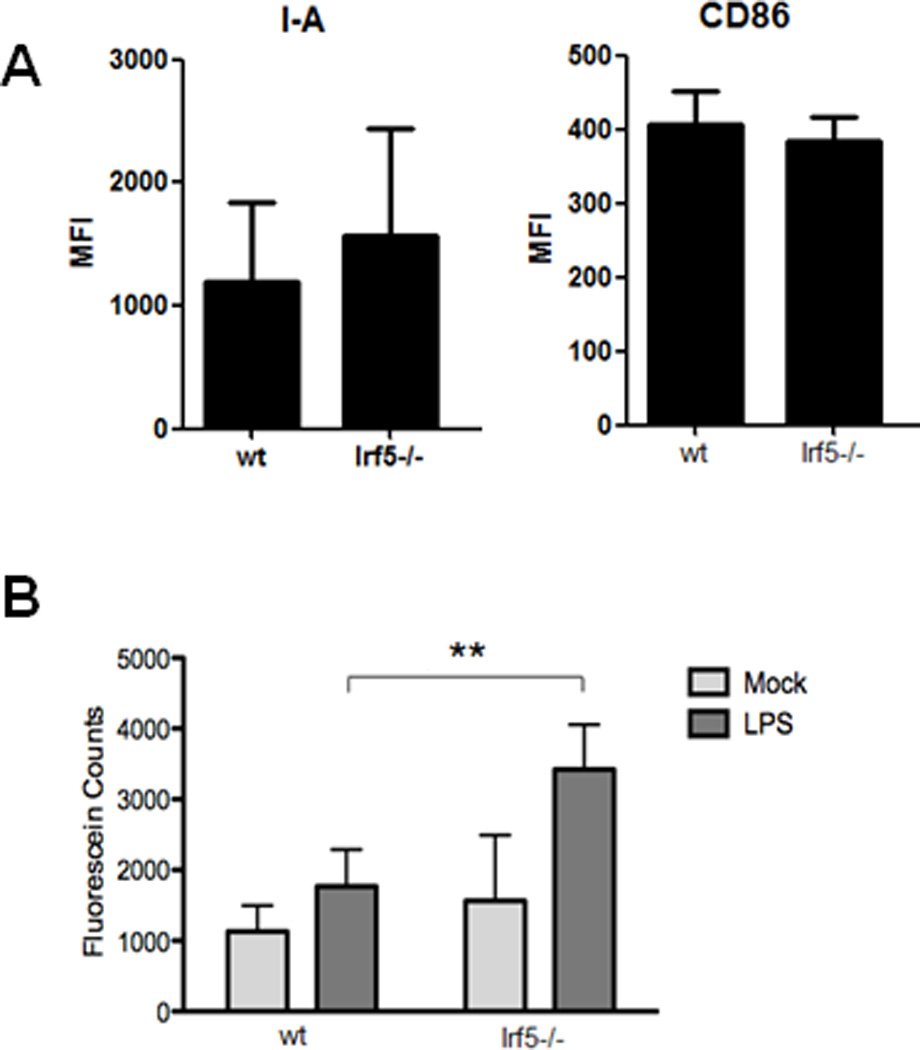
(A) PECs were collected from Irf5+/+ (wt) and Irf5−/− littermates 4 wks post-pristane injection. Expression of activation/maturation surface markers I-A (MHC II) and CD86 were examined on CD11b+Ly6G− monocytes. n = 5 mice per genotype. (B) Splenic macrophages from Irf5+/+ and Irf5−/− mice were isolated by positive selection and in vitro phagocytosis determined by measuring fluorescein counts. n = 4–5 per genotype; **p < 0.01 by unpaired Students t test.
Discussion
The recruitment of monocytes to inflamed tissues is critical for host defense against a variety of pathogens; however, this response can be a double-edged sword creating a state of chronic inflammation if not properly regulated (34). Monocyte trafficking is directed by adhesion proteins, chemokines and their receptors (34), yet the mechanisms and factors controlling these molecules have yet to be fully elucidated. Others and we have recently demonstrated in the pristane-induced model of chronic inflammation that results in the development of lupus-like disease, that mice lacking the transcription factor Irf5 are protected from disease development (19, 26, 36). We found that protection of Irf5−/− mice from lupus onset and pathogenic autoantibody production was due in part to a weakened type I IFN signature and significant skewing of the cytokine milieu towards a more protective Th2-like environment (19). In the current study, we examined the role of IRF5 in the early stages of lupus development that require monocyte trafficking to the PC in response to i.p. injection of pristane. We found a significant reduction in the migration of Ly6C(hi) monocytes to the PC as early as 2 wks post-injection (Fig. 1). These data are reminiscent of findings from pristane-injected IFNAR−/− mice except that we did not observe a concomitant increase in Ly6C(lo) monocytes/macrophages in the PC (7); TLR7−/− and MyD88−/− mice also gave similar decreases in Ly6C(hi) monocyte recruitment to the PC but the Ly6C(lo) subset was not analyzed (45). While preparing this manuscript, a paper by Xu et al. was published that reported the similar finding of reduced Ly6C(hi) monocyte recruitment to the PC of pristane-injected Irf5−/− mice yet, identical to the pristane-injected IFNAR−/− mice, they observed a significant increase in the percentage of Ly6C(lo) cells in the PC at 2 wks post-injection (36). The difference between our findings may in part be explained by the complete abolishment of the type I IFN gene signature in their pristane-injected Irf5−/− mice (36), while we only observed a significant weakening of the IFN signal (19). It was found that accumulation of the Ly6C(lo) subset in the PC of IFNAR−/− mice was due to enhanced maturation of Ly6C(hi) monocytes to Ly6C(lo) rather than the preferential recruitment of the Ly6C(lo) subset (7). We did not observe a significant difference in the expression of activation/maturation markers I-A and CD86 on peritoneal monocytes between Irf5+/+ and Irf5−/− littermates supporting the observed lack of increase in the Ly6C(lo) subset (Fig. 6) (46).
It was recently reported that several independent colonies of Irf5−/− mice in the United States are contaminated with a spontaneous genomic duplication and frameshift mutation in the guanine exchange factor dedicator of cytokinesis 2 (Dock2) gene (24). Dock2, an atypical Rac activator, is essential for TLR7/9-mediated IFN-α induction in PDCs, T and B lymphocyte migration, atrophy of lymphoid follicles and loss of marginal zone B cells (46–48). Previously observed phenotypes of the Irf5−/− mice, such as altered marginal zone B cells, changes in PDC number and function, and type I IFN responses, are now being put into question by these findings (24). It was not reported by Xu and colleagues whether their Irf5−/− mice were screened for the Dock2 mutation that could potentially contribute to the complete abolishment of the type I IFN signature (36). We have reported the genotype of our Irf5−/− mice and they contain homozygous wild-type Dock2 alleles (19). For the most part, results from three independent studies on pristane-injected Irf5−/− mice have been quite similar (19, 26, 36). Nonetheless, it will be critical to our understanding of IRF5 function to replicate and confirm previous findings in Irf5−/− mice that have been genotyped for the Dock2 mutation.
The mechanism(s) of impaired Ly6C(hi) monocyte recruitment to the PC of pristane-injected Irf5−/− mice was not examined in the recent study by Xu and colleagues (36). Here, we found the defect in Ly6C(hi) monocyte recruitment to the PC of Irf5−/− mice to be due, at least in part, to decreased expression of chemokine receptors CCR2 and CXCR4 on Ly6C(hi) monocytes of Irf5−/− mice (Fig. 3). Previous studies have shown that CCR2 is required for monocyte egress from the bone marrow to the circulation during homeostasis and inflammation (49–51). More importantly, it has been shown that the chronic influx of monocytes to the PC by pristane is dependent on CCR2; both Ly6C(hi) and Ly6C(lo) monocyte subsets were largely absent in the PC of ccr2−/− mice (7). We observed normal egress of Ly6C(hi) monocytes from the bone marrow of Irf5−/− mice to the circulation and CCR2 protein expression on Irf5−/− bone marrow monocytes was not changed between Irf5+/+ and Irf5−/− mice (data not shown); however, CCR2 expression was significantly reduced on circulating Irf5−/− blood monocytes (Fig. 3). While little is known of CXCR4 expression and function in the pristane-induced model of murine lupus, expression of CXCR4 has been shown to be upregulated on Ly6C(hi) monocytes of lupus-prone murine strains and accumulation of CXCR4 expressing monocytes/macrophages has been observed in the kidneys of diseased mice (52). CXCR4 antagonism has also been shown to restore monocyte numbers in the circulation following monocyte depletion by CCL2 blocking molecules and administration of CXCR4 antagonists protects B6.Sle1.Yaa mice from lupus (53). Similar to findings of CCR2 expression (7), CXCR4 expression on monocytes was not increased after pristane injection (data not shown); however, CXCR4 protein levels were significantly reduced on bone marrow (Fig. 3F) and blood (data not shown) monocytes from pristane-injected Irf5−/− as compared to Irf5+/+ mice. To put these findings into perspective with the Dock2 mutation, while homing of lymphocytes to secondary lymphoid tissues has been shown to be defective in Dock2−/− mice, the numbers of monocytes in the periphery of Dock2−/− mice were maintained and Dock2−/− monocytes responded normally in vitro to CCL2 and CXCL12 ligands (47). Data in Fig. 3G support that the observed intrinsic defects in CCR2 and CXCR4 expression on Irf5−/− Ly6C(hi) monocytes and their inability to respond to CCL2 and CXCL12 are not due to confounding genotype results as current data support that Dock2 is dispensable for monocyte trafficking (47). This data is further enhanced by the fact that IRF5 can regulate the CCR2 and CXCR4 promoters (Fig. 3H). Thus, these data instead point towards a critical role for IRF5 in regulating the molecules, i.e. chemokine receptors, which are important for pristane-induced monocyte trafficking to the PC.
In vivo data from Irf5−/− mice (Fig. 1) and bone marrow chimerical mice (Fig. 5C) provide clear support for the intrinsic defect in Irf5−/− monocytes. In response to pristane, data from both of these models reveal a defect in the recruitment of Irf5−/− monocytes to the PC; however, data from bone marrow chimerical mice suggests that the primary defect may be in the initial response of Irf5−/− bone marrow monocytes to signals in the blood environment since a significant decrease in the recruitment of Irf5−/− monocytes from the bone marrow to the blood was observed (Fig. 5C). Results from the in vitro chemotaxis assay also support a defect in the response of Irf5−/− bone marrow monocytes to peritoneal lavage fluid from pristane-injected Irf5+/+. Somewhat surprising, however, was the finding of no significant difference between the recruitment of Irf5+/+ and Irf5−/− bone marrow monocytes to peritoneal lavage from Irf5−/− mice (Fig. 2C). This small discrepancy is likely a result of in vivo versus in vitro experimentation and the different signals monocytes are exposed to in each compartment (bone marrow, blood or PC). The in vitro chemotaxis assay simply measured the response of bone marrow monocytes to signals from peritoneal lavage fluid; unlike the in vivo systems, it did not take into account signals from the blood that the bone marrow monocytes would first be exposed to before recruitment to the PC. Another possible explanation is that bone marrow monocytes and blood monocytes lacking Irf5 respond differently to signals from the PC. Further studies will be required to clearly define the role of IRF5 in these two cell populations.
In conclusion, this study defines an essential role for IRF5 in the recruitment of monocytes to the PC in the pristane-induced model of chronic inflammation and murine lupus. Given the recent finding that IRF5 is already activated (nuclear-localized) in human SLE monocytes (22), supporting a pathological function for IRF5 in human disease, it will be interesting to expand these types of studies to examine the functional role of IRF5 in monocytes/macrophages from SLE patients. Together, these findings clearly demonstrate an intrinsic role for IRF5 in the regulation of inflammatory monocyte migration through its ability to control chemotactic responses to CCL2 and CXCL12 and provide significant new insight into how the dysregulation of IRF5 expression and activation may contribute to human SLE pathogenesis.
Supplementary Material
Acknowledgements
We thank Ian Rifkin for providing the Irf5-deficient mice by approval from Tadatsugu Tanaguchi and Tak Mak.
Footnotes
This work was supported in part by funds from UMDNJ New Jersey Medical School and grants from the National Institute of Health (NIH)/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS; 5R03AR054070) and the Arthritis Foundation (to BJB).
References
- 1.Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358:929–939. doi: 10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- 2.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katsiari CG, Liossis SN, Sfikakis PP. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin Arthritis Rheum. 2010;39:491–503. doi: 10.1016/j.semarthrit.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Reefman E, de Jong MC, Kuiper H, Jonkman MF, Limburg PC, Kallenberg CG, Bijl M. Is disturbed clearance of apoptotic keratinocytes responsible for UVB-induced inflammatory skin lesions in systemic lupus erythematosus? Arthritis Res Ther. 2006;8:R156. doi: 10.1186/ar2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee PY, Weinstein JS, Nacionales DC, Scumpia PO, Li Y, Butfiloski E, van Rooijen N, Moldawer L, Satoh M, Reeves WH. A novel type I IFN-producing cell subset in murine lupus. J Immunol. 2008;180:5101–5108. doi: 10.4049/jimmunol.180.7.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES, Nacionales DC, Butfiloski EJ, van Rooijen N, Akira S, Sobel ES, Satoh M, Reeves WH. Type I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am J Pathol. 2009;175:2023–2033. doi: 10.2353/ajpath.2009.090328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, Ortmann WA, Koeuth T, Gonzalez Escribano MF, Pons-Estel B, Petri M, Daly M, Gregersen PK, Martin J, Altshuler D, Behrens TW, Alarcon-Riquelme ME. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 9.Cunninghame Graham DS, Manku H, Wagner S, Reid J, Timms K, Gutin A, Lanchbury JS, Vyse TJ. Association of IRF5 in UK SLE families identifies a variant involved in polyadenylation. Hum Mol Genet. 2007;16:579–591. doi: 10.1093/hmg/ddl469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demirci FY, Manzi S, Ramsey-Goldman R, Minster RL, Kenney M, Shaw PS, Dunlop-Thomas CM, Kao AH, Rhew E, Bontempo F, Kammerer C, Kamboh MI. Association of a common interferon regulatory factor 5 (IRF5) variant with increased risk of systemic lupus erythematosus (SLE) Ann Hum Genet. 2007;71:308–311. doi: 10.1111/j.1469-1809.2006.00336.x. [DOI] [PubMed] [Google Scholar]
- 11.Kozyrev SV, Lewen S, Reddy PM, Pons-Estel B, Witte T, Junker P, Laustrup H, Gutierrez C, Suarez A, Francisca Gonzalez-Escribano M, Martin J, Alarcon-Riquelme ME. Structural insertion/deletion variation in IRF5 is associated with a risk haplotype and defines the precise IRF5 isoforms expressed in systemic lupus erythematosus. Arthritis Rheum. 2007;56:1234–1241. doi: 10.1002/art.22497. [DOI] [PubMed] [Google Scholar]
- 12.Shin HD, Sung YK, Choi CB, Lee SO, Lee HW, Bae SC. Replication of the genetic effects of IFN regulatory factor 5 (IRF5) on systemic lupus erythematosus in a Korean population. Arthritis Res Ther. 2007;9:R32. doi: 10.1186/ar2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, Jonsen A, Rantapaa-Dahlqvist S, Moller B, Kere J, Koskenmies S, Widen E, Eloranta ML, Julkunen H, Kristjansdottir H, Steinsson K, Alm G, Ronnblom L, Syvanen AC. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, Fitzgerald KA, Golenbock DT. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem. 2005;280:17005–17012. doi: 10.1074/jbc.M412584200. [DOI] [PubMed] [Google Scholar]
- 15.Barnes BJ, Richards J, Mancl M, Hanash S, Beretta L, Pitha PM. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J Biol Chem. 2004;279:45194–45207. doi: 10.1074/jbc.M400726200. [DOI] [PubMed] [Google Scholar]
- 16.Barnes BJ, Kellum MJ, Field AE, Pitha PM. Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol Cell Biol. 2002;22:5721–5740. doi: 10.1128/MCB.22.16.5721-5740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J Biol Chem. 2001;276:23382–23390. doi: 10.1074/jbc.M101216200. [DOI] [PubMed] [Google Scholar]
- 18.Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, Taniguchi T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 19.Feng D, Y L, Bi X, Stone RC, Patel P, Barnes BJ. Irf5-deficient mice are protected from pristane-induced lupus via increased Th2 cytokines and altered IgG class switching. European J. Immunol. 2012 doi: 10.1002/eji.201141642. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA. IRF5 promotes inflammatory macrophage polarization and TH1–TH17 responses. Nat Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 21.Feng D, Stone RC, Eloranta ML, Sangster-Guity N, Nordmark G, Sigurdsson S, Wang C, Alm G, Syvanen AC, Ronnblom L, Barnes BJ. Genetic variants and disease-associated factors contribute to enhanced interferon regulatory factor 5 expression in blood cells of patients with systemic lupus erythematosus. Arthritis Rheum. 2010;62:562–573. doi: 10.1002/art.27223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stone RC, Feng D, Deng J, Singh S, Yang L, Fitzgerald-Bocarsly P, Eloranta ML, Ronnblom L, Barnes BJ. Interferon regulatory factor 5 activation in monocytes of systemic lupus erythematosus patients is triggered by circulating autoantigens independent of type I interferons. Arthritis Rheum. 2012;64:788–798. doi: 10.1002/art.33395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30:455–464. doi: 10.1016/j.it.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Purtha WE, Swiecki M, Colonna M, Diamond MS, Bhattacharya D. Spontaneous mutation of the Dock2 gene in Irf5−/− mice complicates interpretation of type I interferon production and antibody responses. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1118155109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305:1153–1157. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 26.Savitsky DA, Yanai H, Tamura T, Taniguchi T, Honda K. Contribution of IRF5 in B cells to the development of murine SLE-like disease through its transcriptional control of the IgG2a locus. Proc Natl Acad Sci U S A. 2010;107:10154–10159. doi: 10.1073/pnas.1005599107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nacionales DC, Kelly KM, Lee PY, Zhuang H, Li Y, Weinstein JS, Sobel E, Kuroda Y, Akaogi J, Satoh M, Reeves WH. Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (pristane) Am J Pathol. 2006;168:1227–1240. doi: 10.2353/ajpath.2006.050125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, von Andrian UH. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med. 2001;194:1361–1373. doi: 10.1084/jem.194.9.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 30.Cancro M, Potter M. The requirement of an adherent cell substratum for the growth of developing plasmacytoma cells in vivo. J Exp Med. 1976;144:1554–1567. doi: 10.1084/jem.144.6.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF. Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood. 1998;91:2005–2009. [PubMed] [Google Scholar]
- 32.Nikolic T, de Bruijn MF, Lutz MB, Leenen PJ. Developmental stages of myeloid dendritic cells in mouse bone marrow. Int Immunol. 2003;15:515–524. doi: 10.1093/intimm/dxg050. [DOI] [PubMed] [Google Scholar]
- 33.de Bruijn MF, Slieker WA, van der Loo JC, Voerman JS, van Ewijk W, Leenen PJ. Distinct mouse bone marrow macrophage precursors identified by differential expression of ER-MP12 and ER-MP20 antigens. Eur J Immunol. 1994;24:2279–2284. doi: 10.1002/eji.1830241003. [DOI] [PubMed] [Google Scholar]
- 34.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auffray C, Fogg DK, Narni-Mancinelli E, Senechal B, Trouillet C, Saederup N, Leemput J, Bigot K, Campisi L, Abitbol M, Molina T, Charo I, Hume DA, Cumano A, Lauvau G, Geissmann F. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J Exp Med. 2009;206:595–606. doi: 10.1084/jem.20081385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Y, Lee PY, Li Y, Liu C, Zhuang H, Han S, Nacionales DC, Weinstein J, Mathews CE, Moldawer LL, Li SW, Satoh M, Yang LJ, Reeves WH. Pleiotropic IFN-Dependent and -Independent Effects of IRF5 on the Pathogenesis of Experimental Lupus. J Immunol. 2012 doi: 10.4049/jimmunol.1103113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabry A, Sheashaa H, El-Husseini A, Mahmoud K, Eldahshan KF, George SK, Abdel-Khalek E, El-Shafey EM, Abo-Zenah H. Proinflammatory cytokines (TNF-alpha and IL-6) in Egyptian patients with SLE: its correlation with disease activity. Cytokine. 2006;35:148–153. doi: 10.1016/j.cyto.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 38.Waszczykowska E, Robak E, Wozniacka A, Narbutt J, Torzecka JD, Sysa-Jedrzejowska A. Estimation of SLE activity based on the serum level of chosen cytokines and superoxide radical generation. Mediators Inflamm. 1999;8:93–100. doi: 10.1080/09629359990586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tokano Y, Morimoto S, Kaneko H, Amano H, Nozawa K, Takasaki Y, Hashimoto H. Levels of IL-12 in the sera of patients with systemic lupus erythematosus (SLE)--relation to Th1- and Th2-derived cytokines. Clin Exp Immunol. 1999;116:169–173. doi: 10.1046/j.1365-2249.1999.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Lee PY, Reeves WH. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Arch Immunol Ther Exp (Warsz) 2010;58:355–364. doi: 10.1007/s00005-010-0093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–1250. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 42.Ren Y, Tang J, Mok MY, Chan AW, Wu A, Lau CS. Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum. 2003;48:2888–2897. doi: 10.1002/art.11237. [DOI] [PubMed] [Google Scholar]
- 43.Moore JM, Rajan TV. Pristane retards clearance of particulate materials from the peritoneal cavity of laboratory mice. J Immunol Methods. 1994;173:273–278. doi: 10.1016/0022-1759(94)90306-9. [DOI] [PubMed] [Google Scholar]
- 44.Calvani N, Caricchio R, Tucci M, Sobel ES, Silvestris F, Tartaglia P, Richards HB. Induction of apoptosis by the hydrocarbon oil pristane: implications for pristane-induced lupus. J Immunol. 2005;175:4777–4782. doi: 10.4049/jimmunol.175.7.4777. [DOI] [PubMed] [Google Scholar]
- 45.Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, Kellner ES, Nacionales D, Barker T, Kelly-Scumpia K, van Rooijen N, Kumar H, Kawai T, Satoh M, Akira S, Reeves WH. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205:2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gotoh K, Tanaka Y, Nishikimi A, Nakamura R, Yamada H, Maeda N, Ishikawa T, Hoshino K, Uruno T, Cao Q, Higashi S, Kawaguchi Y, Enjoji M, Takayanagi R, Kaisho T, Yoshikai Y, Fukui Y. Selective control of type I IFN induction by the Rac activator DOCK2 during TLR-mediated plasmacytoid dendritic cell activation. J Exp Med. 2010;207:721–730. doi: 10.1084/jem.20091776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, Inayoshi A, Noda M, Oike M, Shirai T, Sasazuki T. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature. 2001;412:826–831. doi: 10.1038/35090591. [DOI] [PubMed] [Google Scholar]
- 48.Ippagunta SK, Malireddi RK, Shaw PJ, Neale GA, Walle LV, Green DR, Fukui Y, Lamkanfi M, Kanneganti TD. The inflammasome adaptor ASC regulates the function of adaptive immune cells by controlling Dock2-mediated Rac activation and actin polymerization. Nat Immunol. 2011;12:1010–1016. doi: 10.1038/ni.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi C, Velazquez P, Hohl TM, Leiner I, Dustin ML, Pamer EG. Monocyte trafficking to hepatic sites of bacterial infection is chemokine independent and directed by focal intercellular adhesion molecule-1 expression. J Immunol. 2010;184:6266–6274. doi: 10.4049/jimmunol.0904160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, Pamer EG. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol. 2008;180:6846–6853. doi: 10.4049/jimmunol.180.10.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. The Journal of clinical investigation. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang A, Fairhurst AM, Tus K, Subramanian S, Liu Y, Lin F, Igarashi P, Zhou XJ, Batteux F, Wong D, Wakeland EK, Mohan C. CXCR4/CXCL12 hyperexpression plays a pivotal role in the pathogenesis of lupus. J Immunol. 2009;182:4448–4458. doi: 10.4049/jimmunol.0801920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Cui L, Gonsiorek W, Min SH, Anilkumar G, Rosenblum S, Kozlowski J, Lundell D, Fine JS, Grant EP. CCR2 and CXCR4 regulate peripheral blood monocyte pharmacodynamics and link to efficacy in experimental autoimmune encephalomyelitis. J Inflamm (Lond) 2009;6:32. doi: 10.1186/1476-9255-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.