

Abstract
Human Cytomegalovirus (HCMV) infection is associated with the acceleration of transplant vascular sclerosis (TVS) and chronic allograft rejection (CR). HCMV-negative recipients of latently HCMV infected donor grafts are at highest risk for developing CMV-disease. Using a rat heart transplant CR model, we have previously shown that acute rat CMV (RCMV) infection following transplantation significantly accelerates both TVS and CR. Here, we report that RCMV-naïve recipients of heart allografts from latently RCMV-infected donors undergo acceleration of CR with similar kinetics as acutely infected recipients. In contrast to acutely infected recipients, treatment of recipients of latently infected donor hearts with ganciclovir did not prevent CR or TVS. We observed the formation of tertiary lymphoid structures (TLOs) containing macrophages and T-cells in latently infected hearts prior to transplantation but not in uninfected rats. Moreover, pathway analysis of gene expression data from allografts from latently infected donors, indicated an early and sustained production of TLO-associated genes compared to allografts from uninfected donors. We conclude that RCMV-induced TLO formation and alteration of donor tissue T-cell profiles prior to transplantation in part mediate the ganciclovir-insensitive rejection of latently infected donor allografts transplanted into naïve recipients by providing a scaffold for immune activation.
Keywords: Cytomegalovirus, Chronic Rejection, Transplant Vascular Sclerosis, Latency
Introduction
Human cytomegalovirus (HCMV) infections are life long and the virus persists in a latent state during which time gene expression is limited and infectious virus is not produced. Critical to the ability of HCMV to maintain life-long infection is the capacity to establish cellular sites of viral persistence while avoiding recognition and elimination by the host immune system. Clinical studies have directly associated HCMV infection with the acceleration of solid organ allograft chronic rejection (CR) due to increased transplant vascular sclerosis (TVS). Although all transplant recipients are at risk for developing CMV disease, seronegative recipients of allografts from seropositive donors are at the highest risk for CMV infection and consequently for graft loss due to accelerated TVS [1-3]. When an allograft from a CMV latently infected donor is transplanted into a naïve (uninfected) recipient, the immediate environment is one of inflammation due to ischemia/reperfusion and the obligatory immune suppression therapy given at the time of transplantation to prevent acute allograft rejection. This environment promotes CMV reactivation, which is considered to be the major source of viral infection. Direct evidence for the role of the transplanted organ as a source of HCMV is supported by finding of HCMV genetic material in the vasculature of transplanted organs [4]. In cardiac transplant recipients, HCMV infection either de novo, or by reactivation in the recipient, nearly doubles the 5-year rate of graft failure due to accelerated TVS [5]. Importantly, kidney transplant graft survival was decreased in asymptomatic HCMV infected recipients during the first 100 days post transplantation compared to patients with no HCMV infection, suggesting that the presence of HCMV alone, whether asymptomatic or manifesting overt infection, has a negative impact on graft survival [6, 7]. Supporting the role of HCMV in the development of CR is the observation that treatment of heart transplant recipients with ganciclovir, a potent inhibitor of viral replication, delayed the development time to allograft rejection in comparison to an untreated HCMV seropositive group [8, 9].
Ganciclovir is costly and has significant side effects such as pancytopenia and nephrotoxicity and ganciclovir resistance is common; hence an understanding of the mechanisms associated with HCMV-associated TVS is important. It has been difficult to determine these mechanisms due to the multifactorial etiology of TVS, in addition, HCMV is ubiquitous throughout the population making it nearly impossible to understand how de novo infection vs. reactivation affects TVS. Using rat transplantation models of CR, we have demonstrated that acute infection of the allograft recipient with RCMV dramatically decreases the mean time to develop TVS and rejection [10-16]. Although these observations demonstrate a link between CMV and TVS, the viral characteristics, including source and replication status associated with the acceleration of disease need further clarification. In this report, we examine how the source (donor vs. recipient) and replication status (acute or latent) of RCMV infection influence the progression to the development of TVS and CR. We also evaluate the impact of ganciclovir therapy on accelerated TVS. The findings from this study have the potential to direct further treatment strategies in humans to minimize the risk of CMV associated CR.
Materials and Methods
Heart transplantation
F344 rat donor hearts were transplanted heterotopically into Lewis recipients [17, 18]. All animals were housed in the Portland Veterans Administration Medical Center animal facility, which is AAALAC accredited and complies with the USDA and HHS requirements for animal care. To determine whether status or source of virus infection affected graft survival either the donor (D) or recipient (R) were infected with 1×105 plaque-forming units (PFU) of the Maastricht strain of RCMV [18-20] intraperitoneally (i.p.) at 1 day post-op (POD) (acute R infection), 7 days pre-op (acute D infection) or 6 months pre-op (latent R and D infection). Uninfected allograft R served as controls. Animals were examined daily for overall health and CR was determined by monitoring heartbeat grade, and upon graft failure the hearts were harvested [17]. Paraffin embedded tissue sections were stained with elastic von Gieson. The extent of neointimal formation (TVS) was determined by the neointimal index (NI=(intima area/lumen+intima area) ×100 [18, 21]. To evaluate the impact of anti-viral therapy on accelerated TVS, the recipients in the ARI group and LDI group were treated with ganciclovir (Roche) (20mg/kg/day i.p. for 45 days following transplantation). The severity of TVS in the uninfected compared to the RCMV-infected graft recipients (+/− ganciclovir) were analyzed using Student’s t test. P values <0.05 were considered statistically significant.
RCMV-specific enzyme-linked immunoabsorbent assay (ELISA)
ELISA plates were coated with RCMV lysates (5μg/ml) overnight, washed and blocked with 2% milk in 0.05% Tween-PBS. Triplicate serial dilutions of serum samples were incubated for 2hrs at room temperature followed by 3 washes with 0.05%Tween-PBS. The plates were incubated with horseradish peroxidase-conjugated anti-rat IgM or IgG for 1hr, followed by addition of chromogen OPD substrate to allow detection and quantification of bound antibody molecules. Average antibody titers are calculated from absorbance data (n=3).
TaqMan PCR detection of RCMV
Total genomic DNA was extracted from rat native and graft hearts and submandibular glands using DNAzol. A total of 0.5μg of DNA was analyzed by TaqMan PCR using a probe/primer set recognizing RCMV DNA polymerase sequence [18, 22]. The sensitivity of detection for this assay was <100 copies.
Illumina Microarray Analysis
Allograft gene expression profiles from recipients of latently infected allografts and uninfected controls was analyzed using Illumina RatRef-12 arrays containing >22,000 probes selected from the NCBI RefSeq database. RNA samples from non-transplanted hearts were the baseline controls. Presence and differential expression of a transcript was determined using Genome Studio by Illumina. Heat maps displaying fold change expression data were generated using the Open Source Software R. Pathway analysis was performed using MetaCore by GeneGO. Normalized expression levels for each gene were compared to uninfected allografts and native hearts using Student’s t-test. The p values of <0.05 were considered significant.
Quantitative RT-PCR
Gene expression was quantified using real-time RT-PCR. Primer sets include: CCL19-Fwd: gaccaagcctcccgatctg, Rev: catgctcacactcacgttcaca; CCL21b-Fwd: cacaggcaaagagggagctaga, Rev: gaagcagacaagggtgtgacaa; CXCL13-Fwd: tccctaagaagcagacgcattac, Rev: gtcgaagcattatttccctcatg. Rat fibroblasts and smooth muscle cells were infected with RCMV and RNA was harvested using Trizol at 8, 16, 24 and 36hpi. cDNA was generated using Superscript III (Invitrogen) and analyzed by real-time PCR. Gene amplicons served as quantification standards (sensitivity ≥100 copies/gene). Results were analyzed using ABI StepOne Real-Time PCR system and normalized to L32 [18, 23].
Immunohistochemistry
Antibodies to rat CD8α (BD Biosciences) and ED1 (Serotec) were incubated with the heart tissue sections for 45min at room temperature in a Dako Immunostainer (39). Subsequently, slides were incubated with mouse-specific peroxidase-conjugated secondary antibodies and developed using a MAB kit (Dako). Slides were counterstained with hematoxylin and visualized by microscopy.
Flow Cytometry
Heart, spleen, and blood samples were harvested from control and infected rats at 21 or 180dpi. Following exanguination, the rats were perfused with PBS. The hearts were digested in CollagenaseD (200 unit/ml) at 37°C for 40min. The spleen and digested heart tissue were macerated through a wire mesh and strained through a 70μm nylon cell strainer. Lymphocytes were separated from heart and spleen tissue suspensions and diluted whole blood using Ficoll-Paque Plus (GE Healthcare). After centrifugation, the white blood cell layer was resuspended in RPMI 1640 media (Mediatech). The number of lymphocytes was determined using a Hemavet HV950 Multispecies Hematologic Analyzer (Drew Scientific). For each sample, 2×106 lymphocytes were stained for 20min with fluorescently labeled anti-rat antibodies to CD4 (APC-Cy7, Biolegend), CD8 (PerCP-Cy5.5, BD Biosciences), and CD62L (PE, BD Biosciences). Cell cycle status was determined by measuring Ki-67 expression (BD Biosciences). The cells were analyzed using an LSRII Flow Cytometer and the data was analyzed with FlowJo software (Tree Star).
Results
RCMV Infection Source and Status Influences the Development of TVS/CR
In human transplant recipients the source of virus infection (donor vs. recipient) and the status of viral infection (active vs. latent) can be used to stratify the risk of developing CMV disease and allograft rejection. In this study, we use a rat heart transplant CR model to characterize the importance of source and status of RCMV as it relates to time to chronic allograft rejection and severity of TVS by performing transplants in acutely (A) or latently (L) RCMV-infected donors (D) or recipients (R). For these experiments, F344 rat hearts were transplanted into Lewis recipients (see Table 1). The D or R rats were acutely (R=day 1post-op; D=7 days pre-op) or latently (6 months pre-op for both D and R) infected with RCMV. Upon graft failure, the hearts were harvested and evaluated histologically for TVS (NI, neointimal index). Acutely infected R (ARI) rejected their naïve donor grafts significantly earlier when compared to naïve recipients of grafts from acutely infected D (POD 43 vs. 53, p=0.01, n=6 and 4 respectively), whereas uninfected controls, receiving uninfected hearts, rejected at day 90 (Figure 1 and Table 1). Similarly, at the time of rejection, the ARI also demonstrated increased TVS formation in their grafts compared to those from ADI (NI=74 vs. 50, p=0.01). Importantly, similar to the human scenario, recipients of latently infected D (LDI) (n=8) rejected their grafts significantly earlier than did the latently infected R of CMV naïve grafts (n=7) (POD 54 vs. 63, p=0.01) with more severe TVS at the earlier rejection time (NI=63 vs. 51, p=0.01) (Figure 1 and Table 1). Viral DNA was detected in the graft tissues and/or salivary glands at rejection in all groups except R of grafts from LDI (Table 2). Despite the absence of detectable virus at endpoint, RCMV-specific antibodies were present in all allograft recipients including those receiving hearts from latently infected donors (Table 2). These data demonstrate that the status of CMV infection (acute vs. latent) and the source (donor vs. recipient infection) affect the degree of CMV-acceleration of TVS and CR.
Table 1.
Status and source of RCMV infection is important for the acceleration of TVS in heart allografts.
| Experimental Design | Transplantation Timeline | n= | Donor Infected (day) |
Recipient Infected (day) |
Rejection POD |
Histology | N.I. (±SD) |
|---|---|---|---|---|---|---|---|
| ADI Acute Donor Infection / pre tx day 7 / Naive Recipient |
|
4 | −7 | - | 53 | CR* | 50±10 |
| ARI Acute Recipient Infection / post tx day 1 / Naive Donor |
|
6 | - | +1 | 43+ | CR | 74±7++ |
| LDI Latent Donor Infection / Pre tx day 180 / Naive Recipient |
|
8 | −180 | - | 54# | CR | 63±7## |
| LRI Latent Recipient Infection / pre tx day 180 / Naive Donor |
|
7 | - | −180 | 62 | CR | 51±7 |
| Control |
|
8 | - | - | 90 | CR | 51±7 |
CR = Chronic Rejection,
= p<0.01 vs. ADI,
= p<0.01 vs. ADI,
= p<0.01 vs. LRI,
= p<0.01 vs. LRI
Figure 1. RCMV infection source (donor vs. recipient) and status (acute and latent) are important for virus acceleration of heart allograft chronic rejection.

Shown is a survival curve for heterotopic heart grafts (F344) transplanted into Lewis recipients and monitored over a 120-day period for clinical rejection. Groups included: 1. ADI (POD 51, donor was infected 7 days prior to transplantation, n=4); 2. ARI (POD 43, recipient was infected POD 1, n=6); 3. LDI (POD 55, donor was infected 180 days prior to transplantation, n=8); 4. LRI (POD 63, recipient was infected 180 prior to transplantation, n=7); and 5. Control (uninfected donor and recipient). + = ARI vs. ADI p<0.01, ++ = LDI vs. LRI p<0.01, ADI/ARI/LDI/LRI accelerate graft rejection vs. control p<0.001.
Table 2.
RCMV Detection and serology of allograft recipients at the time of rejection.
| Recipient Serology |
RCMV DNA Copies/μg | |||
|---|---|---|---|---|
| Native Heart | Graft Heart | Salivary Glands | ||
| ACUTE | ||||
| ADI (n=4) | + | n.d. | n.d. | 1×105 |
| ARI (n=6) | + | 3×103 | 3×102 | 3×107 |
| LATENT | ||||
| LDI (n=8) | + | n.d. | n.d. | n.d. |
| LRI (n=7) | + | n.d. | n.d. | 3×105 |
N.D. = not detected, assay detection limit is 100 copies of the RCMV genome.
Ganciclovir Reduces Acute but not Latent RCMV-accelerated TVS/CR
To assess the role of viral replication in acute and latent RCMV-accelerated rejection, we treated allograft recipients with ganciclovir (n=6), a drug that inhibits late viral gene expression. Ganciclovir reduced RCMV-accelerated CR of allografts in ARI by increasing time to rejection from POD 43 to POD 75 (p<0.001) and reducing TVS from NI=80 to NI=65 (p<0.001) (Table 3). This finding indicates that viral replication/late gene expression is critical in the RCMV-accelerated allograft disease processes in ARI. However, ganciclovir treatment of the recipients of allografts from latently infected donors failed to impact the development of graft TVS when compared to that of the graft in the untreated group (NI=65 vs. 63; p=NS, time to rejection was 52 vs. 54; p=NS (Table 3).
Table 3.
Ganciclovir (20mg/kg/day for 30 days) slows RCMV-accelerated TVS inallografts from acutely infected recipients (ARI) but not in allografts from latently infected donors (LDI).
| Group | n= | Infection Day |
POD | Histological Presentation |
Neoinimal Index (TVS)+/−SD |
|---|---|---|---|---|---|
| LDI | 8 | −180 | 54 | Chronic Rejection | 63+/−7 |
| LDI+Ganciclovir | 6 | −180 | 52 | Chronic Rejection | 65+/−3 |
| ARI | 6 | 1 | 43 | Chronic Rejection | 80+/−5 |
| ARI+Ganciclovir | 6 | 1 | 75* | Chronic Rejection | 65+/−4** |
vs. POD ARI p<0.001,
vs. ARI NI p<0.001
This finding indicates that a fundamental difference exists in the mechanisms that define CMV-accelerated rejection between the ARI and LDI groups. To determine whether this difference is in the sequence of events and/or factors that mediate rejection we analyzed the kinetics of TVS formation in ARI and LDI. Assessment of these grafts revealed that RCMV infection in both LDI and ARI groups significantly increased the severity of TVS in heart allografts at POD28, 35 and 45 compared to uninfected controls (n=4 each). In addition, the rate of NI formation between POD21 and rejection for both groups is significantly different when compared to uninfected controls (i.e. line slope: LDI=1.226, R2=0.9725; ARI=1.699, R2=0.9932; and Uninfected=0.3477, R2=0.8695) (Figure 2). To determine whether RCMV reactivated in the LDI group we quantified the levels of viral genomic DNA in the recipient salivary glands at POD 7-28. Viral DNA was first detected in recipient salivary glands at POD21 (Figure 3). We used RCMV-specific ELISA to detect IgM and IgG levels in the LDI group (donors and recipients). The recipients of allografts from latently infected donors had predominately an IgM response at POD21, whereas the donor group (180dpi) had predominately an IgG antibody response (Figure 4) indicating that RCMV recently reactivated in the recipients of hearts from LDI animals.
Figure 2. RCMV infection accelerates the time to develop TVS in graft hearts.

Comparison of the time to allograft rejection and TVS (mean NI) of graft heart vessels of uninfected controls, RCMV-acutely infected recipients (ARI) and recipients of RCMV-latently infected donor hearts (LDI); n=6 for each. The time to rejection for the ARI and LDI was significantly decreased compared to that of the uninfected controls (p<0.05). Similarly, the increase NI parallels that of the time to rejection (p>0.05).
Figure 3. RCMV detection in salivary glands from recipients of latently infected donor hearts.

RCMV genome copy was determined by real time PCR at 7, 14, 21, and 28 days post transplantation in recipients of latently infected donor hearts (n=4 at each time point).
Figure 4. Recipients of RCMV-latently infected donor hearts have predominately an IgM RCMV-specific Antibody Response.

RCMV-specific ELISA plates were coated with lysates from RCMV infected fibroblasts. Plasma samples from allograft recipients of hearts from latently infected rats (LDI) at POD21, latently infected heart donor rats, acutely infected rats (21dpi) and uninfected controls (n=4 for each). Plasma was diluted 1:50. The secondary anti-rat IgM or IgG antibody conjugated to HRP was used for detection. Recipients of RCMV infected donor hearts have predominantly an IgM antibody response, which is statistically significant compared the IgG response (p<0.05).
RCMV Induces TLO-Gene Expression
We have previously shown that genes involved in inflammation, chemotaxis and wound healing are up-regulated in ARI allografts compared to uninfected controls [18, 23, 25]. In the current study, we used DNA microarrays to analyze gene expression in recipient allografts from LDI. The fold change values of the allograft (n=3 for each of the infected and uninfected transplant groups) were compared to uninfected non-transplanted hearts (n=4). In general, allografts from the LDI group had increased expression of a number of genes involved in inflammation and wound healing including a number of cytokines (IL1β, IL6, IL16, IL18, LtB), chemokines (CCLs 2, 3, 4, 5, 7 & 19, CXCLs 1, 2, 9, 10, 11, 12 & 13), cell adhesion molecules (integrins α1, α4, α11, αL, & β2, ICAM-1 and VCAM-1), inflammatory signaling molecules (Hck, Csk, Lck, Apobec 1, IRFs 5 & 7, TLRs 2 & 6, and Jaks 2 & 3) as well as genes that encode proteins with extracellular matrix remodeling activities (MMPs 3, 7, 12, & 14; TIMP-1, cathepsins E, S & W) and growth factor activities (angiogenin, angiopoietin, connective tissue growth factor, endothelin, osteopontin, thrombospondin-2, TGF) (Figure 5). Up-regulation of these genes has the potential to promote allograft rejection by increasing alloreactivity and promoting unchecked wound repair and fibrosis. Interestingly, a few genes were down-regulated in comparison to normal heart tissues and allografts from uninfected recipients. These included a family of fibroblast growth factors (FGF 1, 12, 13 and 16) as well as the TGF-β receptor; however, the implications of down-regulating these genes is yet to be realized, and warrants further investigation. Pathway analysis of the microarray data from latently infected allografts following transplantation indicated an early (day 7) and sustained production of TLO-associated genes compared to uninfected allografts. These genes included the homeostatic chemokines CCL19, CCL21, and CXCL13, lymphocyte adhesion molecules ICAM-1, MAdCAM, and VCAM-1 and Lymphotoxin B (Table 4), suggesting that cardiac allografts from latently infected donors undergo active lymphoid neogenesis, and more specifically the generation of tertiary lymphoid organs (TLOs).
Figure 5. Gene expression profiling identifies differences between allografts from recipients of latently infected donors and uninfected controls.

Microarrays were prepared from total graft and native hearts from recipients of latently infected grafts (LDI) and uninfected controls (Mock) harvested at 7, 14, 21, 28, and 35-post transplantation. Shown is a heat map representing the fold change values of the allograft (n=3) compared to uninfected non-transplanted heart (n=4). The data shown is limited to the identified genes displaying significant dysregulation (up or down) when compared to control hearts (LDI vs. mock infected p<0.05 for at least one of the time points). The data were subdivided into categories based upon predicted biological function using the computer program GeneGo by Metagene.
Table 4. Early and sustained induction of TLO associated genes in allografts from latently infected donors.
Shown are fold change values compared to uninfected non-transplanted hearts (n=3 for each time point).
| Latently Infected Donor (LDI) |
Uninfected Control |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| POD | 7 | 14 | 21 | 28 | 35 | 7 | 14 | 21 | 28 | 35 |
|
|
|
|||||||||
| CCL19 | 4.5* | 6.6 | 14.8 | 17.4 | 11.8 | 1.6 | 2.7 | 7.8 | 10.2 | 7.8 |
| CCL21 | 1.4 | 2.1 | 3.1* | 2.2* | 1.0 | 1.2 | 1.4 | 1.1 | 0.7 | 0.8 |
| CXCL13 | 9.0* | 10.0 | 11.9 | 13.2 | 13.8 | 3.2 | 7.1 | 4.9 | 9.8 | 11.8 |
|
|
|
|||||||||
| ICAM-1 | 3.9* | 3.7 | 4.5 | 5.9 | 5.2 | 1.3 | 3.1 | 2.7 | 3.1 | 5.6 |
| MADCAM1 | 1.1* | 2.0 | 1.3 | 3.0 | 1.7 | 0.8 | 1.2 | 0.8 | 0.9 | 0.9 |
| VCAM-1 | 3.6* | 4.9 | 6.0 | 7.7 | 7.7 | 1.7 | 4.4 | 3.2 | 5.0 | 6.8 |
|
|
|
|||||||||
| Lymphotoxin A | 0.7 | 0.6 | 1.0 | 1.2 | 0.9 | 0.6 | 0.5 | 0.7 | 0.9 | 0.9 |
| Lymphotoxin B | 4.5* | 3.8* | 6.4 | 10.7* | 11.4 | 2.0 | 2.2 | 3.1 | 6.4 | 12.2 |
{p<0.05 infected vs. uninfected}
TLO formation is initiated by lymphoid tissue inducer cells (LTi) that function to up-regulate cell adhesion molecule, cytokine and chemokine expression in stromal cells [26-36]. The production of the homeostatic chemokines CXCL13, CCL21b and CCL19 are crucial for the formation of TLOs [32, 37]. Typically, a number of different cell types can act as stromal cells including fibroblasts, endothelial cells, immune cells, and pericytes that can differentiate into smooth muscle cells (SMC). We next determined whether RCMV infection directly promotes the activation of a TLO phenotype in rat fibroblasts and SMC by measuring homeostatic chemokine gene synthesis. While low levels of CXCL13 and CCL19 are present in uninfected SMC, RCMV infection dramatically increased expression of CCL19, CCL21b and CXCL13 (Figure 6). In fibroblasts, RCMV infection also induced CCL21b and CXCL13 but did not increase expression of CCL19. Expression of these TLO-associated chemokines is critical for the initial stages of lymphoid neogenesis leading to the recruitment of lymphocytes and additional LTi’s [38].
Figure 6. RCMV Infection Induces SMC and Fibroblast Expression of TLO Associated Chemokines.

Quantitative RT-PCR was used to quantify CCL19 (A), CCL21b (B), and CXCL13 (C) chemokine expression in samples of RCMV infected primary fibroblasts and aortic SMC. RNA samples were analyzed in triplicate and normalized to expression of L32 (cellular ribosomal protein), and the relative copy number was determined using amplicon standards to each gene.
Latent RCMV Infected Pre-Transplant Hearts Contain Increased Levels of Macrophages And T-cells
Given that latent RCMV infection induces the expression of TLO associated genes, we performed immunohistochemistical staining for T-cells and macrophages on sections from latently infected (180 dpi) non-transplanted hearts as well as uninfected control hearts. We detected increased numbers of resident ED1+ macrophages and CD8α+ T-cells within the hearts from each of the latently infected animals but not in uninfected controls (n=3) (Figure 7). In addition, within the hearts from the three latently infected animals, both macrophages and CD8α positive T-cells were observed in large clusters. In contrast, clusters of macrophages and/or T-cells were absent in hearts from uninfected animals.
Figure 7. Hearts from RCMV latently infected rats contain macrophages and T cells.
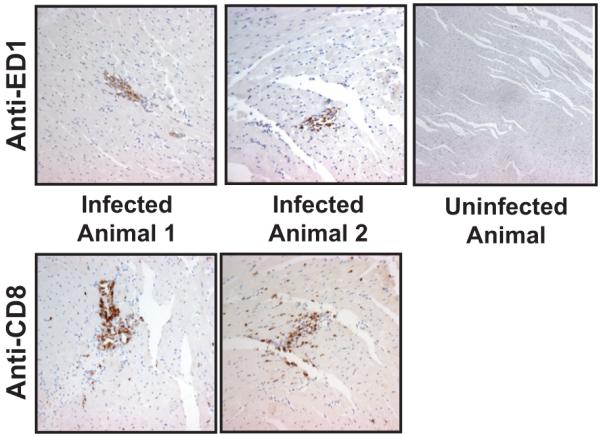
Paraffin embedded heart sections from latently infected rats or controls were probed using antibodies directed against ED1 (Mac) and CD8 (T-cell). (mag=40x).
We then characterized the T-cell subsets from PBMC, splenocytes, and heart-derived lymphocytes from RCMV-infected donor hearts at 21 and 180dpi using flow cytometry, and compared them to cells harvested from uninfected control rats (n=4 for each group). We found the frequency of CD4 T-cells is increased in hearts from latently infected donors compared to both uninfected controls and tissues from rats infected for 21 days (27.8% vs. 20.4% and 20.4%; respectively) (Figure 8). However, the percentage of CD4 T-cells in the blood and spleen from latently infected rats is significantly reduced compared to both uninfected controls and at 21dpi (blood: 39.1% vs. 49.5% and 51.3%, respectively; p<0.001 and spleen: 23.9% vs. 31.5% and 35.6%, respectively; p<0.001) (Table 5). Frequency of CD8 T-cells is similarly increased in the infected hearts compared to uninfected controls with the percentage escalating from 19.5% at 21dpi to 37.1% at 180dpi (Figure 8). In contrast to CD4 T-cells, the percentage of CD8+ T-cells also increases in both blood (20.4% vs. 20.9% and 26%, respectively) and spleen (13.3% vs. 15.5% and 24.6%, respectively) of latently infected animals.
Figure 8. Latently infected rat hearts contain activated CD4 and CD8 T cells.

Peripheral blood was harvested from uninfected rats or rats infected with RCMV at either 21 or 180 dpi (n=4 for each). Total lymphocytes were isolated from the rat hearts and these cells were stained for the cell surface markers CD4, CD8, and CD62 as well as for the intracellular marker of cellular proliferation, Ki67 and then analyzed by flow cytometry. Hearts from latently infected rats contain a significantly increased level of activated CD4+ and CD8+ effector T-cells compared to hearts from uninfected controls (p<0.05).
Table 5.
Phenotyping of T cells from spleen and blood of RCMV infected rats at 21 and 180 dpi. n=6
| Uninfected Control |
Infected 21 dpi |
Infected 180 dpi |
||
|---|---|---|---|---|
| CD4+ (% lymphocytes) |
Blood | 51.3 ± 3.6 | 49.5 ± 1.5 | 39.1 ± 3.5 |
| Spleen | 35.6 ± 2.1 | 31.5 ± 2.2 | 23.9 ± 3.3 | |
|
|
||||
| CD4+CD62Llow (% CD4) |
Blood | 7.2 ± 1.5 | 18.7 ± 1.7 | 17.3 ± 3.1 |
| Spleen | 16.4 ± 5.2 | 27.8 ± 5 | 40.9 ± 5.5 | |
|
|
||||
| CD4+CD62LlowKi67+ (% CD4) |
Blood | 1.8 ± 0.4 | 2.6 ± 0.2 | 3.7 ± 1.1 |
| Spleen | 2.8 ± 0.6 | 5.1 ± 1.3 | 7.4 ± 1.4 | |
|
|
||||
| CD8 (% lymphocytes) |
Blood | 20.4 ± 1.1 | 20.9 ± 1.6 | 26.0 ± 3.8 |
| Spleen | 13.3 ± 1.9 | 15.5 ± 1.9 | 24.6 ± 3.2 | |
|
|
||||
| CD8+CD62Llow (% CD8) |
Blood | 3.3 ± 1.8 | 16.1± 2.3 | 11.7 ± 5.5 |
| Spleen | 11.3 ± 1.9 | 19.7 ± 4.7 | 18.1 ± 3.3 | |
|
|
||||
| CD8+CD62LlowKi67+ (% CD8) |
Blood | 37.1 ± 8.2 | 46.7 ± 4.8 | 40.2 ± 12.0 |
| Spleen | 25.9 ± 2.9 | 35.3 ± 6.5 | 39.0 ± 7.2 | |
We also found that the frequency of CD4 and CD8 effector memory cells increased in a time-dependent fashion, with the latently infected donors exhibiting significantly higher levels in spleen (Table 5) and heart (Figure 8) compared to those from rats at 21dpi (p<0.001). In fact, 93.6% of CD4+ T cells and 88.7% of CD8+ T-cells are of the effector memory phenotype in the heart tissues from rats at 180dpi (Figure 8). Interestingly, both the CD4+ and CD8+ T-cells isolated from the infected heart tissues were actively proliferating prior to the time of harvest as evidenced by the presence of Ki67, a marker of recent entry into the cell cycle (Figure 8). These findings demonstrate that the RCMV latently infected donor hearts contain a substantial number of activated T-cells prior to transplantation.
Discussion
The donor and recipient HCMV serologic status may be the most important factor in determining the development of severe HCMV disease and consequently accelerated allograft CR. In the current report, we examined the effects of RCMV infection status and source on the acceleration of TVS and CR using a rat heart transplant model. We show the source of infection (donor vs. recipient) and the infection status (acute vs. latent) prior to transplantation had a major impact on allograft survival. The ARI group rejected with the most rapid kinetics (POD43) and most severe TVS (NI=74). While both the ADI and LDI groups rejected at nearly the same time (POD53 vs. 54, respectively), the allograft TVS associated with the LDI was significantly more severe than the ADI group (NI=63 vs. 50, respectively). As expected from the experience with human solid organ transplants, the latently infected recipient group (LRI) rejected with the slowest kinetics (POD62) with a degree of TVS similar to uninfected controls at POD90 (NI=50 vs. 51, respectively). The overall effect of status and source of virus on RCMV-accelerated rat heart allograft rejection parallels the human transplantation scenario [5].
The best evidence supporting a role for HCMV in the development of TVS/CR is that treatment with ganciclovir delays the time to allograft rejection [8, 9]. In our study, inhibiting active viral replication by treating the ARI group with ganciclovir substantially increased the time to rejection from POD42 to 75. This was in contrast to treatment of the LDI group, which was not affected by ganciclovir and developed allograft vasculopathy similar to the untreated group. This novel finding suggests the mechanisms of rejection and requirements for viral replication differ between the two groups. In the ARI group, virus replication is crucial for the infection of the allograft heart, which potentiates the host immune response to both virus and graft leading to an early rejection process. Whereas in the LDI model, persistently infected cells already exist within the allograft and viral immediate early and/or early genes are, for the most part, responsible for inducing CR. In addition, the donor immune response to the latent virus may prime the allograft for rejection by recruiting immune cells into the allograft prior to transplantation. We found that hearts from RCMV latently infected rats have an abundance of macrophages and T cells organized in TLOs. Uninfected rats were devoid of these structures and they did not harbor activated cardiac T-lymphocytes. TLOs are aggregates of immune cells that form under conditions of chronic inflammation in tissues that do not normally contain lymphoid organs. Similar to peripheral lymphoid structures, TLOs are scaffolds for immune activation. TLOs form during many different chronic inflammatory diseases including arthritis, atherosclerosis, cancer, and following various viral infections [26-36]. TLOs initially form through the recruitment of LTi’s that stimulate the surrounding stromal cells and up-regulate expression of cell adhesion molecules and homeostatic chemokines, which effectively attract lymphocytes. A genome-wide microarray analysis of heart allografts from infected (LDI) and uninfected transplant groups revealed up-regulation of genes involved in inflammation and tissue remodeling processes. These genes included a number of chemokines such as CCL2, 3, 4, 5, 7 and 19 as well as CXCL 9, 10, and 13, known to recruit inflammatory cells. Similarly, adhesion molecule expression was more highly induced in the allografts from latently infected donors compared to uninfected allografts including integrins, selectins as well as ICAM-1 and V-CAM-1. These adhesion molecules promote the extravasation and tissue localization of the chemoattracted immune cells. While a number of cytokines were up-regulated in the allografts from the LDI group compared to uninfected controls the most highly expressed included IL-1β, IL-6, IL-16, IL-18 and lymphotoxin β. These cytokines are mediators of cell-mediated immunity and could thus enhance the allo-response in the latently infected donors. Importantly, we found that by POD7, heart allografts from latently infected donors demonstrated a significant increase in TLO chemokine, adhesion molecule and LTβ expression compared to allografts from uninfected donor rats. It is thought that expression of the homeostatic chemokines, alone, can initiate TLO formation by attracting lymphocytes and LTi’s, this is particularly true of CXCL13 [38]. RCMV infection of SMC and fibroblasts, which are potential cardiac stromal cells, increases expression of the homeostatic chemokines including CXCL13. However, RCMV infection did not affect LTα, LTβ, or LTβR expression (data not shown). Interestingly, mouse aortic SMC have been shown to differentiate into LTi’s upon treatment with both TNF and an agonistic anti-LTβR mAb, and the production of these cells is associated with the development of atherosclerosis in mice [46]. We and others have demonstrated that MCMV accelerates atherosclerosis; however, the effects of CMV on TLO formation in this model were never specifically addressed. Along these lines, Vleigen et al. using similar models, found that MCMV promotes the accumulation of T cells and macrophages in the atherosclerotic vasculature [47-51]. It is quite likely that persistent CMV infections associated with the vasculature or heart tissues can promote TLO formation by activating genes involved in lymphoid neogenesis, which ultimately enhances inflammatory processes that are specifically associated with chronic disease.
TLOs have also been associated with allograft rejection [44, 45] by supporting naïve T cell activation and the generation of effector and memory T-cells [34]. In deed, we detected the presence of T-cell and macrophage foci in latently infected hearts but not in the hearts from uninfected rats. This finding implies that latent RCMV infections, while asymptomatic, are persistent in the heart tissues and that these tissues harbor active anti-viral reactive immune cells. Approximately, 90% of the CD4+ and CD8+ T cells present in the hearts of latently infected rats were of the effector phenotype, whereas, those found within hearts of control animals or those infected for 21 days were not. Both CD4+ and CD8+ T-cells present in latently infected hearts were Ki67+ suggesting they were actively replicating. Interestingly, CMV infections are known to promote long-term alterations of T-cell profiles including T-cell memory inflation and the accumulation of effector memory CD8 T-cells [40-43]. These data suggest that the presence of effector CD4+ and CD8+ T-cells promote graft rejection via enhancing the local alloreactive immune response even in the absence of replicating virus (ganciclovir treated LDI group).
In summary, we show that, similar to the clinical transplant scenario, naïve recipients of heart allografts from latently infected donors undergo CR acceleration with similar kinetics to acutely infected recipients. Interestingly, treatment of recipients of latently infected donor hearts with ganciclovir did not prevent TVS and CR, which differs from its effect on rejection in ARI. We show that TLOs form in hearts of latently infected rats and transplantation of these hearts into naïve recipients significantly accelerates the time to rejection. Our data suggest that latent CMV infection induces TLO formation in the graft prior to procurement and this subsequently leads to enhancement of immune activation following transplantation. Further studies defining the mechanisms of this complex interplay between virus and TLO formation are necessary to develop appropriate therapeutic strategies to prevent and treat this important clinical transplantation problem.
Acknowledgements
This work was supported by research grants to S.L. Orloff from the Department of Veterans Affairs and from the National Institutes of Health (HL 66238-01) and grants from the National Institutes of Health to D.N. Streblow (HL083194) and J.A. Nelson (HL 088603). D.N. Streblow and I. Messaoudi are supported by AHA Scientist Development Grants.
References
- 1.Almond PS, Matas A, Gillingham K, Dunn DL, Payne WD, Gores P, Gruessner R, Najarian JS. Risk factors for chronic rejection in renal allograft recipients. Transplant. 1993;55(4):752–6. doi: 10.1097/00007890-199304000-00013. discussion 756-7. [DOI] [PubMed] [Google Scholar]
- 2.Isoniemi H, Nurminen M, Tikkanen MJ, von Willebrand E, Krogerus L, Ahonen J, Eklund B, Hockerstedt K, Salmela K, Hayry P. Risk factors predicting chronic rejection of renal allografts. Transplantation. 1994;57(1):68–72. doi: 10.1097/00007890-199401000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Fitzgerald JT, Gallay B, Taranto SE, McVicar JP, Troppmann C, Chen X, McIntosh MJ, Perez RV. Pretransplant recipient cytomegalovirus seropositivity and hemodialysis are associated with decreased renal allograft and patient survival. Transplantation. 2004;77(9):1405–11. doi: 10.1097/01.tp.0000122184.97674.20. [DOI] [PubMed] [Google Scholar]
- 4.Gnann JW, Jr., Ahlmen J, Svalander C, Olding L, Oldstone MB, Nelson JA. Inflammatory cells in transplanted kidneys are infected by human cytomegalovirus. Am J Pathol. 1988;132(2):239–48. [PMC free article] [PubMed] [Google Scholar]
- 5.Grattan MT, Moreno-Cabral CE, Starnes VA, Oyer PE, Stinson EB, Shumway NE. Cytomegalovirus infection is associated with cardiac allograft rejection and atherosclerosis. JAMA. 1989;261(24):3561–6. [PubMed] [Google Scholar]
- 6.Sagedal S, Hartmann A, Nordal KP, Osnes K, Leivestad T, Foss A, Degre M, Fauchald P, Rollag H. Impact of early cytomegalovirus infection and disease on long-term recipient and kidney graft survival. Kidney Int. 2004;66(1):329–37. doi: 10.1111/j.1523-1755.2004.00735.x. [DOI] [PubMed] [Google Scholar]
- 7.Wu TC, Hruban RH, Ambinder RF, Pizzorno M, Cameron DE, Baumgartner WA, Reitz BA, Hayward GS, Hutchins GM. Demonstration of cytomegalovirus nucleic acids in the coronary arteries of transplanted hearts. Am J Pathol. 1992;140(3):739–47. [PMC free article] [PubMed] [Google Scholar]
- 8.Merigan TC, Renlund DG, Keay S. A controlled trial of ganciclovir to prevent cytomegalovirus disease after heart transplantation. N Engl J Med. 1992;326:1182–6. doi: 10.1056/NEJM199204303261803. e. al. [DOI] [PubMed] [Google Scholar]
- 9.Valantine HA, Gao SZ, Santosh G. Impact of prophylactic immediate post-transplant ganciclovir on development of transplant atherosclerosis. A post-hoc analysis of a randomized, placebo-controlled study. Circulation. 1999;100:61–66. doi: 10.1161/01.cir.100.1.61. e. al. [DOI] [PubMed] [Google Scholar]
- 10.Soule JL, Streblow DN, Andoh TF, Kreklywich CN, Orloff SL. Cytomegalovirus accelerates chronic allograft nephropathy in a rat renal transplant model with associated provocative chemokine profiles. Transplant Proc. 2006;38(10):3214–20. doi: 10.1016/j.transproceed.2006.10.187. [DOI] [PubMed] [Google Scholar]
- 11.Lautenschlager I, Soots A, Krogerus L, Inkinen K, Kloover J, Loginov R, Holma K, Kauppinen H, Bruggeman C, Ahonen J. Time-related effects of cytomegalovirus infection on the development of chronic renal allograft rejection in a rat model. Intervirology. 1999;42(5-6):279–84. doi: 10.1159/000053961. [DOI] [PubMed] [Google Scholar]
- 12.Lautenschlager I, Soots A, Krogerus L, Kauppinen H, Saarinen O, Bruggeman C, Ahonen J. CMV increases inflammation and accelerates chronic rejection in rat kidney allografts. Transplant. Proc. 1997;29:802–3. doi: 10.1016/s0041-1345(96)00109-1. [DOI] [PubMed] [Google Scholar]
- 13.Martelius T, Krogerus L, Hockerstedt K, Bruggeman C, Lautenschlager I. Cytomegalovirus infection is associated with increased inflammation and severe bile duct damage in rat liver allografts. Hepatology. 1998;27(4):996–1002. doi: 10.1002/hep.510270415. [DOI] [PubMed] [Google Scholar]
- 14.Orloff SL, Yin Q, Coreless CL, Loomis CB, Rabkin JM, Wagner CR. A rat small bowel transplant model of chronic rejection: histopathologic characteristics. Transplantation. 1999;68(6):766–79. doi: 10.1097/00007890-199909270-00008. [DOI] [PubMed] [Google Scholar]
- 15.Orloff SL. Elimination of donor-specific alloreactivity by bone marrow chimerism prevents cytomegalovirus accelerated transplant vascular sclerosis in rat small bowel transplants. J. Clin. Virol. 1999;12(2):142. (G3-25) [Google Scholar]
- 16.Orloff SL, Yin Q, Corless CL, Orloff MS, Rabkin JM, Wagner CR. Tolerance induced by bone marrow chimerism prevents transplant vascular sclerosis in a rat model of small bowel transplant chronic rejection. Transplantation. 2000;69(7):1295–303. doi: 10.1097/00007890-200004150-00015. [DOI] [PubMed] [Google Scholar]
- 17.Orloff SL, Streblow DN, Soderberg-Naucler C, Yin Q, Kreklywich C, Corless CL, Smith PA, Loomis CB, Mills LK, Cook JW, Bruggeman CA, Nelson JA, Wagner CR. Elimination of donor-specific alloreactivity prevents cytomegalovirus-accelerated chronic rejection in rat small bowel and heart transplants. Transplantation. 2002;73(5):679–88. doi: 10.1097/00007890-200203150-00005. [DOI] [PubMed] [Google Scholar]
- 18.Streblow DN, Kreklywich C, Yin Q, De La Melena VT, Corless CL, Smith PA, Brakebill C, Cook JW, Vink C, Bruggeman CA, Nelson JA, Orloff SL. Cytomegalovirus-mediated upregulation of chemokine expression correlates with the acceleration of chronic rejection in rat heart transplants. J Virol. 2003;77(3):2182–94. doi: 10.1128/JVI.77.3.2182-2194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruggeman CA, Schellekens H, Grauls G, Debie WM, van Boven CP. Rat cytomegalovirus: induction of and sensitivity to interferon. Antiviral Res. 1983;3(5-6):315–24. doi: 10.1016/0166-3542(83)90039-6. [DOI] [PubMed] [Google Scholar]
- 20.Bruggeman CA, Meijer H, Bosman F, van Boven CP. Biology of rat cytomegalovirus infection. Intervirology. 1985;24(1):1–9. doi: 10.1159/000149612. [DOI] [PubMed] [Google Scholar]
- 21.Armstrong AT, Strauch AR, Starling RC, Sedmak DD, Orosz CG. Morphometric analysis of neointimal formation in murine cardiac allografts. Transplant. 1997;63:941–7. doi: 10.1097/00007890-199704150-00006. [DOI] [PubMed] [Google Scholar]
- 22.Streblow DN, Kreklywich CN, Smith P, Soule JL, Meyer C, Yin M, Beisser P, Vink C, Nelson JA, Orloff SL. Rat cytomegalovirus-accelerated transplant vascular sclerosis is reduced with mutation of the chemokine-receptor R33. Am J Transplant. 2005;5(3):436–42. doi: 10.1111/j.1600-6143.2004.00711.x. [DOI] [PubMed] [Google Scholar]
- 23.Streblow DN, Kreklywich CN, Andoh T, Moses AV, Dumortier J, Smith PP, Defilippis V, Fruh K, Nelson JA, Orloff SL. The role of angiogenic and wound repair factors during CMV-accelerated transplant vascular sclerosis in rat cardiac transplants. Am J Transplant. 2008;8(2):277–87. doi: 10.1111/j.1600-6143.2007.02062.x. [DOI] [PubMed] [Google Scholar]
- 24.Britt WJ, Alford CA. Cytomegalovirus. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. Lippincott-Raven Publishers; Philadelphia: 1996. pp. 2493–523. [Google Scholar]
- 25.Streblow DN, Dumortier J, Moses AV, Orloff SL, Nelson JA. Mechanisms of cytomegalovirus-accelerated vascular disease: induction of paracrine factors that promote angiogenesis and wound healing. Curr Top Microbiol Immunol. 2008;325:397–415. doi: 10.1007/978-3-540-77349-8_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grabner R, Lotzer K, Dopping S, Hildner M, Radke D, Beer M, Spanbroek R, Lippert B, Reardon CA, Getz GS, Fu YX, Hehlgans T, Mebius RE, van der Wall M, Kruspe D, Englert C, Lovas A, Hu D, Randolph GJ, Weih F, Habenicht AJ. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J Exp Med. 2009;206(1):233–48. doi: 10.1084/jem.20080752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Pare PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–53. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 28.Kendall PL, Yu G, Woodward EJ, Thomas JW. Tertiary lymphoid structures in the pancreas promote selection of B lymphocytes in autoimmune diabetes. J Immunol. 2007;178(9):5643–51. doi: 10.4049/jimmunol.178.9.5643. [DOI] [PubMed] [Google Scholar]
- 29.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14(2):164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148(1-2):11–23. doi: 10.1016/j.jneuroim.2003.10.056. [DOI] [PubMed] [Google Scholar]
- 31.Marinkovic T, Garin A, Yokota Y, Fu YX, Ruddle NH, Furtado GC, Lira SA. Interaction of mature CD3+CD4+ T cells with dendritic cells triggers the development of tertiary lymphoid structures in the thyroid. J Clin Invest. 2006;116(10):2622–32. doi: 10.1172/JCI28993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Kusser K, Randall TD. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc Natl Acad Sci U S A. 2007;104(25):10577–82. doi: 10.1073/pnas.0700591104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. 2006;116(12):3183–94. doi: 10.1172/JCI28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasr IW, Reel M, Oberbarnscheidt MH, Mounzer RH, Baddoura FK, Ruddle NH, Lakkis FG. Tertiary lymphoid tissues generate effector and memory T cells that lead to allograft rejection. Am J Transplant. 2007;7(5):1071–9. doi: 10.1111/j.1600-6143.2007.01756.x. [DOI] [PubMed] [Google Scholar]
- 35.Baddoura FK, Nasr IW, Wrobel B, Li Q, Ruddle NH, Lakkis FG. Lymphoid neogenesis in murine cardiac allografts undergoing chronic rejection. Am J Transplant. 2005;5(3):510–6. doi: 10.1111/j.1600-6143.2004.00714.x. [DOI] [PubMed] [Google Scholar]
- 36.GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D, Osterhaus AD, Hendriks R, Rimmelzwaan GF, Lambrecht BN. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J Exp Med. 2009;206(11):2339–49. doi: 10.1084/jem.20090410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rangel-Moreno J, Moyron-Quiroz J, Kusser K, Hartson L, Nakano H, Randall TD. Role of CXC chemokine ligand 13, CC chemokine ligand (CCL) 19, and CCL21 in the organization and function of nasal-associated lymphoid tissue. J Immunol. 2005;175(8):4904–13. doi: 10.4049/jimmunol.175.8.4904. [DOI] [PubMed] [Google Scholar]
- 38.Honda K, Nakano H, Yoshida H, Nishikawa S, Rennert P, Ikuta K, Tamechika M, Yamaguchi K, Fukumoto T, Chiba T, Nishikawa SI. Molecular basis for hematopoietic/mesenchymal interaction during initiation of Peyer’s patch organogenesis. J Exp Med. 2001;193(5):621–30. doi: 10.1084/jem.193.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tu W, Potena L, Stepick-Biek P, Liu L, Dionis KY, Luikart H, Fearon WF, Holmes TH, Chin C, Cooke JP, Valantine HA, Mocarski ES, Lewis DB. T-cell immunity to subclinical cytomegalovirus infection reduces cardiac allograft disease. Circulation. 2006;114(15):1608–15. doi: 10.1161/CIRCULATIONAHA.105.607549. [DOI] [PubMed] [Google Scholar]
- 40.Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, Phillips RE, Klenerman P. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J Immunol. 2003;170(4):2022–9. doi: 10.4049/jimmunol.170.4.2022. [DOI] [PubMed] [Google Scholar]
- 41.Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J Immunol. 2006;177(1):450–8. doi: 10.4049/jimmunol.177.1.450. [DOI] [PubMed] [Google Scholar]
- 42.Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity. 2008;29(4):650–9. doi: 10.1016/j.immuni.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snyder CM, Loewendorf A, Bonnett EL, Croft M, Benedict CA, Hill AB. CD4+ T cell help has an epitope-dependent impact on CD8+ T cell memory inflation during murine cytomegalovirus infection. J Immunol. 2009;183(6):3932–41. doi: 10.4049/jimmunol.0900227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thaunat O, Nicoletti A. Lymphoid neogenesis in chronic rejection. Curr Opin Organ Transplant. 2008;13(1):16–9. doi: 10.1097/MOT.0b013e3282f3df15. [DOI] [PubMed] [Google Scholar]
- 45.Thaunat O, Patey N, Morelon E, Michel JB, Nicoletti A. Lymphoid neogenesis in chronic rejection: the murderer is in the house. Curr Opin Immunol. 2006;18(5):576–9. doi: 10.1016/j.coi.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 46.Lotzer K, Dopping S, Connert S, Grabner R, Spanbroek R, Lemser B, Beer M, Hildner M, Hehlgans T, van der Wall M, Mebius RE, Lovas A, Randolph GJ, Weih F, Habenicht AJ. Mouse Aorta Smooth Muscle Cells Differentiate Into Lymphoid Tissue Organizer-Like Cells on Combined Tumor Necrosis Factor Receptor-1/Lymphotoxin {beta}-Receptor NF-{kappa}B Signaling. Arterioscler Thromb Vasc Biol. 2010;30(3):395–402. doi: 10.1161/ATVBAHA.109.191395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burnett MS, Gaydos CA, Madico GE, Glad SM, Paigen B, Quinn TC, Epstein SE. Atherosclerosis in apoE knockout mice infected with multiple pathogens. J Infect Dis. 2001;183(2):226–231. doi: 10.1086/317938. [DOI] [PubMed] [Google Scholar]
- 48.Streblow DN, Orloff SL, Nelson JA. Do pathogens accelerate atherosclerosis? J Nutr. 2001;131(10):2798S–2804S. doi: 10.1093/jn/131.10.2798S. [DOI] [PubMed] [Google Scholar]
- 49.Vliegen I, Duijvestijn A, Grauls G, Herngreen S, Bruggeman C, Stassen F. Cytomegalovirus infection aggravates atherogenesis in apoE knockout mice by both local and systemic immune activation. Microbes Infect. 2004;6(1):17–24. doi: 10.1016/j.micinf.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 50.Vliegen I, Duijvestijn A, Stassen F, Bruggeman C. Murine cytomegalovirus infection directs macrophage differentiation into a pro-inflammatory immune phenotype: implications for atherogenesis. Microbes Infect. 2004;6(12):1056–62. doi: 10.1016/j.micinf.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 51.Vliegen I, Stassen F, Grauls G, Blok R, Bruggeman C. MCMV infection increases early T-lymphocyte influx in atherosclerotic lesions in apoE knockout mice. J Clin Virol. 2002;25(Suppl 2):S159–71. doi: 10.1016/s1386-6532(02)00095-1. [DOI] [PubMed] [Google Scholar]