

Abstract
Objectives
This study aimed at developing a murine model of surgically induced acute aortic dissection type A (AAD) for investigation of the formation and progression of AAD, and to test whether this system could be used for biomarker discovery.
Methods
Adult fibrillin-1 deficient, Fbn1C1039G/+ mice and wild-type mice were anesthetized, ventilated and the ascending aorta exposed via hemisternotomy. We hypothesized that AAD could be induced either by injecting autologous blood into the aortic wall or by injury to the wall with aortic clamping. Echocardiography was done preoperatively, and serum samples collected before and 30 minutes after surgery, and analyzed by ELISA.
Results
Echocardiography revealed larger aortic root diameters in Fbn1C1039G/+ compared with wild-type mice (P=0.001). Histology showed that aortic clamp injury but not injection of blood leads to large intimal tears, disruption of aortic wall structures and localized dissection of the aortic media in Fbn1C1039G/+ mice. AAD developed in 4 out of 5 Fbn1C1039G/+ mice versus 0 out of 5 wild-type mice after aortic clamping (P<0.01). Elastin staining showed higher elastic fiber fragmentation and disarray in Fbn1C1039G/+ compared with wild-type mice. ELISA analysis revealed elevated circulating TGFβ1 concentrations after inducing AAD in Fbn1C1039G/+ mice (P=0.02, 150±61 ng/ml vs. 456±97 ng/ml), but not in wild-type or sham-operated mice.
Conclusions
Aortic clamp injury can induce AAD in Fbn1C1039G/+, but not in wild-type mice. This murine model of surgically induced AAD is highly reproducible and non-lethal in the short-term. Using this system, we revealed that circulating TGFβ1 is a promising biomarker for AAD.
Introduction
Acute dissections involving the ascending aorta (AAD) are life-threatening conditions with high morbidity and mortality even after emergent surgery [1, 2]. AAD develops often in people older than 40 years reflecting their increased rates of arterial hypertension and atherosclerosis [3]. Other predisposing factors, especially in younger patients, include preexisting aortic aneurysm, aortic wall trauma, and connective tissue disorders as Marfan syndrome (MFS) [4]. MFS is an autosomal dominant inherited disorder caused by mutations in the gene encoding the extracellular matrix protein fibrillin-1 and dysregulated transforming growth factor β (TGFβ) [5, 6]. Numerous theories about the process that initiates an AAD exist, however it is uncertain whether the initiating event is a primary rupture of the intima with secondary dissection of the media, or hemorrhage within the media and subsequent rupture of the overlying intima. Further understanding of molecular mechanisms leading to and associated with the formation and progression of an AAD are needed to gather novel insights and improve diagnostic and therapeutic strategies. The current signalment for preventive surgical intervention is an aortic root diameter measuring 5 cm. However, a majority of patients which develop aortic dissection or rupture never reach the 5 cm diameter standard [7]. Therefore, there is an urgent need for the development of new parameters or surrogate markers to more accurately identify impending aortic dissection or rupture. Studying AAD in humans is limited because most patients are seen after the dissection has occurred, and patients are mostly at different stages of the disease. A general difficulty in human studies is the wide inter-individual variation in tissue specimens (e.g. aortic tissue, serum and plasma) and in external conditions which preclude a precise analysis of disease mechanisms. Small animal models for AAD could be used to exclude those factors and facilitate a more exact, standardized approach to studying the development and propagation of aortic dissection. Several murine and rat models of spontaneous aortic dissection have been described, but these systems are either transgenic or chemically induced and do not allow prediction of the time point at which the dissection occurs [8-12]. We hypothesized that such a prediction could only be made if the AAD was surgically induced. A murine model of MFS which is heterozygous for a mutant fibrillin-1 allele, Fbn1C1039G/+, and develops progressive aortic root dilatation with sporadic dissections throughout life, might be predisposed to develop an AAD after surgical manipulation [5, 13]. The goal of our study was to develop a reproducible and in the short-term non-lethal murine model of a surgically induced AAD that could be used to discover candidate biomarkers relevant to dissection.
Materials and Methods
Animals
The animals used in this study were 10–month-old female mice harboring Fbn1 mutations, Fbn1C1039G/+ (n=10) as described previously [13, 14], and age-matched wild-type littermates (n=10). All analyses on Fbn1C1039G/+ mice were done after back-crossing this mutation into the C57BL/6J background (>9 generations). In addition, adult Sprague Dawley (CD) rats (Charles River) (n=3) were used. All animals were treated and cared for in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” (National Research Council, Washington, 1996), and all protocols were approved by the Animal Care and Use Committee at the Johns Hopkins University School of Medicine, Baltimore, USA.
Surgical Procedure
Anesthesia was induced by placing the animal in a closed induction chamber supplied with 2.5% isoflurane. Rats were not intubated but breathed spontaneously through a mask connected to a vaporizor (VetEquip, Inc). The mice, once anesthetized, were transferred to the operating table (Rodent WorkStand, Braintree Scientific Inc., MA) in a supine position and orally intubated (Braintree Scientific Inc., MA) using microsurgical loops with 3.5 magnification (Design for Vision, Ronkonkoma, NY). The endotracheal tube was connected to a rodent ventilator (Hugo-Sachs, March, Germany) set at 250 respirations/minute with a 200 ul tidal volume. Isoflurane was set at 2.5% in a 100% oxygen mix for the remainder of the procedure for mice and rats. For all surgical procedures a surgical microscope (Microsurgery Instruments Inc., Bellaire, TX) was used. First, the right femoral vein was dissected and prepared for later collection of pre-clamping blood samples. A small upper sternal incision was made, the muscle layers on the right parasternal side were divided sharply and a transverse sternotomy was performed at the height of the third intercostal space. The mediastinum was entered and a small retractor was positioned to provide appropriate access. The pericardium was opened, and the ascending aorta was exposed (Figure 1A). Blood samples were then collected from the right femoral vein. In a first attempt to develop a surgical technique to induce the AAD, autologous blood was injected into the ascending aortic wall of Fbn1C1039G/+ and wild-type mice and rats using microinjection needles (33 to 36 gauge; World Precision Instruments, Sarasota, FL). Alternatively, mice received an aortic clamp injury just above the sino-tubular junction using a microsurgical needleholder (Microsurgery Instruments Inc., Bellaire, TX) (Figure 1A). The injury was made by partially clamping the aortic wall for 0.5 to 1 second until a hematoma was visible (Figure 1B). The sternum was then closed with 5-0 silk intercostal sutures, and the remaining divided muscle layers and skin were closed with a running 6-0 silk suture. After 30 minutes under anesthesia, the sternum was reopened and a post-procedure blood sample was collected from the right atrium. All mice and rats were humanely euthanized. Two Fbn1C1039G/+ and two wild-type mice underwent a sham operation with blood sample collection as controls.
Figure 1.

(A) Intraoperative view through the dissecting microscope (Microsurgery Instruments Inc., Bellaire, TX) showing the surgical approach through an upper partial sternotomy and the opened pericardium in a Fbn1C1039G/+ mouse. The ascending aorta (tip of the arrow) was partially clamped with a microsurgical needleholder. (B) Macroscopic view of the ascending aorta 30 minutes after the aortic clamp injury in the Fbn1C1039G/+ mouse. The aortic dissection was induced by clamping in the middle part of the ascending aorta (*). After the clamp injury, the hematoma and partially the dissection extended towards the aortic root and the distal ascending aorta (arrows). Ao=ascending aorta; Cr=cranial; RV=right ventricle.
Tissue and Histology
After the animals were sacrificed, the thoracic aorta and the attached heart were harvested and immediately fixed in 10 % buffered formalin. For further analysis, the fixed aorta was processed and embedded in paraffin and sectioned at 5 μm, stained with hematoxylin and eosin (H-E) or Verhoeff-van Gieson (VVG; elastin) for analysis by conventional light microscopy. VVG stained sections of the ascending aorta in Fbn1C1039G/+ mice at 1 month, 4 months and 9 months of age were analyzed for comparative histology over time.
Serum
All blood samples, immediately before and 30 minutes after the aortic injury, were allowed to clot for 2 hours at room temperature before being centrifuged for 20 minutes at 2000 g [15]. Serum was removed, divided into aliquots and immediately frozen and stored at −80°C until further analysis.
ELISA for TGFβ
TGFβ1 concentrations were measured in serum samples by ELISA using the Quantikine TGFβ1 immunoassay (from R&D Systems, Minneapolis, MN, catalog no. MB100M). The minimum detectable dose of TGFβ1 in this assay ranges from 1.7 to 15.4 pg/ml, and there is less than 1% (MB100M) cross-reactivity with the latent TGFβ1 complex. Total TGFβ1 concentrations were measured by first acid activating (with 1N HCl) latent TGFβ1 to immunoreactive TGFβ1. The ELISA immunoassay was performed according to the manufacturer’s protocol. Briefly, the wells are pre-coated with capture monoclonal antibody specific for TGFβ1. Standards, controls and samples were added and incubated for 2 hours. After washing, an enzyme-linked polyclonal detection antibody specific for TGFβ1 was added. After another washing, samples were incubated with a substrate solution for 30 minutes and the color reaction stopped by addition of a dilute HCl solution. The optical density of each well was measured immediately with a microplate reader at 450 nm; the wavelength correction was set to 570 nm. All samples were run in duplicate. We considered the results valid when recovery (expected concentration divided by calculated concentration multiplied by 100) of the standards/calibrators was 100 ± 20%, the coefficient of variation (CV) was < 20%, the intra-assay CV was < 10% and the interassay CV was < 20%. A run was considered valid when > 85% of samples were within these specifications.
Echocardiography
Transthoracic echocardiograms (TTE) were performed on awake, unsedated 10-month-old Fbn1C1039G/+ mice (n=10) and age-matched wild-type mice (n=10) using the VisualSonics Vevo 660 V1.3.6 imaging system and a 40- or 60-MHz transducer (model RMV603, VisualSonics Inc., Toronto, Canada). The aorta was imaged in the parasternal long-axis view, and 3 measurements were obtained at the level of the sinus of Valsalva (SOV) by an observer blinded to genotype. All echocardiographic studies were performed by 2 individuals with extensive experience in mouse echocardiography.
Statistical Analysis
For statistical analysis the Student’s t test and the Fisher’s exact test were used as appropriate. Data are presented as mean±SEM, and two-sided P values of less than 0.05 were considered to indicate statistical significance for all statistical tests and models. SPSS statistical software 9.0 was used for all analyses.
Results
Operative observations
Injection of autologous blood into the aortic wall of Fbn1C1039G/+ and wild-type mice was difficult because of the small aortic wall dimensions (despite the 36-gauge microinjection needles) and the rapid pulse rate. In consequence, the reproducibility of the aortic wall hematoma was inconsistent, and there were only small intimal tears at the site of the injection. These aortic wall lesions did not resemble AAD as seen in humans. Injection of autologous blood into the aortic wall of adult Sprague Dawley rats was much easier, and resulted in reproducible and extensive periaortic wall hematomas. The aortic clamp injury performed surgically in Fbn1C1039G/+ and wild-type mice was highly reproducible. While the clamping led in wild-type mice (n=5) only to a localized aortic wall hematoma, in Fbn1C1039G/+ mice (n=5) the aortic clamp injury resulted in massive localized aortic wall hematomas that separated the tunica media extending into the aortic root and the proximal part of the aortic arch (Figure 1B). The extent of these lesions increased in proportion with the aortic root diameter. In addition, there were large intimal tears at the site of the clamping in Fbn1C1039G/+ mice, but wild-type mice showed only small intimal lesions. No aortic rupture occurred in Fbn1C1039G/+ and wild-type mice within 30 minutes after surgery.
Histology
H-E stained aortic sections showed that injection of autologous blood into the aortic wall of WT rats results in large, but strictly periaortic wall hematomas. There was no aortic media dissection in the histological sections examined (not shown). After the aortic clamp injury, there were small intimal lesions in wild-type mice, and 2 out of 5 wild-type mice developed transmural lesions with blood within the adventitial layer, but no aortic media dissection occurred (Figure 2A). In contrast, the aortic clamping in Fbn1C1039G/+ mice resulted in large intimal tears, a massive disruption of aortic wall structures and in 4 out of 5 Fbn1C1039G/+ mice a break-up of the aortic medial layer with subsequent localized dissection and blood within the aortic media (Figure 2B–2D).
Figure 2.

Histological sections of the ascending aorta stained with hematoxylin and eosin. (A) Aortic section in a wild-type mouse after the aortic clamp injury which led to transmural tears (arrows) and periadventitial hematoma, however no aortic media dissection occurred. (B-D) In contrast, the clamp injury in Fbn1C1039G/+ mice resulted in large intimal tears, disruption and break-up of the aortic medial layer with subsequent localized dissection and blood within the aortic media (arrows). WT=wild-type, Lu=lumen of the vessel.
Staining for elastin showed extensive disruption of elastic lamellae and elastin fibers at the site of the aortic clamping with large intimal tears in Fbn1C1039G/+ mice (Figure 3A–3D). There were localized aortic dissections with blood within the fragmented and disarrayed elastic fibers of the media layer (Figure 3A–3D), resembling those seen in human Marfan-like AAD [3]. All surgically induced aortic dissections in Fbn1C1039G/+ mice were localized around the aortic clamping area and limited to the ascending aorta. Comparative histology with VVG staining of the ascending aorta in Fbn1C1039G/+ mice at 1 month, 4 months and 9 months of age showed progressive fragmentation and disarray of elastic fibers, and increase in matrix deposition within the aortic media over time (Figure 4A-4D). VVG staining showed marked elastin fiber fragmentation and disarray in 10 month old Fbn1C1039G/+ compared with age-matched wild-type mice (not shown).
Figure 3.

Verhoeff-van Gieson (VVG; elastin) stained sections of the ascending aorta in Fbn1C1039G/+ mice. (A–D) The aortic clamp injury led to extensive intimal tears, disruption of elastic fibers and localized dissection with blood within the aortic medial layer (arrows). Lu=lumen of the vessel.
Figure 4.

Verhoeff-van Gieson (VVG; elastin) stained sections of the ascending aorta in Fbn1C1039G/+ mice at 1 month, 4 months and 9 months of age. Histological sections show progressive fragmentation and disarray of elastic fibers (arrows), and increased matrix deposition within the aortic media over time. While severe structural aortic wall changes already exist in 4 month old Fbn1C1039G/+ mice, these lesions translate into progressive aortic root dilatation with increasing age and subsequently a predisposition for developing aortic dissection. Lu=lumen of the vessel.
Aortic Root Diameters
TTE revealed significantly larger sinus of Valsalva (SOV) diameters in Fbn1C1039G/+ (n=10) compared with age-matched wild-type (n=10) mice (P=0.001; mean±SEM 2.12±0.08 mm vs. 1.77±0.05 mm).
Circulating TGFβ
Circulating TGFβ1 concentrations in serum samples from Fbn1C1039G/+ mice (n=5) significantly increased by 30 minutes after inducing the AAD (P=0.02 (paired t test), mean±SEM 150±61 ng/ml vs. 456±97 ng/ml) (Figure 5). In contrast, wild-type mice (n=5) showed no significant increase in circulating TGFβ1 levels after the aortic clamp injury (P=0.7, 82±54 ng/ml vs. 112±48 ng/ml); similar results were found in sham operated Fbn1C1039G/+ (n=2) and sham operated wild-type (n=2) mice (P=0.7, 219±160 ng/ml vs. 198±126 ng/ml).
Figure 5.
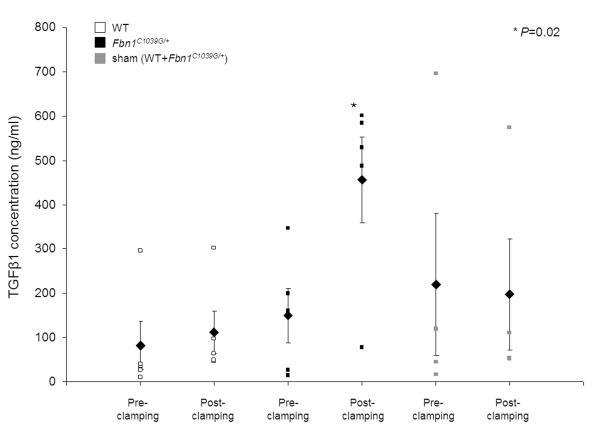
Circulating TGFβ1 serum concentrations in Fbn1C1039G/+ and age-matched wild-type mice before (pre-clamping) and 30 minutes after (post-clamping) the aortic clamp injury. Circulating TGFβ1 levels significantly increased after inducing the AAD in Fbn1C1039G/+ mice (P=0.02), but not in wild-type mice and sham operated litters. The graph shows each data point (square), mean values (rhombus) and SEM.
Discussion
Several models of spontaneous aortic dissection in mice and rats have been described [8-12,16]. Mice with hypomorphic or mutant fibrillin-1 alleles develop proximal thoracic aortic aneurysm, aortic dissection and rupture [16]. Mice with a biglycan deficiency show dissection and rupture of the thoracic and abdominal aorta within the first 3 months of life [8]. Biglycans play a critical role in the control of collagen fibrillogenesis and in the proper assembly of the extracellular matrix. In utero exposure to semicarbazide, an inhibitor of the vascular enzyme semicarbazide-sensitive amine oxidase, results in newborn rats in thoracic aortic aneurysm and dissection, but no rupture [9]. The postnatal administration of β-aminopropionitrile monofumarate, which induces vascular medial degeneration, fragmentation of elastic fibers and apoptosis of vascular smooth muscle cells, leads to aortic dissection in rats by inhibiting the cross linking of collagen fibers [10]. Other authors report aneurysm formation and media dissection induced by angiotensin II infusion in apo E-deficient mice or salt-sensitive aortic dissection in mice with overproduction of angiotensin [12]. While all these non-surgical means to induce aortic dissection provide new insights into specific pathomolecular mechanisms of the disease, they are less useful for prospective studies of the formation and progression of AAD. In addition, discovery of a specific biomarker might be difficult as non-surgical manipulations will result in a variety of alterations at the serum or plasma protein level not related to the aortic dissection.
We hypothesized that a small animal model could most consistently be achieved by inducing the AAD surgically. The initial hypothesis was that injecting autologous blood into the ascending aortic wall might be the most physiological way to induce an AAD. We tested this technique using wild-type mice and mice heterozygous for a mutant Fbn1 allele, Fbn1C1039G/+, which is the most common class of mutation causing MFS in humans [13, 17]. Fbn1C1039G/+ mice show enlarged aortic root and ascending aortic dimensions beginning at 2 weeks of age with progression throughout life, but only sporadically develop aortic dissection later in life. In contrast, homozygous Fbn1-/- mice uniformly die of aortic dissection and rupture prior to postnatal day fifteen [13, 18]. In the present study, right-sided upper partial sternotomy provided excellent exposure of the ascending aorta. However, injection of autologous blood into the aortic wall of Fbn1C1039G/+ and wild-type mice was technically difficult because of the small blood vessels and high pulse rates, and did not lead to a reproducible aortic wall hematoma or dissection. Aortic wall lesions were small and did not resemble those seen in patients with AAD. We hypothesized that this technique might be much easier in Sprague Dawley rats with their larger dimensions and slower pulse rates. Using this technique in the rats, we were able to inject autologous blood into the aortic wall, resulting in extensive and reproducible aortic wall hematomas. Unfortunately, histological analysis revealed that those hematomas were most often in the periaortic tissue, and no dissection of the aortic media or intimal tearings occurred. The aortic wall tissue of rats was apparently too compact and stable to dissect after the manipulation. While the injection approach did not initiate an aortic dissection in mice or rats, it led to the hypothesis that aortic clamp injury at the ascending aorta might induce AAD. The induction of dissection in humans after aortic clamping has been reported, though rarely [4]. Using the approach described above, we applied a short and partial aortic clamp injury to the ascending aorta in Fbn1C1039G/+ and wild-type mice (Figure 1A). This procedure was technically easy and consistently resulted in massive localized intramural aortic wall hematomas extending towards the aortic root and the proximal part of the aortic arch in Fbn1C1039G/+ mice (Figure 1B). Wild-type mice, however, were resistant to this type of intramural hematoma formation. At the site of the aortic clamp injury, there were large intimal tears in Fbn1C1039G/+ mice, but not in their wild-type counterparts. Histological sections revealed that the aortic clamping in Fbn1C1039G/+ mice resulted in a massive localized disruption of the intimal and aortic medial layer with subsequent break-up of the aortic media and localized dissection with blood within the fragmented and disarrayed elastic fibers in 4 out of 5 Fbn1C1039G/+ mice (Figure 2B-2D). These findings resembled those seen in patients with MFS-like AAD [3]. Interestingly, the Fbn1C1039G/+ mouse in which no dissection could be induced showed the smallest aortic root diameter among the studied Fbn1C1039G/+ litters. In wild-type mice, there were some intimal tearings and transverse aortic lesions, but no dissection within the aortic media occurred. All surgically induced AADs in Fbn1C1039G/+ mice were localized around the aortic clamping area and limited to the ascending aorta. There was no extension of the dissection to the aortic arch and the descending aorta which might be due to the short post-clamping time period studied (30 minutes). No Fbn1C1039G/+ or wild-type mice died due to rupture at the site of the aortic clamping. Fbn1C1039G/+ mice might be predisposed to developing dissection after surgical manipulation because of their abnormalities within the aortic media [13]. Histological analysis showed higher elastin fiber fragmentation and disarray in Fbn1C1039G/+ mice compared to age-matched wild-type mice, and those structural wall changes progress over time (Figure 4A-4D) leading to enlarged aortic root diameter with increasing age [13]. Further, longer-term survival studies are needed to analyze this novel murine model of surgically induced AAD, in particular to look at the progression of the dissection and whether it will extend to the descending and abdominal aorta. If so, this system would closely mimick aortic dissections seen in people with MFS.
We tested whether the murine model could be used to discover novel, much needed candidate biomarkers for diagnosis and monitoring of AAD. Serum samples were collected before and 30 minutes after the aortic clamp injury in Fbn1C1039G/+ and wild-type mice, and sham operated littermates. It was recently shown that dysregulated TGFβ plays a causal role in many of the multi-organ manifestations of MFS including aortic root dilatation [13, 19]. There is excessive free TGFβ and TGFβ signaling within the aortic root tissue of Fbn1C1039G/+ mice, which was also found in aortic specimen from patients with MFS [5, 13]. We hypothesized that the aortic clamp injury which leads to large intimal tearings and a break-up of the aortic medial layer with subsequent localized aortic dissection in Fbn1C1039G/+ mice, might result in elevated circulating TGFβ concentrations. If so, circulating TGFβ levels should not increase in wild-type and sham operated mice. Serum samples were analyzed by ELISA. The results revealed that circulating TGFβ1 levels indeed significantly increased 30 minutes after inducing the AAD in Fbn1C1039G/+ mice (Figure 5). Interestingly, the Fbn1C1039G/+ mouse in which no dissection could be induced showed the lowest pre- and post-clamp circulating TGFβ1 concentrations among the Fbn1C1039G/+ mice. Wild-type mice and sham operated mice showed no change in circulating TGFβ1 levels. Therefore, it seems possible that TGFβ is a promising biomarker for AAD in MFS. This would be intriguing since TGFβ is a direct effector in pathomolecular mechanisms involved in aortic root disease in MFS [13]. Further studies are clearly needed to validate circulating TGFβ as a candidate biomarker relevant to dissection, determine whether it has the potential to improve diagnosis, and possibly even be used to predict aortic dissection in MFS. Other biomarkers for dissection have been described, e.g. lactate dehydrogenase, smooth-muscle myosin heavy chain, soluble elastin fragments, C-reactive protein or D-dimer [20-23]. Unfortunately, the sensitivity and specificity of those markers is limited and no diagnostic blood test for aortic dissection is in routine clinical use to date. Therefore, we decided not to analyze those markers in the murine model. Our study presents circulating TGFβ as a novel and promising biomarker for aortic dissection in MFS, and we report a murine model of surgically induced AAD which may assist further investigation and analysis in this field.
Acknowledgments
Peter Matt thanks the Swiss National Foundation, the Novartis Foundation and the Hippocrate Foundation Basel for financial support. Jennifer E. Van Eyk is supported by grants from the National Heart, Lung, and Blood Institute Proteomic Initiative (contract NOHV-28120), the Daniel P. Amos Family Foundation and the Institute for Clinical and Translational Science Award (Grant NO 1U54RR023561-01A1). Harry C. Dietz is supported by the National Institutes of Health, the Howard Hughes Medical Institute, the William S. Smilow Center for Marfan Syndrome Research and the National Marfan Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hagan PG, Nienaber CA, Isselbacher EM, Bruckman D, Karavite DJ, Russman PL, et al. The International Registry of Acute Aortic Dissection (IRAD): new insights into an old disease. Jama. 2000;283:897–903. doi: 10.1001/jama.283.7.897. [DOI] [PubMed] [Google Scholar]
- 2.Masuda Y, Yamada Z, Morooka N, Watanabe S, Inagaki Y. Prognosis of patients with medically treated aortic dissections. Circulation. 1991;84:III7–13. [PubMed] [Google Scholar]
- 3.Marsalese DL, Moodie DS, Lytle BW, Cosgrove DM, Ratliff NB, Goormastic M, et al. Cystic medial necrosis of the aorta in patients without Marfan’s syndrome: surgical outcome and long-term follow-up. J Am Coll Cardiol. 1990;16:68–73. doi: 10.1016/0735-1097(90)90458-2. [DOI] [PubMed] [Google Scholar]
- 4.Svensson LG, Crawford ES. Aortic dissection and aortic aneurysm surgery: clinical observations, experimental investigations, and statistical analyses. Part II. Curr Probl Surg. 1992;29:913–1057. doi: 10.1016/0011-3840(92)90003-l. [DOI] [PubMed] [Google Scholar]
- 5.Matt P, Habashi J, Carrel T, Cameron DE, Van Eyk JE, Dietz HC. Recent advances in understanding Marfan syndrome: should we now treat surgical patients with losartan? J Thorac Cardiovasc Surg. 2008;135:389–94. doi: 10.1016/j.jtcvs.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 6.Murdoch JL, Walker BA, Halpern BL, Kuzma JW, McKusick VA. Life expectancy and causes of death in the Marfan syndrome. N Engl J Med. 1972;286:804–8. doi: 10.1056/NEJM197204132861502. [DOI] [PubMed] [Google Scholar]
- 7.Pape LA, Tsai TT, Isselbacher EM, Oh JK, O’Gara PT, Evangelista A, et al. Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD) Circulation. 2007;116:1120–7. doi: 10.1161/CIRCULATIONAHA.107.702720. [DOI] [PubMed] [Google Scholar]
- 8.Heegaard AM, Corsi A, Danielsen CC, Nielsen KL, Jorgensen HL, Riminucci M, et al. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation. 2007;115:2731–8. doi: 10.1161/CIRCULATIONAHA.106.653980. [DOI] [PubMed] [Google Scholar]
- 9.Gong B, Trent MB, Srivastava D, Boor PJ. Chemical-induced, nonlethal, developmental model of dissecting aortic aneurysm. Birth Defects Res A Clin Mol Teratol. 2006;76:29–38. doi: 10.1002/bdra.20222. [DOI] [PubMed] [Google Scholar]
- 10.Nagashima H, Uto K, Sakomura Y, Aoka Y, Sakuta A, Aomi S, et al. An angiotensin-converting enzyme inhibitor, not an angiotensin II type-1 receptor blocker, prevents beta-aminopropionitrile monofumarate-induced aortic dissection in rats. J Vasc Surg. 2002;36:818–23. [PubMed] [Google Scholar]
- 11.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–12. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishijo N, Sugiyama F, Kimoto K, Taniguchi K, Murakami K, Suzuki S, et al. Salt-sensitive aortic aneurysm and rupture in hypertensive transgenic mice that overproduce angiotensin II. Lab Invest. 1998;78:1059–66. [PubMed] [Google Scholar]
- 13.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–21. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–81. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matt P, Fu Z, Fu Q, Van Eyk JE. Biomarker discovery: proteome fractionation and separation in biological samples. Physiol Genomics. 2008;33:12–7. doi: 10.1152/physiolgenomics.00282.2007. [DOI] [PubMed] [Google Scholar]
- 16.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. 1999;96:3819–23. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–9. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 18.Ramirez F, Sakai LY, Dietz HC, Rifkin DB. Fibrillin microfibrils: multipurpose extracellular networks in organismal physiology. Physiol Genomics. 2004;19:151–4. doi: 10.1152/physiolgenomics.00092.2004. [DOI] [PubMed] [Google Scholar]
- 19.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–92. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki T, Katoh H, Watanabe M, Kurabayashi M, Hiramori K, Hori S, et al. Novel biochemical diagnostic method for aortic dissection. Results of a prospective study using an immunoassay of smooth muscle myosin heavy chain. Circulation. 1996;93:1244–9. doi: 10.1161/01.cir.93.6.1244. [DOI] [PubMed] [Google Scholar]
- 21.Shinohara T, Suzuki K, Okada M, Shiigai M, Shimizu M, Maehara T, et al. Soluble elastin fragments in serum are elevated in acute aortic dissection. Arterioscler Thromb Vasc Biol. 2003;23:1839–44. doi: 10.1161/01.ATV.0000085016.02363.80. [DOI] [PubMed] [Google Scholar]
- 22.Makita S, Ohira A, Tachieda R, Itoh S, Moriai Y, Yoshioka K, et al. Behavior of C-reactive protein levels in medically treated aortic dissection and intramural hematoma. Am J Cardiol. 2000;86:242–4. doi: 10.1016/s0002-9149(00)00869-9. [DOI] [PubMed] [Google Scholar]
- 23.Eggebrecht H, Naber CK, Bruch C, Kroger K, von Birgelen C, Schmermund A, et al. Value of plasma fibrin D-dimers for detection of acute aortic dissection. J Am Coll Cardiol. 2004;44:804–9. doi: 10.1016/j.jacc.2004.04.053. [DOI] [PubMed] [Google Scholar]