

Abstract
Huntington's disease (HD) is caused by a mutation in the huntingtin (htt) gene encoding an expansion of glutamine repeats at the N terminus of the Htt protein. Proteolysis of Htt has been identified as a critical pathological event in HD models. In particular, it has been postulated that proteolysis of Htt at the putative caspase-6 cleavage site (at amino acid Asp-586) plays a critical role in disease progression and pathogenesis. However, whether caspase-6 is indeed the essential enzyme that cleaves Htt at this site in vivo has not been determined. To evaluate, we crossed the BACHD mouse model with a caspase-6 knock-out mouse (Casp6−/−). Western blot and immunocytochemistry confirmed the lack of caspase-6 protein in Casp6−/− mice, regardless of HD genotype. We predicted the Casp6−/− mouse would have reduced levels of caspase-6 Htt fragments and increased levels of full-length Htt protein. In contrast, we found a significant reduction of full-length mutant Htt (mHtt) and fragments in the striatum of BACHD Casp6−/− mice. Importantly, we detected the presence of Htt fragments consistent with cleavage at amino acid Asp-586 of Htt in the BACHD Casp6−/− mouse, indicating that caspase-6 activity cannot fully account for the generation of the Htt 586 fragment in vivo. Our data are not consistent with the hypothesis that caspase-6 activity is critical in generating a potentially toxic 586 aa Htt fragment in vivo. However, our studies do suggest a role for caspase-6 activity in clearance pathways for mHtt protein.
Introduction
Huntington's disease (HD) is a dominantly inherited neurodegenerative disorder characterized by progressive decline of cognitive and motor function due to loss of striatal and cortical neurons. HD is caused by a mutation in the huntingtin (Htt) gene encoding a glutamine repeat (CAG expansion) at the N terminus of Htt (Huntington's Disease Collaborative Research Group, 1993). A neuropathological hallmark of HD is the accumulation of N-terminal Htt fragments, suggesting that Htt proteolysis may be a critical event in pathogenesis (Goldberg et al., 1996; Davies et al., 1997; DiFiglia et al., 1997; Martindale et al., 1998; Gutekunst et al., 1999; Gafni and Ellerby, 2002; Gafni et al., 2004).
Htt is cleaved by multiple proteases, including aspartyl proteases, calpains, γ-secretase, and caspases (Goldberg et al., 1996; Wellington et al., 1998; Gafni and Ellerby, 2002; Lunkes et al., 2002; Gafni et al., 2004; Kegel et al., 2010). Indeed Htt was the first neuronal protein identified as a caspase substrate (Goldberg et al., 1996), and studies have defined the Htt cleavage sites for caspase-3 at amino acids Asp-513 and Asp-552, for caspase-2 at amino acid Asp-552, and for caspase-6 at the IVLD amino acid Asp-586 site (Wellington et al., 1998, 2000; Hermel et al., 2004). Caspase-6 binds to Htt, and dominant-negative caspase-6 blocks HD striatal neuronal degeneration (Hermel et al., 2004). Furthermore, the processed form of caspase-6 can be found activated in mouse and human HD striatum (Hermel et al., 2004; Graham et al., 2010). Proteolytic cleavage of Htt at the caspase-6 site at amino acid Asp-586 may be an important event in the pathogenesis of HD (Graham et al., 2006). Interestingly, in a YAC128 mouse model of HD, mutation of the potential caspase-6 cleavage site at Asp-586 (YAC128 C6R; note also mutated Asp-589 site) in mHtt did not recapitulate behavioral phenotypes or a number of key pathological changes apparent in YAC128 mice that do not carry that mutation (Graham et al., 2006). However, it is not clear whether these differences in YAC128 C6R transgenic mice are due to the ablation of the caspase-6 cleavage site, altered expression levels, or to structural changes within Htt caused by the mutations at Asp-586 and Asp-589. Furthermore, it remains to be tested whether caspase-6 is the protease responsible for cleavage of Htt at Asp-586 and whether it is required for disease progression in vivo.
Caspase-6 activation has been implicated in other neurological conditions and diseases; it is activated early in sporadic and familial forms of Alzheimer's disease (AD) with active caspase-6 localized to neuropil threads, neuritic plaques, and neurofibrillary tangles (Guo et al., 2004; Albrecht et al., 2009). Caspase-6 activation in AD models leads to neuritic beading, axonal degeneration, and eventually cell death, which can be prevented by incubation with caspase-6 inhibitors or dominant-negative caspase-6 (Nikolaev et al., 2009; Sivananthan et al., 2010). Active caspase-6 and caspase-6-cleaved tau have also been observed in noncognitively impaired aged individuals that have the lowest cognitive scores, suggesting that caspase-6 activation may be an early step in cognitive decline (Albrecht et al., 2007).
Since caspase-6 activation has been identified in a number of neurological conditions and may play an important role in HD pathogenesis, we crossed the well characterized BACHD mouse model (Gray et al., 2008; Menalled et al., 2009) with a caspase-6 knock-out mouse (Casp6−/−) to test whether caspase-6 deficiency would prohibit production of the putative caspase-6-derived amino acid 586 Htt fragment and lead to altered levels of full-length mutant or wild-type Htt. In addition, limited measures were followed to track the effects of caspase-6 knock-out on behavior and histopathology in the BACHD mouse. We found that, counter to expectations, the amino acid 586 Htt fragment remained abundant in the BACHD Casp6−/− mice, and, moreover, the amount of full-length mHtt was markedly decreased. We report preliminary findings that suggest clearance pathways in the BACHD Casp6−/− mouse brain may be invoked to clear full-length mHtt, including activation of other proteases, macroautophagy, a lysosomal quality control pathway, or other ubiquitin-mediated protein degradation pathways.
Materials and Methods
Caspase-6 knock-out mouse
Knock-outs of the mouse caspase-6 gene were made at Taconic/Xenogen in C57BL/6 mice using homologous recombination in mouse embryonic stem cells and subsequent blastocyst injection of the appropriate targeted ES cells to create the gene targeted mice. The RP23-35M15 BAC clone was used to generate the homology arms and the conditional knock-out region for the gene targeting vector, as well as the Southern probes to screen for targeted events. All recombinant plasmids were confirmed by restriction digestion and end-sequencing, and all exons were sequence confirmed. The final vector was obtained by standard molecular cloning. Thirty micrograms of the NotI-linearized final KO vector DNA was electroporated into ∼107 C57BL/6 ES cells. The cells were selected with 200 μg/ml G418 and 192 ES clones were picked for screening. Based on this Southern analysis using the 5′, 3′, and neo probes, four clones were confirmed to be correctly targeted and to have a single neo insertion. Clones were injected to produce 30 male chimeras, which were bred with wild-type females to generate heterozygotes. Initial characterization of the Casp6−/− mice is reported (Uribe et al., 2012).
Transgenic mouse model expressing Htt145Q(1–586)
The RMCE ES cell line [derived from mouse strain C57BL/6-Gt(ROSA)26Sor tm596Arte] was grown on a mitotically inactivated feeder layer comprised of mouse embryonic fibroblasts in DMEM high-glucose medium containing 20% FBS (PAN) and 1200 U/ml leukemia inhibitory factor (Millipore; ESG 1107). For manipulation, 2 × 105 ES cells were plated on 3.5 cm dishes in 2 ml of medium. For transfection, 3 μl of Fugene6 Reagent (Roche; catalog #1 814 443) was mixed with 100 μl of serum-free medium (OptiMEM I with Glutamax I; Invitrogen; catalog #51985-035) and incubated for 5 min at room temperature (RT). One hundred microliters of the Fugene/OptiMEM solution was added to the DNA mixture containing 2 μg of circular vector and 2 μg of CAGGS-Flp plasmid. This transfection complex was incubated for 20 min at RT and then added dropwise to the cells. From day 2 onward, the medium was replaced daily with medium containing 250 μg/ml G418 (Geneticin; Invitrogen; catalog #10131-019). Seven days later, single clones were isolated, expanded, and analyzed by Southern blotting according to standard procedures. After administration of hormones, superovulated BALB/c females were mated with BALB/c males. Blastocysts were isolated from the uterus at day post-coitum 3.5. For microinjection, blastocysts were placed in a drop of DMEM with 15% FCS under mineral oil. A flat tip, piezo actuated microinjection pipette with an internal diameter of 12–15 μm was used to inject 10–15 targeted C57BL/6 N.tac ES cells into each blastocyst. After recovery, eight injected blastocysts were transferred to each uterine horn of 2.5 d post-coitum, pseudopregnant NMRI females. Chimerism was measured in chimeras (G0) by coat color contribution of ES cells to the BALB/c host (black/white). Highly chimeric mice were bred to strain C57BL/6 females. The C57BL/6 mating partners were nonmutant or mutant for the presence of a recombinase gene. Germline transmission was identified by the presence of black, strain C57BL/6, offspring (G1).
Hdh knockdown mice
Three-month-old Hdh KD mice (inducible knockdown of mouse Htt with shRNA) were fed 3000 ppm doxycycline pellets for 4 weeks and then killed. Samples were analyzed for Western analysis as described below.
Mouse development and behavioral evaluation
Subjects.
Experimental female mice used in this study were bred at Taconic by crossing BACHD mutant female mice (FVB/N) (Gray et al., 2008) with male caspase-6 knock-out mice (C57BL/6) generated by Taconic/Xenogen. Mice arrived at PsychoGenics around 11–14 weeks of age. Mice were handled on 2 consecutive days (1 min each day), and then tail tattooed and chipped (Datamars) for identification purposes. Mice were housed in Optimice cages containing wood shavings, play tunnels, shredded paper, and plastic bones. Mice had ad libitum access to water and unaltered Purina 5001 food diet pellets. On the day of the experiment, mice were allowed to acclimate to the testing room for at least 1 h before the beginning of the experiment. Mice were transported from the colony room to the experimental room in their home cages.
Body weight and general health.
Mice were weighed weekly throughout the course of the experiment, and general health was monitored on a daily basis. At PsychoGenics, all procedures were approved by the Institutional Animal and Use Committee of PsychoGenics (PHS OLAW Animal Welfare Assurance number A4471-01), an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International Accredited Institution (unit number 001213). At the Buck Institute for Research on Aging, an AAALAC International Accredited Institution (unit number 001070), all procedures were approved by the Institutional Animal and Use Committee (A4213-01).
Rotarod.
Mice were tested over 3 consecutive days. Each daily session included a training trial of 5 min at 4 rpm on the rotarod apparatus. One hour later, the animals were tested for three consecutive accelerating trials of 5 min with the speed changing from 0 to 40 rpm over 300 s and an intertrial interval of at least 30 min. The latency to fall from the rod on each of the three testing trials was recorded. Mice remaining on the rod for >300 s were removed, and their time was scored as 300 s. Animals were evaluated at 16, 20, 24, 37, and 51 weeks of age.
Open field.
Activity chambers (MED Associates; 27 × 27 × 20.3 cm) were equipped with infrared beams. Mice were placed in the center of the chamber, and their behavior was recorded for 30 min. Quantitative analysis was performed on the following five dependent measures: total locomotion, locomotion in the center of the open field, rearing rate in the center, total rearing frequency, and velocity. Animals were evaluated at 16, 20, 24, 37, and 51 weeks of age.
Data analysis of weight and behavior.
An α level of 0.05 was selected for all inferential statistics. The repeated-measures analysis (age as dependent factor) was performed with SAS (SAS Institute, Inc.) using mixed-effect models. This approach is based on likelihood estimation, which is more robust to missing values than moment estimation. The models were fitted using the procedure PROC MIXED (Singer, 1998). The BACHD, BACHD Casp6+/−, and BACHD Casp6−/− groups were compared to evaluate the effect of the caspase-6 deficiency on the BACHD mice. Factors for analyses included caspase-6 genotype and age.
MRI analysis of BACHD and BACHD Casp6−/− mice.
Mice were perfused transcardially with heparinized 0.9% saline followed by 4% PFA. Brains were removed from the skull and left in 4% PFA until measured by MRI. Before imaging, the brains were rinsed with saline and embedded in perfluoropolyether (Fomblin). T2-weighted MRI was performed using a horizontal 7 T magnet with inner bore diameter of 160 mm (Magnex Scientific Ltd.) equipped with actively shielded Magnex gradient set (maximum gradient strength, 400 mT/m) interfaced to a Varian DirectDrive console (Varian). Linear RF volume-coil was used for transmission and surface phased array coil for receiving (Rapid Biomedical). For determination of ex vivo total brain, striatal, and cortical volumes, T2-weighted continuous multislice images covering the whole brain were acquired using fast spin-echo sequence with the following: TR, 4500 ms; echo train length, 4; effective TE, 37.7 ms; matrix size, 512 × 256 (zeropadded to 512 × 512); FOV, 30*30 mm2; slice thickness, 0.6 mm; number of slices, 21 and four averages. The acquired coronal images were analyzed for total brain, striatal, and cortical volumes using in-house written analysis program run under MATLAB (Mathworks) environment. All values are presented as mean ± SEM, and differences are considered to be statistically significant at the p < 0.05 level. Statistical analysis was performed using StatsDirect statistical software (StatsDirect). Values of p were not corrected for multiple comparison.
Biochemical analysis
Huntingtin neoepitope antibody production.
Antibodies specific for the C-terminal ends of Htt caspase cleavage product ending at amino acid 586 was prepared using the immunizing peptide KLH-CPSDSSEIVLD. Peptide sequences were injected into rabbits, antibody was purified to the injected peptide, and a bridging peptide was used to remove antibodies reacting to full-length Htt (Open Biosystems). Antibodies were affinity purified as previously described with minor modifications (Gervais et al., 1999; Wellington et al., 2002; Leyva et al., 2010).
Western blot analysis.
One-half of the cortex and striatum from 13- and 15-month-old animals were lysed in Tissue Protein Extraction Reagent (TPER) (Thermo Fisher Scientific) with protease inhibitors (Complete Mini; Roche Applied Science) and 1 μm epoxymycin at 10 ml/g. In some cases, phosphatase inhibitors (Phosphatase Inhibitor Cocktail Set II; Calbiochem) and HDAC inhibitors (50 μm TSA, 30 μm sodium butyrate, 30 mm nicotinamide) were also included. Tissue was homogenized, sonicated, and spun at 14,000 × g for 20 min. Supernatant and pellets were kept for analysis. Protein concentration was determined using a BCA assay (Pierce), and 40–60 μg of protein was run on a polyacrylamide gel and transferred to a 0.45 μm nitrocellulose (Whatman) or 0.2 μm polyvinylidene difluoride (Pall Life Sciences) membrane. Polyclonal primary antibodies used were as follows: cleaved caspase-6 9761 (1:75; Cell Signaling), procaspase-6 9762 (1:125; Cell Signaling), procaspase-3 9662 (1:200; Cell Signaling), cleaved caspase-3 9661S (1:100; Cell Signaling), cleaved caspase-7 9491S (1:100; Cell Signaling), procaspase-9 9504 (1:100; Cell Signaling), cleaved caspase-9 9509 (1:100; Cell Signaling), cleaved lamin A/C 2031s (1:50; Cell Signaling), neoHtt586 90000188 (1:50; CHDI), LC3 P.M.036 (1:1000; MBL), LC3 AP1802a (1:100; Abgent), ubiquitin UG9510 (1:250; Enzo), and β-actin 4967 (1:1000; Cell Signaling). Monoclonal antibodies used were as follows: Htt 2166 (1:500; Millipore), Htt 5490 (1:250; Millipore), Htt 1574 (1:1000; Millipore), caspase-2 3507 (1:100; Millipore), procaspase-7 56063 (1:200; Santa Cruz Biotechnology), p62 610833 (1:1000; BD Biosciences Transduction Laboratories), GAPDH 10R-G109A (1:1000; Fitzgerald), and fodrin 1622 (1:500; Millipore). Immunoblots were developed with a peroxidase-conjugated secondary antibody and enhanced chemiluminescence. For positive controls, lysates were treated with caspase-6 (400 U; Enzo) for 2 h at 37°C. It should be noted that, while both LC3 antibodies were made against the LC3B isoform, there is no evidence they are specific for this LC3 subtype (Zois et al., 2011). Densitometry was performed using ImageQuant TL, version 2005, software. Statistics was performed using GraphPad Prism, version 4.0c. For ANOVA, Tukey's multiple-comparison post hoc test was used.
Formic acid pellet extraction.
Pellets from lysing protocol were incubated with 1 ml of 70% formic acid for 1 h with shaking and then spun at 14,000 × g for 20 min. A volume of 750 μl of supernatant was transferred to a new tube and formic acid was evaporated in a vacuum dryer. Remaining solid was resuspended in 50 μl of resuspension buffer [1× NuPAGE LDS buffer (Invitrogen), 2% SDS, 5 μm DTT], boiled for 10 min, sonicated for 5 min, and reboiled for 10 min. A volume of 20 μl was spun down at 16,100 × g for 30 min, and 15 μl was loaded on a polyacrylamide gel. Proteins were transferred to 0.45 μm nitrocellulose membrane (Whatman), and blots were probed as specified above (see Western blot analysis).
Immunohistochemistry.
Formalin-fixed female and male WT, BACHD, and BACHD Casp6−/− mice were paraffin-embedded, sectioned, and deparaffinized with xylene. For cleaved caspase-6, neoHtt586, and ubiquitin immunohistochemistry, antigen retrieval was performed by microwaving sections in 10 mm citrate buffer, pH 6.0, for 2 min at maximum power and then 20% power for an additional 5 min (cleaved caspase-6, neoHtt586) or 5 min at 40% power (ubiquitin). Sections were incubated overnight at 4°C with polyclonal cleaved caspase-6 antibody 9761 (1:100; Cell Signaling) and polyclonal ubiquitin antibody 1690 (1:1000; Millipore) and with polyclonal neoHtt586 antibody (2.5 μg/ml; CHDI) for 3 h at 37°C. Biotinylated anti-rabbit IgG secondary antibody (6 μg/ml; Vector) was incubated 1 h at RT followed by signal amplification with avidin/biotin (Vectastain Elite ABC Reagent; Vector) and diaminobenzedine (DAB Peroxidase Substrate Kit; Vector) visualization. Mayer's hematoxylin (American MasterTech) was used as a counterstain.
For LC3 immunohistochemistry, a variation of the procedure used by Osmond et al. (2006) was performed. Briefly, following deparaffinization and rehydration, samples were formic acid treated twice for 10 min each time and then incubated in 1% sodium borohydride for 20 min. Tissue was permeabilized with 0.4% Triton X-100 and then polyclonal LC3 (PM036 antibody; 1:10,000; MBL) was applied overnight at 4°C. Biotinylated anti-rabbit IgG (6 μg/ml; Vector) was incubated for 1.5 h at RT followed by avidin–biotin (1:400; Vectastain Elite ABC Kit; Vector) application for 1 h at RT. Amplification of signal was performed by incubating in biotinylated tyramine for 10 min followed by repeating avidin–biotin incubation. Sections were developed with diaminobenzedine and counterstained with hematoxylin as described above.
For ubiquitin/Htt double immunofluorescence, antigen retrieval was performed in 10 mm citrate buffer by microwaving for 5 min at 40% power. Chicken anti-mouse IgG (2 μg/ml; Aves Labs) was added to block buffer, and slides were incubated with polyclonal ubiquitin 1690 (1:100; Millipore) and monoclonal Htt 2166 (1:200; Millipore) overnight at 4°C. For p62/acetyl-Htt-Lys-444 double immunofluorescence, antigen retrieval was performed in 10 mm citrate buffer by microwaving for 10 min at 40% power and then sequentially incubated with polyclonal p62 (1:100; Progen) and polyclonal acetyl-Htt-Lys-444 (1:50; Open Biosystems) for 3 h at RT. After primary antibody incubation, secondary antibodies Alexa Fluor 488 or 555 goat anti-rabbit IgG, Alexa Fluor 555 goat anti-guinea pig IgG, and/or Alexa Fluor 488 goat anti-mouse IgG (1:500; Invitrogen) were incubated for 1 h at RT followed by coverslipping with Prolong Gold containing DAPI (Invitrogen). The custom Htt acetylation antibody (acetyl-Htt-Lys-444; Open Biosystems) was produced by injecting synthesized peptide corresponding to Htt acetylated at lysine 444 into rabbits. At day 58, bleeds were collected and serum was separated from plasma and tested for signal by Western blot analysis. Those with positive results were purified by affinity binding and stored for use (Cong et al., 2011).
For polyclonal aggregate antibody S830 single labeling (7.0 μg/ml; gift from A. Osmond, University of Tennessee Graduate School of Medicine Research Center, Knoxville, TN, and G. Bates, King's College London, UK), antigen retrieval was performed by microwaving four times for 5 min each time at 40% power in 10 mm citrate buffer and then treating with Proteinase K (20 μg/ml; Eastman Kodak Scientific) for 10 min at 37°C. Duplicate photos were taken from the same area of cortex and striatum at identical magnification and exposure settings. Aggregates and DAPI-stained nuclei were quantified using Imaris X64 7.3.0 software. Aggregate number was normalized to the number of DAPI-stained nuclei in the field.
Primary cultures and immunocytochemistry.
E17–E18 embryos were obtained from timed pregnant C57BL/6 and Casp6−/− mice. Briefly, striatal tissue was dissociated with scalpels, trypsinized for 5–8 min (0.25% Trypsin/EDTA; Cellgro), and triturated with a pipette 15–20 times. After centrifugation and rinsing with fresh 20% FBS in DMEM (Cellgro), cells were resuspended in Neurobasal complete media [Neurobasal A media, 1 mm Glutamax, 2% B27, and 1% penicillin/streptomycin (Invitrogen)] and filtered through a 70 μm nylon strainer. Cells were plated on eight-chamber, poly-d-lysine-coated slides (BD Biosciences) at 400,000 cells/chamber in Neurobasal complete media supplemented with 1% FBS. After 72 h, cells were fixed with 4% paraformaldehyde in PBS for 20 min and permeabilized in 0.1–0.5% Triton in TBS for 15 min RT. Cells were incubated in polyclonal cleaved caspase-6 9761 (1:50; Cell Signaling) and monoclonal MAP2 M4403 (1:500; Sigma-Aldrich) overnight at 4°C. Fluorescent secondary antibodies Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 555 goat anti-mouse IgG (1:500; Invitrogen) were incubated for 1 h at RT and slides were coverslipped with Prolong Gold containing DAPI (Invitrogen).
Quantitative RT-PCR for Htt and caspase-6 cDNAs.
Total RNA from 12-month-old C57BL/6 and Casp6−/− striatum was extracted using the miRNeasy Mini Kit (QIAGEN) and homogenized by triturating with a 27.5 gauge needle. cDNAs were made from 1.0 μg of RNA samples using Message Sensor RT kit (Ambion), and then 75 ng of each cDNA was preamplified for 14 cycles using the Applied Biosystems TaqMan PreAmp master mix and protocol to generate PCR templates. Forward and reverse primers for the three genes of interest (Htt, caspase-6, and β-actin) were designed using the Roche Universal Probe Library Assay Design Center and ordered from Operon and Sigma-Aldrich. For Htt, nucleotides 4944–5004, 9301–9367, and 4822–4890 were amplified. For caspase-6, nucleotides 33–133, 150–262, and 264–373 were amplified, and results were averaged. The real-time quantitative PCR was performed with SYBR Green (Applied Biosystems) on a Light Cycler 480 (Roche) according to the manufacturer's instructions.
Results
Caspase-6 is activated in BACHD mouse brains
Caspase-6 is activated during aging and in postmortem HD and AD brain (Hermel et al., 2004; Graham et al., 2006; Albrecht et al., 2007). Furthermore, characterization of the YAC128 mouse model of HD shows an age-dependent increase in caspase-6 activation (Graham et al., 2010). To confirm that caspase-6 is activated in BACHD mice, sections of WT and BACHD were immunostained with an antibody that detects active mouse caspase-6 at 9 months of age (n = 3). As predicted, WT mice at this age had detectable active caspase-6 in the striatal medium-spiny and cortical neurons by 9 months (Fig. 1A). The staining was dendritic and nuclear in localization. Consistent with previous work (Graham et al., 2010), the BACHD mouse displayed an increase in activation of caspase-6 when compared with WT mice both in the striatum and cortex (Fig. 1A). This was also confirmed by Western blot analysis of these mice at 9 months of age with a 2.5-fold increase in active caspase-6 in the striatum (Fig. 1B).
Figure 1.

Cleaved caspase-6 is increased in BACHD mice. A, Immunohistochemistry of cleaved caspase-6 in 9-month-old WT and BACHD striatum and cortex. Tissue is counterstained with hematoxylin. B, Western blot of cleaved caspase-6 levels in 9-month-old WT and BACHD striatum (left panel). Densitometry of cleaved caspase-6 normalized to GAPDH (right panel; n = 3; t test, ***p < 0.001). Error bars indicate SD.
Caspase-6 knock-out abolishes the expression of caspase-6
To evaluate the role of caspase-6 in HD, we generated caspase-6 knock-out mice using homologous recombination at Taconic/Xenogen. A BAC clone was used for generating the homology arm and targeting vector. The targeting vector (Fig. 2A) was electroporated into C57BL/6 ES cells, and Southern analysis identified several clones used for microinjection to generate caspase-6 knock-out mice. The Casp6−/− mice were viable, bred well, and had no obvious phenotype. To determine whether caspase-6 levels were eliminated in the caspase-6 knock-out mice, we performed Western blot analysis (Fig. 2B) and evaluated active caspase-6 immunoreactivity in primary striatal neurons derived from WT and Casp6−/− mice (Fig. 2C). Western blotting and immunocytochemistry demonstrated the elimination of procaspase-6 and active caspase-6, respectively.
Figure 2.
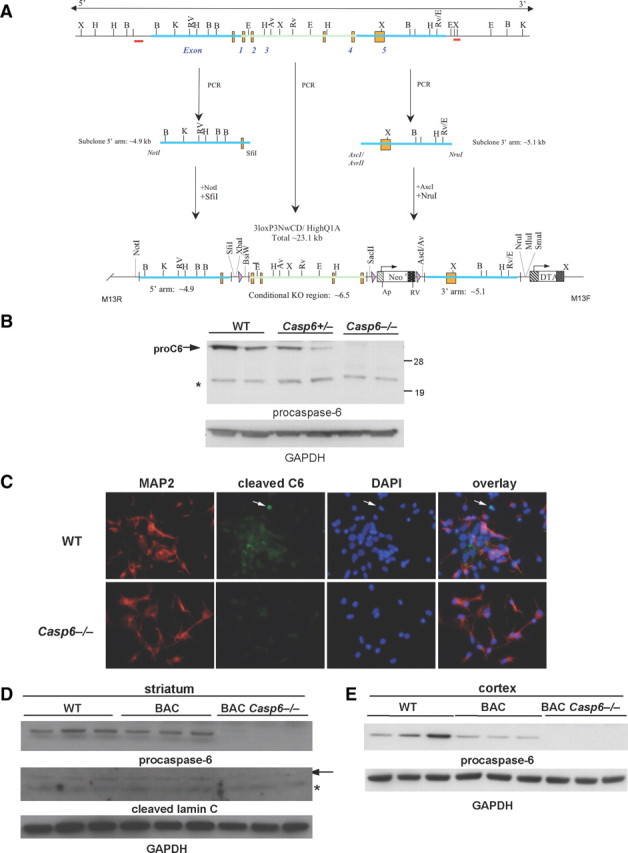
Knocking out caspase-6 eliminates caspase-6 expression. A, Vector scheme for caspase-6 knock-out mouse. B, Western blot of procaspase-6 protein in 15-month-old WT, Casp6+/−, and −/− cortex. The band to the right of the asterisk is nonspecific. C, Immunocytochemistry of cleaved caspase-6 in WT and Casp6−/− striatal neuronal cultures. D, Western blot of procaspase-6 and cleaved lamin C protein production in 13-month-old WT, BACHD, and BACHD Casp6−/− striatal lysates. The arrow points to cleaved lamin C, and the asterisk indicates nonspecific band. E, Western blot of procaspase-6 levels in 13-month-old WT, BACHD, and BACHD Casp6−/− cortical lysates.
Having established that the Casp6−/− mice lacked the targeted protein, we crossed BACHD with Casp6−/− mice. Western blot analysis of WT (littermate controls), BACHD, and BACHD Casp6−/− at 13 months of age demonstrates the elimination of the inactive p32 proform of caspase-6 in both the striatum and cortex of BACHD Casp6−/− mice (Fig. 2D,E). Furthermore, we observed a decrease in the proform of caspase-6 when comparing WT with BACHD mice in both the striatum and cortex (Fig. 2D,E) consistent with the activation of this enzyme in HD mouse models. As expected, cleavage of the caspase-6 substrate, lamin C, is eliminated in the BACHD Casp6−/− mice, suggesting a functional knock-out of caspase-6 (Fig. 2D).
BACHD caspase-6 knock-out has reduced Htt levels in the striatum
Given that Htt is predicted to be a substrate of caspase-6, we examined whether the proteolysis of Htt was altered in the absence of caspase-6 both in the striatum (Fig. 3) and cortex (data not shown) of WT, Casp6−/−, BACHD, and BACHD Casp6−/−. We predicted that blocking caspase-6 cleavage would reduce the level of fragmentation of Htt and preserve full-length Htt protein. As shown in Figure 3A, we found the levels of full-length Htt reduced in the BACHD Casp6−/− striatum when compared with BACHD mice. Furthermore, there was a twofold decrease in the level of poly(Q) expanded Htt fragments in the BACHD Casp6−/− when compared with BACHD mice (Fig. 3B) as detected by the 1C2 antibody. The change in levels of full-length Htt can be largely attributed to a decrease in the level of poly(Q)-expanded Htt as shown in Figure 3C. Indeed, the levels of Htt with 97 glutamine repeats was reduced fourfold in the BACHD Casp6−/− when compared with BACHD mice (Fig. 3C). The wild-type levels of full-length Htt appear reduced as well, but the change in expression between BACHD and BACHD Casp6−/− did not reach statistical significance (Fig. 3C). When expanded full-length Htt levels were quantified relative to wild type, we observed a twofold decrease in the mutant/normal Htt ratio in BACHD Casp6−/− relative to BACHD mice (data not shown) (n = 3; t test, **p < 0.01). We also evaluated the proteolysis of Htt in the cortex and found similar but less robust changes in the level of expression of Htt in BACHD Casp6−/− when compared with BACHD mice (data not shown). In the cortex, we also observed a significant 1.5-fold decrease in expanded full-length Htt levels relative to wild-type levels in the BACHD Casp6−/− (data not shown) (n = 3; t = test, *p < 0.05). A relatively trivial explanation for the changes in protein levels could be that the knock-out of caspase-6 altered the levels of htt mRNA. However, as shown in Figure 3D, the levels of mRNA for htt were not altered in the Casp6−/− mice when compared with WT. As a control, the levels of mRNA for caspase-6 were measured and as expected are absent in the Casp6−/− mice (Fig. 3D).
Figure 3.

Full-length Htt and Htt fragments are reduced in BACHD Casp6−/− striatal lysates. A, Western blot of striatal lysates from 13-month-old animals probed with Htt2166 antibody, an antibody that detects both wild-type and expanded Htt. B, Western blot of 13-month-old striatal lysates probed with an antibody that preferentially detects expanded Htt (1C2) (left panel). Densitometry of Htt fragments between 98 and 188 kDa in left panel (left of line) normalized to GAPDH (right panel; n = 3; ANOVA, ***p < 0.001). C, Further separation of full-length wild-type and expanded striatal Htt visualized with Htt2166 antibody using Western blot analysis (left panel). Densitometry of full-length expanded Htt relative to wild-type Htt from left panel (right panel; n = 3; WT, ANOVA, *p < 0.05; expanded, t test, **p < 0.01). D, Quantitative RT-PCR of Htt and caspase-6 in 12-month-old WT and Casp6−/− striatum. The numbers on the x-axis indicate region of nucleotide amplification. Error bars indicate SD.
Huntingtin aggregates are reduced in BACHD caspase-6 knock-out mice
Htt aggregates are associated with disease pathology in both human HD patients and HD animal models (Lee and Kim, 2006; Hoffner et al., 2007). While some studies identify Htt aggregates as toxic, others suggest that they are protective in nature (Hodgson et al., 1999; Sánchez et al., 2003; Zhang et al., 2008; Richards et al., 2011). We were interested in determining levels of Htt aggregates in BACHD Casp6−/− mice, relative to BACHD mice, not only because of their relationship to disease progression, but also to determine whether the decrease in soluble full-length Htt in BACHD Casp6−/− mice is due to relocalization of Htt to inclusion bodies. Thus, we performed immunohistochemistry using a specific antibody that detects aggregated Htt, S830 (Steffan et al., 2000). We found Htt aggregates were dramatically decreased in the striatum and cortex of BACHD Casp6−/− mice relative to BACHD mice, with a fourfold decrease in the striatum and an eightfold decrease in the cortex (Fig. 4A,B). Virtually no staining was observed in WT mice showing the specificity of the antibody (Fig. 4A,B).
Figure 4.

Aggregate formation is reduced in BACHD Casp6−/− striatum and cortex. A, Immunofluorescence of 13-month-old cortex and striatum using the S830 Htt aggregate antibody counterstained with DAPI. B, Quantification of aggregates shows a dramatic decrease in number in BACHD Casp6−/− (n = 3–6; ANOVA, *p < 0.05). Error bars indicate SD.
BACHD caspase-6 knock-out mice do not show a reduction in the proteolysis of Htt at amino acid 586
Next, we evaluated whether caspase-6 is the protease solely responsible for the cleavage of Htt at amino acid 586. We found that there was no change in the cleavage products for both wild-type and mutant Htt in WT, BACHD, and BACHD Casp6−/− cortex and striatum that migrated at a molecular weight similar in size to the amino acid 586 Htt cleavage product (see Fig. 6A–C).
Figure 6.

Fragments similar in size to Htt586 are present in BACHD Casp6−/− cortex and striatum. A, Western blot of cortical lysates from 13-month-old animals with caspase-6-, caspase-3-, and caspase-2-treated lysates probed with Htt2166 antibody. The arrows indicate Htt586 products. B, Western blot of 13-month-old cortical lysates probed with 1C2 antibody. Fodrin shows equal loading. The arrow indicates size of expanded Htt586. C, Western blot of 15-month-old striatal lysates probed with Htt2166 antibody. Fodrin shows equal loading. The arrows indicate size of Htt586 products. D, N-terminal Htt antibody (Htt115–129) detects Htt586 fragments in 13-month-old solubilized striatal pellet Western blot. The arrows indicate size of Htt586 products. E, A neoHtt586 antibody detects Htt586 fragments in 13-month-old solubilized striatal pellets. The arrows indicate size of Htt586 products. F, Immunohistochemistry with a neoHtt586 antibody in 15-month-old cortex and striatum shows an increase in staining in BACHD and a subsequent decrease in staining in BACHD Casp6−/− most likely reflecting lower Htt levels overall.
To detect the amino acid 586 Htt cleavage products, we generated a series of neoepitope rabbit antibodies to the site; the polyclonal serum only detects the amino acid 586 Htt cleavage product and not others (Fig. 5A,B). The purified polyclonal neoHtt antibody appears to be selective to cleaved Htt since in a HD transgenic mouse overexpressing the Htt586 fragment, the immunoreactivity is greatly increased relative to WT, and in an inducible Htt RNAi knockdown model (SFN2009) (Menalled et al., 2009), the signal decreases with the reduction of mouse Htt (Fig. 5B,C). When we evaluated these antibodies using the soluble lysates described in Figure 6A–C, we were not able to detect Htt immunoreactive to neoHtt586 in older animals. However, when we formic acid extracted proteins from the pellets, we were able to detect both wild-type and expanded Htt reactive to the neoHtt586 antibody (Fig. 6D,E), suggesting that the product is insoluble. The cleavage product was not reduced in the BACHD Casp6−/− striatum, suggesting it is possible another protease cleaves Htt at this site. In addition, performance of caspase activity assays on striatal tissue and cultures using the caspase-6 substrate, VEID, showed an increase in VEIDase activity in Casp6−/− lysates, again suggesting another protease may be involved in Htt586 production (data not shown). It should be noted that VEID is also a caspase-3 substrate, which may be a reason for the increase in VEIDase activity observed (McStay et al., 2008).
Figure 5.

NeoHtt586 antibody is specific for the caspase-6 cleaved product of Htt. A, Lysates from 293T cells overexpressing full-length Htt 23Q treated with caspase-2, caspase-3, caspase-6, and caspase-7, and Htt23Q(1–513), Htt23Q(1–536), Htt23Q(1–552), and Htt23Q(1–586) were used to verify neoHtt586 specificity. Western blots were probed with an N-terminal Htt antibody (left panel) and a neoHtt586 antibody (right panel). B, Western blot of cortical lysates from WT mice and mice expressing expanded Htt constructs with 171, 469, 536, 552, and 586 aa probed with noeHtt586 antibody (left panel). NeoHtt586 immunohistochemistry of WT and D586 cortex (right panel). C, Cortical and striatal lysates from Htt knockdown mice with and without doxycyclin treatment analyzed by Western blot. Control cortical lysates were treated with recombinant caspase-6 as a positive control (+C6). Samples were probed with a Htt antibody (left panel) and an antibody that specifically interacts with the caspase-6 produced Htt fragment (neoHtt586) (right panel). Fodrin was used as a loading control.
To further characterize whether Htt is cleaved at amino acid 586 in vivo, we performed immunohistochemistry of WT, BACHD, and BACHD Casp6−/− in cortex and striatum using neoHtt586. We found increased immunoreactivity of neoHtt586 in the BACHD compared with WT (n = 3; Fig. 6F). We detected decreased immunoreactivity of neoHtt586 in the BACHD Casp6−/− mouse brain, which may be related to the clearance of Htt described above.
Characterization of behavioral phenotype of BACHD caspase-6 knock-out
Behavioral evaluation and general health
Systematic behavioral analysis has previously been used to phenotype numerous mouse models of HD, including the BACHD mice (Menalled et al., 2009). Studies using two different strains of BACHD mice showed impaired rotarod performance, hypoactivity in the open-field test, and an increased body weight compared with WT from an early age (Gray et al., 2008; Menalled et al., 2009). To determine whether knocking out caspase-6 protects against health and behavioral deficits, body weight, brain volume, and motor performance were measured in BACHD and BACHD Casp6−/− animals. BACHD Casp6+/− mice were included to study the effects of gene dosage.
Body weight
To determine whether there were changes in body weight, mice were weighed weekly up to 51 weeks of age. The body weight of the BACHD mice examined in this study was comparable with previous findings (Gray et al., 2008; Menalled et al., 2009). BACHD Casp6−/− weighed significantly less than BACHD mice at 16 weeks and from 19 to 40 and from 42 to 45 weeks of age (Fig. 7A; caspase-6 knock-out genotype by age interaction, F(80,915) = 1.51, p < 0.01; caspase-6 knock-out genotype main effect, F(2,23) = 4.44, p < 0.05; age main effect, F(40,915) = 11.53, p < 0.0001; simple main effects, values of p < 0.05). BACHD Casp6−/− were also found to be significantly smaller than BACHD Casp6+/− at 51 weeks of age (values of p < 0.05), and BACHD Casp6+/− were significantly smaller than BACHD mice from 48 to 51 weeks of age (values of p < 0.05) (Fig. 7A).
Figure 7.
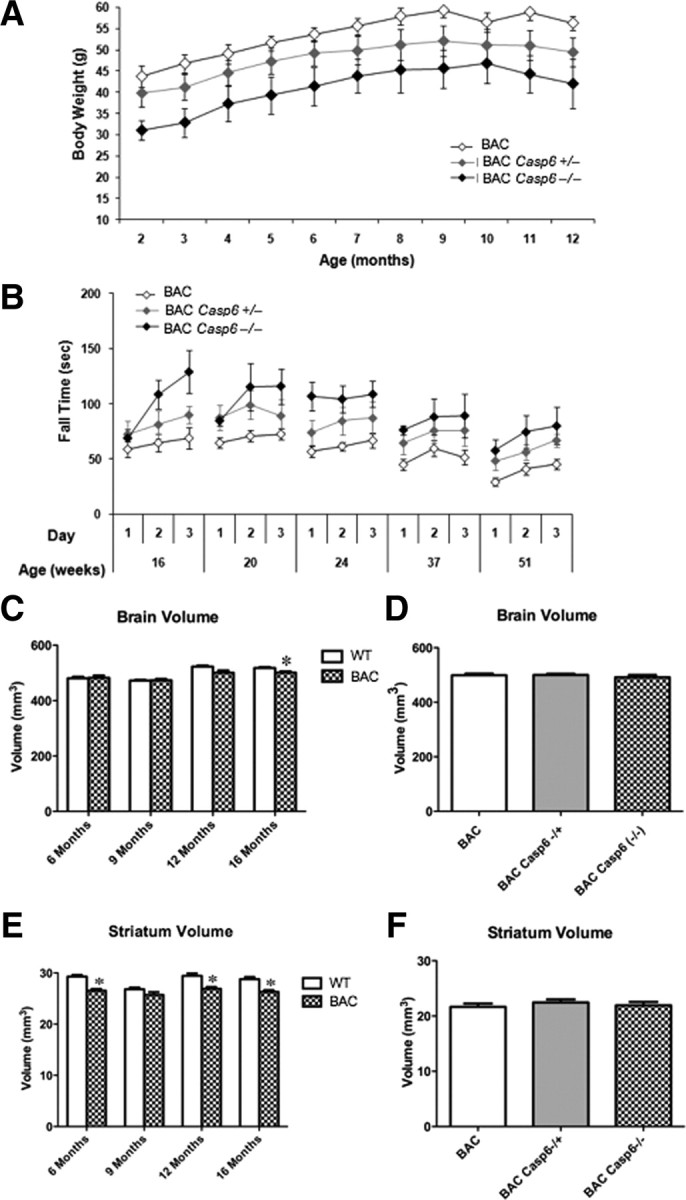
Effect of caspase-6 deletion on development and behavior. A, Knock-out of caspase-6 produced a significant reduction in body weight of BACHD mice when compared with WT animals (values of p < 0.05). B, Deletion of caspase-6 improves rotarod performance in the BACHD mice (values of p < 0.05). For statistical analysis, see Materials and Methods, Data analysis of weight and behavior. C, BACHD mice had significantly smaller whole-brain volume at 16 months of age (t test, *p < 0.05, compared with WT mice). E, BACHD mice had significantly smaller striatal volumes at 6, 12, and 16 months of age (t test, *p < 0.05, compared with WT mice). D, F, Brain and striatal volumes were unaffected in BACHD Casp6+/− and BACHD Casp6−/− mice relative to BACHD. Error bars indicate SEM.
Rotarod and open-field test
Next, we evaluated the motor performance of the BACHD relative to BACHD Casp6−/− mice at 16, 20, 24, 37, and 51 weeks of age using conditions previously published (Menalled et al., 2009). The performance of the BACHD mice examined in this study is comparable with previous findings (Gray et al., 2008; Menalled et al., 2009). BACHD mice show rotarod deficits as early as 16 weeks of age, and the deficit is progressive with age. As shown in Figure 7B, knock-out of caspase-6 resulted in only a modest improvement in rotarod performance with both BACHD Casp6−/− and BACHD Casp6+/− showing an increase in latency to fall relative to BACHD mice at all time points tested (caspase-6 knock-out genotype main effect, F(2,23) = 7.40, p < 0.01; age main effect, F(4,92) = 12.02, p < 0.0001; caspase-6 knock-out genotype by age interaction, F(8,92) = 0.89, p = 0.5253). Behavior in the open field was also assessed at 16, 20, 24, 37, and 51 weeks of age. When the total locomotion, locomotion in the center of the open field, rearing rate in the center, total rearing frequency, and average velocity were evaluated, no behavioral rescue was observed in the BACHD Casp6−/− mice relative to BACHD in any of the aforementioned test parameters (data not shown).
MRI analysis of WT, BACHD, and BACHD Casp6−/−
To assess whether there are changes in tissue volume, MRI analyses of WT, BACHD, BACHD Casp6+/−, and BACHD Casp6−/− were performed. While BACHD mice had smaller whole-brain volume than WT at 16 months of age, they had a smaller striatal volume at 6, 12, and 16 months of age (Fig. 7C,E). In contrast, there were no differences in cerebellar volume between BACHD and WT mice at any age (data not shown). When BACHD, BACHD Casp6+/−, and BACHD Casp6−/− whole-brain and striatal volumes were compared at 13 months of age, no significant differences were observed (Fig. 7D,F). This suggests that knock-out of caspase-6 does not have a profound effect on brain neuropathology.
Analysis of potential mHtt clearance pathways in BACHD Casp6−/− mice
Given the observation that Htt levels are altered in the BACHD Casp6−/− mice, we decided to evaluate known pathways involved in cleaving Htt or clearing the protein. It is well established for a number of caspase knock-out mice that compensatory upregulation and activation of other family members can contribute to the lack of phenotype in these models (Zheng et al., 2000; MacFarlane, 2001). To explore the potential compensatory pathways in the Casp6−/− mice, we evaluated the expression of caspase-2, caspase-3, caspase-7, caspase-8, caspase-9, and caspase-12. We did not detect any measurable changes in the striatum, but did detect a significant increase in procaspase-3 in the BACHD Casp6−/− cortex when compared with BACHD cortex (Fig. 8; data not shown). We subsequently looked at cleaved caspase-2, caspase-3, caspase-7, and caspase-9 levels in both the striatum and cortex and did not observe any significant differences (data not shown). DEVD (caspase-3/7 substrate), VEID (caspase-3/6 substrate), and IETD (caspase-3/6/8/10 substrate) substrate cleavage in cortical and striatal lysates also was not significantly different (data not shown). These results suggest it is unlikely that the clearance of Htt observed in BACHD Casp6−/− is due to activation of other caspase family members.
Figure 8.

Compensatory changes in caspase levels are modest in BAC Casp6−/− mice. A, Western blot of procaspase-3 and procaspase-7 protein levels in 13-month-old striatum (left panel). Densitometry of striatal caspases normalized to GAPDH (right panel). B, Western blot of procaspase-3 and procaspase-7 protein levels in 13-month-old cortex (left panel). Densitometry of cortical caspases normalized to GAPDH (right panel; n = 3; ANOVA, **p < 0.01). Error bars indicate SD.
To investigate other possible mechanisms responsible for the reduction in soluble mHtt and aggregates in the BACHD Casp6−/− mice, we measured the expression of LC3 and p62, proteins known to be involved in macroautophagy, as well as the acetylated form of Htt at Lys-444, which has been recently shown to target mHtt to the autophagosome (Jeong et al., 2009). In addition, we analyzed protein ubiquitination, which is involved in the ubiquitin proteasome system (UPS) clearance pathway.
We found that the LC3 levels are increased and localization is more concentrated to the cortical neuropil in BACHD Casp6−/− when compared with BACHD and WT mice (Fig. 9A,B). p62 levels were evaluated and may be decreased in BACHD Casp6−/− cortex relative to BACHD (Fig. 9C) (2.2-fold decrease; n = 3–5; t test, p = 0.055). Since clearance of Htt has been shown to be regulated by acetylation at K444 in Htt (Jeong et al., 2009) and is critical for autophagy-regulated clearance mechanisms, we used double immunofluorescence of 13-month-old BAC Casp6−/− cortex to show a cytosolic colocalization of acetylated Htt and p62 (Fig. 9D).
Figure 9.
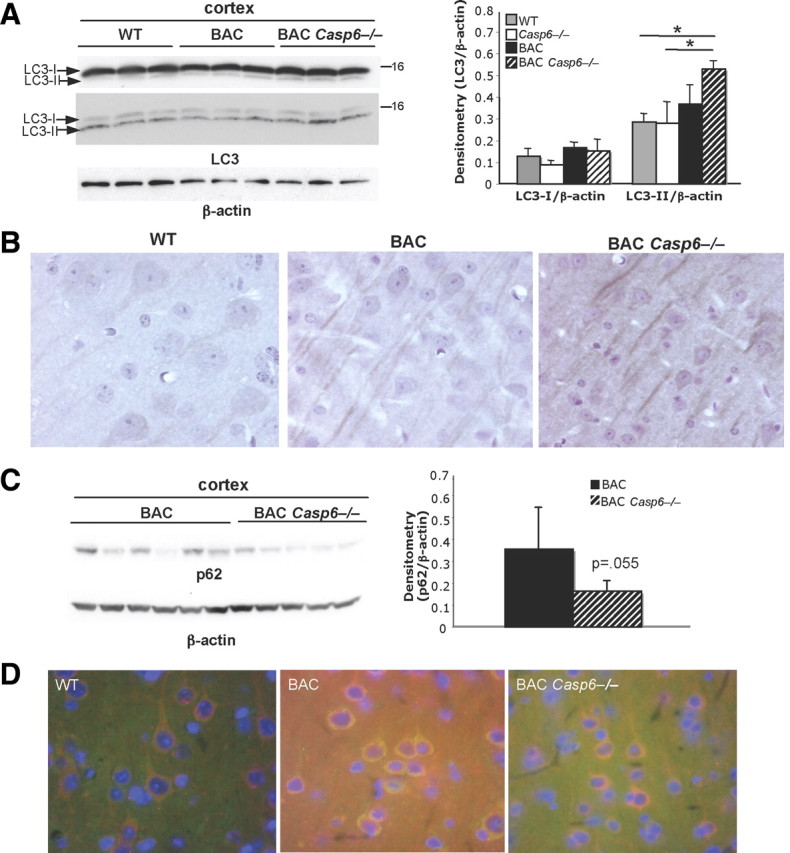
Autophagy-associated proteins are altered in 13-month-old BACHD Casp6−/− mice. A, Western analysis shows an increase in LC3-II levels in BACHD Casp6−/− cortex with antibodies from MBL (top left panel) and Abgent (middle left panel). Densitometry of cortical LC3 levels (Abgent antibody) normalized to β-actin (right panel; n = 3; ANOVA, *p < 0.05). B, Immunohistochemistry shows an increase in LC3 immunoreactivity and altered localization in the 13-month-old BACHD Casp6−/− cortex relative to both WT and BACHD. C, p62 protein levels decrease in BACHD Casp6−/− cortex relative to BACHD (left panel; 13- and 15-month-old cortex) using Western blot analysis. Densitometry of p62 normalized to β-actin (right panel; n = 5–6; t test). D, Immunohistochemistry shows colocalization of p62 (red) and Htt acetylated at amino acid K444 (green) in 13-month-old cortex. Error bars indicate SD.
Ubiquitinated proteins often build up in HD mouse models, and so, given the enhanced clearance of the Htt protein in the BACHD Casp6−/− mice, we analyzed ubiquitin and Htt distribution in the cortex of BAC Casp6−/− mice. We observed an overall decrease in ubiquitin staining in BAC Casp6−/− mice (Fig. 10A) and redistribution of ubiquitin staining from the nucleus in the BACHD mice to perinuclear localization in the BACHD Casp6−/− mice (Fig. 10B). We also observed an increased colocalization of Htt with ubiquitin in the BACHD Casp6−/− when compared with BACHD mice (Fig. 10C).
Figure 10.

Ubiquitination levels increase in BACHD and BACHD Casp6−/− mice. A, Western analysis shows an increase in overall protein ubiquitination levels in 13-month-old cortex (left panel). Densitometry of total ubiquitinated protein normalized to β-actin (right panel; n = 3). B, Immunohistochemistry shows changes in localization of ubiquitinated proteins in 15-month-old BACHD Casp6−/− cortex. C, Double immunofluorescence of ubiquitin (red) and huntingtin (green) confirms colocalization in 13-month-old cortex. DAPI nuclear stain is in blue. Error bars indicate SD.
Discussion
HD is caused by a mutation encoding a polyglutamine expansion at the N terminus of Htt protein. It has been postulated that proteolytic processing of Htt leads to toxic fragments that drive disease progression, so the enzymes involved are potential therapeutic targets for HD. Previously, mice expressing mHtt that is not cleaved by caspase-6 but is cleaved by caspase-3 were shown to have normal neuronal function and were not susceptible to neurodegeneration (Graham et al., 2006). We therefore investigated whether the putative protease responsible for cleavage of Htt at Asp-586 was disease modifying in vivo. Specifically, we crossed BACHD mice with Casp6−/− mice.
Caspase-6 was suggested to be the key protease that cleaves Htt at amino acid 586 (Graham et al., 2006), and so a homozygous caspase-6 knock-out would be predicted to eliminate production of this Htt fragment. While we found that knocking out caspase-6 reduced expanded full-length Htt levels and overall Htt fragment levels, surprisingly it did not eliminate production of the amino acid 586 Htt cleavage product. Interestingly, the YAC128 C6R mouse that showed full neurological rescue (Graham et al., 2006) was mutated at both the amino acid 586 and amino acid 589 sites, suggesting that either cleavage site may be involved in the phenotypic rescue. Alternatively, like the secretase pathways in which mutation of the amyloid precursor protein site changes activity of the secretases, the rescue of YAC128 C6R could be due to altered protease activity (Golde et al., 2011). Analysis of a conditional caspase-6 knock-out crossed to a HD mouse models is required to eliminate the possibility of compensatory pathways explaining our results. Further experiments will need to be conducted to confirm the cleavage site and key protease involved in Htt fragmentation.
No HD-like phenotypic abnormalities have been reported in the YAC128C6R mouse model (Graham et al., 2006) in which a YAC128 mHtt transgene harboring the caspase-6 motif mutations (amino acid 586 and amino acid 589) was introduced into the mouse by transgenesis. Given the hypothesis that absence of caspase-6 cleavage at the mutated site in the YAC128 Htt protein leads to mice lacking an HD phenotype, we sought to independently investigate the effects of caspase-6 ablation on the general health, motor, and MRI phenotypes of the BACHD mouse model. BACHD Casp6−/− mice showed significant improvement in rotarod motor performance compared with BACHD mice. The reduction in both soluble and insoluble mHtt aggregates may be responsible for this motor improvement, since reduction in expanded Htt levels has been shown to have a protective effect in HD (Ravikumar et al., 2002, 2004; Sánchez et al., 2003). However, it should be noted that recent studies show that body weight can in part modulate rotarod performance in the BACHD line (A. E. Kudwa, S. Oakeshott, C. Murphy, R. Mushlin, J. Fitzpatrick, S. Miller, K. McConnell, R. Port, J. Torello, D. Howland, S. Ramboz, D. Brunner, L. Menalled, unpublished results), making it possible that the improvement in rotarod performance in BACHD Casp6−/− mice is at least partly due to their reduced body weight. Interestingly, the reduced body weight in BACHD Casp6−/− and BACHD Casp6+/− animals may be a consequence of the unexpected reduction of full-length expanded Htt since it is known that full-length Htt levels in YAC128 mice can modulate body weight (Van Raamsdonk et al., 2006).
We have previously found that body weight also modulates the open-field performance of mice, with smaller mice tending to present higher locomotor and rearing activity (A. E. Kudwa, S. Oakeshott, C. Murphy, R. Mushlin, J. Fitzpatrick, S. Miller, K. McConnell, R. Port, J. Torello, D. Howland, S. Ramboz, D. Brunner, L. Menalled, unpublished results). Consequently, the lack of effects of the caspase-6 knock-out on locomotor and rearing activity exhibited by experimental mice in the open-field task might be considered surprising given that they are significantly smaller than their WT counterparts. It is possible, however, that the knock-out of caspase-6, independent of the BACHD genotype, induces a reduction of locomotor activity that is then masked by weight-related increases in activity; this possibility should be further investigated in the Casp6−/− mouse.
Our data do not support a neuroprotective effect of eliminating caspase-6 activity on mHtt-mediated whole-brain and striatal atrophy, as measured in the BACHD mouse. Although these atrophies are modest in the BACHD mice, we have no evidence that caspase-6 knock-out prevents such changes.
A surprising finding of this study is the decrease in expanded full-length mHtt and mHtt fragments. We present preliminary evidence that soluble mHtt lowering and loss of mHtt aggregates may be modulated through enhanced protein clearance pathways in BACHD Casp6−/− mice. Interestingly, it has recently been shown that beclin, p62, and hAtg3—each involved in autophagy—are substrates for caspase-6 cleavage (Norman et al., 2010). This connection between caspase-6 and protein clearance pathways led us to hypothesize that the Casp6−/− mice might exhibit altered levels of autophagy or UPS activation. Our immunohistochemical analyses of BACHD Casp6−/− brain tissue suggests increased LC3 expression, with a redistribution in cortical neurons, as well as a decrease in p62 is consistent with the hypothesis that caspase-6 may be modulating autophagic clearance of mHtt. Furthermore, in cortical neurons of BACHD Casp6−/− mice, we found evidence for increased association of p62 and K444-acetylated Htt relative to BACHD mice; the latter is a substrate for autophagy-mediated clearance (Jeong et al., 2009), and our results suggest it is altered in the homozygote Casp6−/− mice. Further work is required for a detailed understanding of the specific mechanisms engaged in the BACHD Casp6−/− mice that lead to lower levels of soluble mHtt and insoluble inclusion bodies. Autophagy would seem a viable candidate, but our data showing a redistribution of ubiquitin staining and colocalization of Htt and ubiquitin in BAC Casp6−/− mouse brain raises the possibility that the UPS may be involved.
In summary, we show that elimination of caspase-6 protein and activity in the BACHD mouse model does not prevent the production of a 586 aa Htt proteolytic fragment in the brain. These data suggest that generation of this fragment in vivo is not due exclusively to caspase-6 activity and raises serious concerns about pursuing caspase-6 as a therapeutic target for HD in the context of the proteolytic fragment hypothesis. Studies using the HdhQ150 knock-in model (Lin et al., 2001) crossed to the Casp6−/− mice showed no change in the general pattern of Htt fragmentation or level of the Htt586 product, further supporting this conclusion (G. P. Bates, personal communication). Further investigation into cleavage of Htt at the amino acid 586 and amino acid 589 sites is required to verify the key cleavage site, identify the protease responsible, and explore whether protease inhibition is a viable therapeutic strategy. An siRNA screen monitoring Htt586 or Htt589 cleavage levels as the endpoint may be an unbiased way to identify other potential proteases involved in this pathway. Future work directed at dissecting the role of caspase-6 in HD may require the use of conditional knockdown strategies to resolve the contributions of clearance pathways and Htt proteolysis.
Footnotes
This work was supported by National Institutes of Health Grant NS40251 (L.M.E.) and CHDI Foundation (L.M.E.). Neo Htt epitope antibodies and the Htt acetylation antibody were produced in collaboration with Jacqueline Duke and Matt Baker at Open Biosystems. We thank both of them for their expert advice and help. Many thanks to Richard Mushlin for the statistical support and to Judy Watson-Johnson and Melinda Ruiz at PsychoGenics, Inc., for their technical assistance. We thank Gill Bates for the S829 and S830 Htt antibodies. We thank Dr. A. Osmond for protocols for immunostaining. We thank Akilah Bonner, Khan Zafar, and Caitlin Rugani for perfusion and tissue dissection.
References
- Albrecht S, Bourdeau M, Bennett D, Mufson EJ, Bhattacharjee M, LeBlanc AC. Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol. 2007;170:1200–1209. doi: 10.2353/ajpath.2007.060974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht S, Bogdanovic N, Ghetti B, Winblad B, LeBlanc AC. Caspase-6 activation in familial alzheimer disease brains carrying amyloid precursor protein or presenilin I or presenilin II mutations. J Neuropathol Exp Neurol. 2009;68:1282–1293. doi: 10.1097/NEN.0b013e3181c1da10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong X, Held JM, DeGiacomo F, Bonner A, Chen JM, Schilling B, Czerwieniec GA, Gibson BW, Ellerby LM. Mass spectrometric identification of novel lysine acetylation sites in huntingtin. Mol Cell Proteomics. 2011;10:M111.009829. doi: 10.1074/mcp.M111.009829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Gafni J, Ellerby LM. Calpain activation in Huntington's disease. J Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Hermel E, Young JE, Wellington CL, Hayden MR, Ellerby LM. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem. 2004;279:20211–20220. doi: 10.1074/jbc.M401267200. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13:442–449. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- Golde TE, Schneider LS, Koo EH. Anti-Aβ therapeutics in Alzheimer's disease: the need for a paradigm shift. Neuron. 2011;69:203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Carroll J, Vaid K, Cowan C, Pouladi MA, Metzler M, Bissada N, Wang L, Faull RL, Gray M, Yang XW, Raymond LA, Hayden MR. Cleavage at the 586 amino acid caspase-6 site in mutant huntingtin influences caspase-6 activation in vivo. J Neurosci. 2010;30:15019–15029. doi: 10.1523/JNEUROSCI.2071-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, Li XJ, Levine MS, Yang XW. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer's disease. Am J Pathol. 2004;165:523–531. doi: 10.1016/S0002-9440(10)63317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermel E, Gafni J, Propp SS, Leavitt BR, Wellington CL, Young JE, Hackam AS, Logvinova AV, Peel AL, Chen SF, Hook V, Singaraja R, Krajewski S, Goldsmith PC, Ellerby HM, Hayden MR, Bredesen DE, Ellerby LM. Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ. 2004;11:424–438. doi: 10.1038/sj.cdd.4401358. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Hoffner G, Souès S, Djian P. Aggregation of expanded huntingtin in the brains of patients with Huntington disease. Prion. 2007;1:26–31. doi: 10.4161/pri.1.1.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Jeong H, Then F, Melia TJ, Jr, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Sapp E, Alexander J, Reeves P, Bleckmann D, Sobin L, Masso N, Valencia A, Jeong H, Krainc D, Palacino J, Curtis D, Kuhn R, Betschart C, Sena-Esteves M, Aronin N, Paganetti P, Difiglia M. Huntingtin cleavage product A forms in neurons and is reduced by gamma-secretase inhibitors. Mol Neurodegener. 2010;5:58. doi: 10.1186/1750-1326-5-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ST, Kim M. Aging and neurodegeneration. Molecular mechanisms of neuronal loss in Huntington's disease. Mech Ageing Dev. 2006;127:432–435. doi: 10.1016/j.mad.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Leyva MJ, Degiacomo F, Kaltenbach LS, Holcomb J, Zhang N, Gafni J, Park H, Lo DC, Salvesen GS, Ellerby LM, Ellman JA. Identification and evaluation of small molecule pan-caspase inhibitors in Huntington's disease models. Chem Biol. 2010;17:1189–1200. doi: 10.1016/j.chembiol.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haïem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10:259–269. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- MacFarlane M. Compensatory caspase activation: a cautionary tale. Trends Pharmacol Sci. 2001;22:60. doi: 10.1016/s0165-6147(00)01630-8. [DOI] [PubMed] [Google Scholar]
- Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- McStay GP, Salvesen GS, Green DR. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15:322–331. doi: 10.1038/sj.cdd.4402260. [DOI] [PubMed] [Google Scholar]
- Menalled L, El-Khodor BF, Patry M, Suárez-Fariñas M, Orenstein SJ, Zahasky B, Leahy C, Wheeler V, Yang XW, MacDonald M, Morton AJ, Bates G, Leeds J, Park L, Howland D, Signer E, Tobin A, Brunner D. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol Dis. 2009;35:319–336. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Norman JM, Cohen GM, Bampton ET. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy. 2010;6:1042–1056. doi: 10.4161/auto.6.8.13337. [DOI] [PubMed] [Google Scholar]
- Osmand AP, Berthelier V, Wetzel R. Imaging polyglutamine deposits in brain tissue. Methods Enzymol. 2006;412:106–122. doi: 10.1016/S0076-6879(06)12008-X. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Richards P, Didszun C, Campesan S, Simpson A, Horley B, Young KW, Glynn P, Cain K, Kyriacou CP, Giorgini F, Nicotera P. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington's disease. Cell Death Differ. 2011;18:191–200. doi: 10.1038/cdd.2010.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez I, Mahlke C, Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421:373–379. doi: 10.1038/nature01301. [DOI] [PubMed] [Google Scholar]
- Singer J. Using SAS PROC MIXED to fit multilevel models, hierarchical models, and individual growth models. J Educ Behav Stat. 1998;23:323. [Google Scholar]
- Sivananthan SN, Lee AW, Goodyer CG, LeBlanc AC. Familial amyloid precursor protein mutants cause caspase-6-dependent but amyloid beta-peptide-independent neuronal degeneration in primary human neuron cultures. Cell Death Dis. 2010;1:e100. doi: 10.1038/cddis.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe V, Wong BK, Graham RK, Cusack CL, Skotte NH, Pouladi MA, Xie Y, Feinberg K, Ou Y, Ouyang Y, Deng Y, Franciosi S, Bissada N, Spreeuw A, Zhang W, Ehrnhoefer DE, Vaid K, Miller FD, Deshmukh M, Howland D, et al. Rescue from excitotoxicity and axonal degeneration accompanied by age-dependent behavioral and neuroanatomical alterations in caspase-6-deficient mice. Hum Mol Genet. 2012;21:1954–1967. doi: 10.1093/hmg/dds005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Gibson WT, Pearson J, Murphy Z, Lu G, Leavitt BR, Hayden MR. Body weight is modulated by levels of full-length huntingtin. Hum Mol Genet. 2006;15:1513–1523. doi: 10.1093/hmg/ddl072. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE, et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273:9158–9167. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, Cattaneo E, Hackam A, Sharp A, Thornberry N, Nicholson DW, Bredesen DE, Hayden MR. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275:19831–19838. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Ellerby LM, Gutekunst CA, Rogers D, Warby S, Graham RK, Loubser O, van Raamsdonk J, Singaraja R, Yang YZ, Gafni J, Bredesen D, Hersch SM, Leavitt BR, Roy S, Nicholson DW, Hayden MR. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J Neurosci. 2002;22:7862–7872. doi: 10.1523/JNEUROSCI.22-18-07862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li Q, Graham RK, Slow E, Hayden MR, Bezprozvanny I. Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington's disease. Neurobiol Dis. 2008;31:80–88. doi: 10.1016/j.nbd.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–1247. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]
- Zois CE, Giatromanolaki A, Sivridis E, Papaiakovou M, Kainulainen H, Koukourakis MI. “Autophagic flux” in normal mouse tissues: focus on endogenous LC3A processing. Autophagy. 2011;7:1371–1378. doi: 10.4161/auto.7.11.16664. [DOI] [PubMed] [Google Scholar]