

Abstract
Obesity and cardiovascular disease are among the world's leading causes of death, especially in Western countries where consumption of high caloric food is commonly accompanied by low physical activity. This lifestyle often leads to energy imbalance, obesity, diabetes and their associated metabolic disorders, including cardiovascular diseases. It has become increasingly recognized that obesity and cardiovascular disease are metabolically linked, and a better understanding of this relationship requires that we uncover the fundamental genetic mechanisms controlling obesity-related heart dysfunction, a goal that has been difficult to achieve in higher organisms with intricate metabolic complexity. However, the high degree of evolutionary conservation of genes and signalling pathways allows researchers to use lower animal models such as Drosophila, which is the simplest genetic model with a heart, to uncover the mechanistic basis of obesity-related heart disease and its likely relevance to humans. Here, we discuss recent advances made by using the power of the Drosophila as a powerful model to investigate the genetic pathways by which a high fat diet may lead to heart dysfunction.
Keywords: triglycerides, obesity, heart dysfunction, genetic control
Introduction
Vertebrate animal models (rodents, frogs and fish) have contributed greatly to our understanding of human biology and physiology. However, these models are genetically and metabolically complex, which limits their ability to provide insight into fundamental processes. Invertebrate model organisms, such as Saccharomyces cerevisiae, Caenorhabditis elegans and Drosophila melanogaster are genetically simpler, have less redundancy, and less complex metabolism; they have thus been instrumental in dissecting fundamental pathways and mechanisms. Among the invertebrate models, Drosophila is the only one with a heart, it has a short lifespan, and there are established genetic tools for analysing heart function. Importantly, most gene families and signalling pathways are highly conserved between flies and humans 12, making Drosophila the organism of choice among the invertebrate models to perform heart function-related studies. The Drosophila heart develops in a homologous fashion to vertebrate hearts, including that of humans 13, and functions in an analogous manner to pump the blood (haemolymph) through the body cavity in an open circulatory system 14. Therefore, despite the fact that the mature Drosophila and vertebrate hearts are quite different morphologically, the initial developmental processes and the basic functional properties and molecular constituents are remarkably conserved. This is exemplified by the tinman gene, first discovered in the fly 16, which turned out to be the first gene found critical for heart determination in all species. Since the discovery of tinman, Drosophila has been established as a model of many forms of congenital heart disease, cardiomyopathies and of cardiac ageing. Here, we discuss the use of Drosophila as a model for studying obesity and heart dysfunction as a consequence of excess dietary fat intake.
High fat diet feeding of Drosophila causes excess fat accumulation
The critical organs for glucose and lipid metabolism in vertebrates and mammals also have counterparts in insects, including Drosophila. For example, the functions of the liver and adipose tissue in mammals are assumed by the fat body and specialized oenocyte cells in the fly. The fly also has insulin producing cells and cells producing the glucagon-like hormone AKH 24, which are analogous to mammalian pancreatic beta and alpha cells respectively. Collectively, these tissues integrate genetic, environmental and dietary signals to control energy balance and metabolic homeostasis in the fly. This suggests that the central mechanisms controlling fat and glucose metabolism are preserved in Drosophila and likely participate in the development of obesity and obesity-related heart dysfunction.
The worldwide epidemic of diabetes and its associated metabolic disorders are at least partly caused by consumption of high caloric fat- and sugar-enriched foods. Recently, we established an obesity model, in which Drosophila are fed a high fat diet (HFD; 30% coconut oil-enriched food) for several days [28]. Like humans, flies on HFD display symptoms of obesity that include increased levels of triglyceride (TG), the main form of cellular lipid storage, as well as increased levels of glucose and insulin, a hallmark of insulin resistance. The HFD-induced rise in TG levels is accompanied by increased signalling through the target of rapamycin (TOR) pathway, decreased expression of adipose triglyceride lipase (ATGL, encoded by the Bmm gene in flies) and a corresponding increase in fatty acid synthase (FAS) expression (Fig. 1; [28]). Although the mechanism by which high fat-supplemented food induces symptoms of obesity and the metabolic syndrome remains to be determined, it is interesting to note recent findings that a high sugar diet also induces excess TG accumulation and associated heart defects in flies ([29]; J. Na, R.B., K. Ocorr and R. Cagan., unpubl. data).
Fig 1.
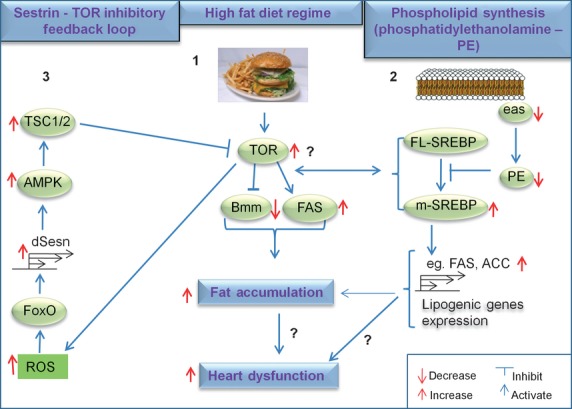
Impaired lipid metabolism induced by HFD or genetic manipulation induces heart dysfunction. Different mechanisms that initiate fat accumulation also lead to heart dysfunction in Drosophila. In 1 (centre), a HFD regime activates the TOR pathway, lowers ATGL/Bmm lipase and increases FAS levels. This leads to fat accumulation and heart dysfunction [28]. In 2 (right), dysregulation of phospholipid synthesis in easily shocked (eas) mutant flies (decreased production of phosphatidylethanolamine, PE) augments SREBP processing, which leads to accumulation of active (nuclear) SREBP (m-SREBP) and increased expression of lipogenic genes (e.g. FAS, ACC). This process also results in fat accumulation and heart dysfunction [33]. Both direct and indirect genetic mechanisms are implicated in fat accumulation and heart dysfunction, demonstrating the need to identify new factors (?) that might reveal how fat accumulation causes heart dysfunction. In 3 (left), a new feedback inhibitory loop involving Sestrin (dSesn in flies) is involved in modulating TOR activity. Increased TOR signaling augments ROS production, which activates the transcription factor FoxO via Jun-N-terminal kinase. In turn, FoxO activates dSesn, which inhibits TOR activity by activating the AMPK/TSC axis. In dSesn mutant flies, this negative feedback loop is diminished, leading to hyperactivation of the TOR pathway and consequently to fat accumulation and heart dysfunction [32].
Excess fat accumulation is associated with heart dysfunction in Drosophila
After two centuries of scientific observation and debate (reviewed in [30]), it has become clear that obesity and diabetes in humans promote the development of secondary diseases, including heart dysfunction, high blood pressure, atherosclerosis and fatty liver disease, collectively known as the me[31] has revealed some answers. HFD feeding reduces cardiac output (fractional shortening), causes partial conduction block and induces regions of non-contractility (reminiscent of an infarct-like state) in the majority of these ‘fatty’ hearts [28], strongly suggesting that HFD also significantly impair heart function in flies. These HFD-induced changes in the regular heartbeat pattern are illustrated in the M-mode traces from SOHA: under HFD feeding conditions the heart rate is faster and more erratic than observed under normal feeding conditions (Fig. 2 tabolic syndrome. How obesity causes symptoms of metabolic syndrome, in particular cardiomyopathies, is still largely unknown. The recently developed fly models of obesity-related heart dysfunction have begun to reveal some of the genetic mechanisms involved.
Fig 2.

Heart M-modes from wildtype Drosophila fed a normal or high fat diet. The figure shows M-mode traces from high-speed movies of Drosophila hearts using the SOHA program [[11], [31]]. The upper panel shows a cardiac M-mode from a young (2-week-old) fly fed a normal food diet, which shows a regular heart rhythm. The lower panel shows an M-mode from a fly fed a high-fat diet; these hearts beat faster and with an irregular, erratic rhythm. Heartbeat duration (heart period) is represented by the black bars.
Recent advances in the measurement of heart function in adult Drosophila have made it possible to determine whether obesity induces heart dysfunction in Drosophila, as it does in humans. Indeed, the Semi Automated Optical Heart Analysis (SOHA) method developed by Ocorr and colleagues). These findings demonstrate that, as for humans, flies exposed to prolonged HFD feeding (days in flies) exhibit severe cardiac dysfunction, which suggests that the underlying mechanisms by which dietary intake affects metabolic homeostasis and heart function may indeed be conserved between Drosophila and humans.
The next step to understanding the relationship between dietary fat intake and cardiovascular disease is to investigate the molecular mechanisms that link altered fat metabolism to dysregulated heart function. In this regard, Birse et al. 2010 have shown that the insulin-TOR pathways are key factors in mediating fat accumulation in the whole organism and in the heart itself, and the subsequent development of heart dysfunction (Fig. 1). For example, genetic manipulations that reduce insulin-TOR pathway activity prevent the HFD-induced accumulation of fat, and importantly, these interventions also protect against heart dysfunction. Moreover, when insulin-TOR signalling was diminished only in the fly's heart, excess fat still accumulated in the rest of the body, but the HFD-induced cardiac abnormalities were abolished. These findings suggest that cardiac-specific interventions of major metabolic regulators (such as the insulin-TOR pathways) may represent new therapeutic avenues for targeting obesity-related heart dysfunction.
Two other studies, also using a genetic approach to modulate fat accumulation, have uncovered new factors in the relationship between fat metabolism and heart dysfunction in Drosophila [32,33]. In one study a new feedback inhibitory loop for TOR signalling was found to involve the transcriptional regulation of the fly homolog of the Sestrins (dSesn) through TOR-dependent activation of the FoxO transcription factor. dSesn seems to act as a rheostat to stimulate AMPK activity, which in turn inhibits TOR function. Thus, in the absence of dSesn TOR was hyperactivated, leading to excess fat accumulation and ensuing heart defects; these effects were reversed (rescued) by addition of the TOR inhibitor rapamycin or the AMPK activator AICAR (Fig. 1; [32]).
In the second study, ablation of the gene easily shocked (eas), which encodes a key enzyme in the synthesis of the membrane phospholipid phosphatidylethanolamine (PE), also caused excess fat accumulation and cardiac defects [33], a phenotype reminiscent of dSesn mutants. eas mutant flies have low PE levels, and exhibit increased processing of the transcription factor SREBP to its active mature (nuclear) form m-SREBP (Fig. 1; [34]). m-SREBP activates transcription of lipogenic genes which results in elevated TG levels and impaired heart function [33]. Remarkably, hearts from eas mutants display a similar phenotype to those of HFD-fed flies including tachycardia [28]. In addition, overexpression of m-SREBP in the heart mimics the cardiac dysfunction seen in eas mutants, whereas cardiac-specific knockdown of SREBP or its lipogenic targets rescues the eas cardiac phenotype [33]. Therefore, perturbation of PE synthesis interferes with fat metabolism through SREBP processing, which in turn contributes to heart dysfunction.
Collectively, these studies have shown that both dietary and genetic causes of impaired or altered lipid metabolism can increase fat concentrations and lead to detrimental effects on heart function (Figs 1 and 2). Our understanding of these relationships will be facilitated by studying possible links between the insulin-TOR and SREBP/lipid metabolism pathways, and discovering new mechanisms that link fat accumulation and cardiac dysfunction. Indeed, a recent study in mammalian cells suggested that mTOR complex 1 regulates the SREBP pathway through the nuclear localization of the phosphatidic acid phosphatase Lipin 1 and subsequent nuclear shape [35]. Collectively, these studies will accelerate the identification of new therapeutic targets to protect the heart against the detrimental effects of obesity.
New players in the fat–heart relationship
To identify new modifiers of obesity-associated heart dysfunction in Drosophila, we have conducted a screen of genes that are implicated in various aspects of lipid metabolism, to find new gene functions that modulate the degree of fat accumulation and have potentially detrimental effects on heart function. Several candidates are emerging as interesting players, among them a potential fly homolog of the PPARγ Coactivator-1 (PGC-1) genes. In mammals, PGC-1 genes (PGC-1α, PGC-1β and PRC) are involved in numerous metabolic functions, including mitochondrial biogenesis and function, fatty acid oxidation, muscle fibre determination, gluconeogenesis and thermogenesis (Fig. 3; [36,37,38,39,40,41]). In mammals, the functions of PGC-1 family members in mitochondrial biogenesis and fatty acid oxidation have been shown to be essential in satisfying the high energy needs in the neonatal and the adult heart [42,43,,44]
Fig 3.
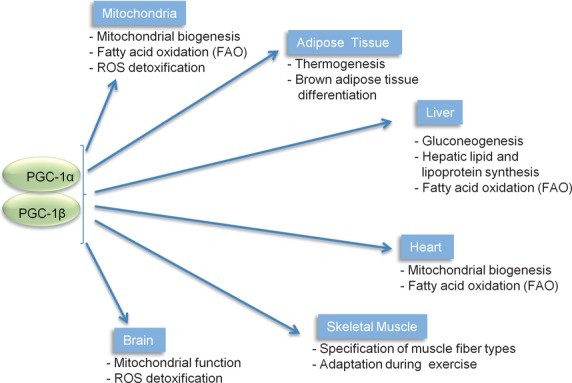
Summary of PGC-1 metabolic functions in mammals. PGC-1 family members play essential roles in mitochondrial biogenesis, as well as in the control of the electron transport chain and fatty acid oxidation (FOA), both occurring inside the mitochondria.
In Drosophila, only one gene has been identified as a bona fide homolog of the PGC-1 genes; Spargel [45]. This offers the opportunity to study its role in flies without the complications of potential redundancies and functional complexities found in mammals. Spargel has recently been implicated in nutrient responses during larval development in Drosophila, where it appears to be an important player in mitochondrial function, as it is in mammals. [45,46]
Interestingly, flies with partial loss-of-function of Spargel exhibit even more fat accumulation than do wildtype flies in response to a HFD, whereas flies with ubiquitous overexpression of Spargel are leaner than wildtype flies and are protected from excessive fat accumulation when fed a HFD (S.B. Diop and R. Bodmer, unpubl. data). Flies with reduced Spargel function also exhibit the same heart defects as are induced by a HFD (S.B. Diop and R. Bodmer, unpubl. data). Therefore, increased fat accumulation in flies with decreased Spargel function is correlated with an increase in heart defects, whereas the heart is protected from the effects of obesity when Spargel is overexpressed, suggesting that this co-activator plays an important role in HFD-induced obesity and heart dysfunction. It will be interesting to see how Spargel function relates to other modulators of the fat–heart relationship, as we discussed above.
Concluding remarks
The studies discussed in this review have shown that the mechanisms by which a high caloric diet induces heart dysfunction is remarkably conserved from mammals to Drosophila. Genetic studies showed the insulin-TOR and SREBP pathways play a central role in this process, and new genes are currently being investigated using the genetic and analytical tools now available in Drosophila. This model system is well suited to dissect genetic interactions and identify new players that link impairment of lipid metabolism to a dysfunctional heart. This will ultimately help design new therapeutic strategies to protect the heart against the deleterious effects of high caloric diets or genetic causes of obesity.
Acknowledgments
We thank Anne O'Rourke for editorial assistance in preparing the manuscript. We thank Ryan Birse for comments on the manuscript. S.B.D. is supported by a diversity supplement from NHLBI at NIH. R.B. is supported by grants from NHLBI and NIA at NIH, and from the Ellison Medical Foundation.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- Hunziker W, Kiener TK, Xu J. Vertebrate animal models unravel physiological roles for zonula occludens tight junction adaptor proteins. Ann N Y Acad Sci. 2009;1165:28–33. doi: 10.1111/j.1749-6632.2009.04033.x. [DOI] [PubMed] [Google Scholar]
- Burggren WW, Warburton S. Amphibians as animal models for laboratory research in physiology. ILAR J. 2007;48:260–9. doi: 10.1093/ilar.48.3.260. [DOI] [PubMed] [Google Scholar]
- Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8:353–67. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- Chianese R, Chioccarelli T, Cacciola G, et al. The contribution of lower vertebrate animal models in human reproduction research. Gen Comp Endocrinol. 2011;171:17–27. doi: 10.1016/j.ygcen.2010.12.011. [DOI] [PubMed] [Google Scholar]
- Botstein D, Chervitz SA, Cherry JM. Yeast as a model organism. Science. 1997;277:1259–60. doi: 10.1126/science.277.5330.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumby AM, Richardson HE. Using Drosophila melanogaster to map human cancer pathways. Nat Rev Cancer. 2005;5:626–39. doi: 10.1038/nrc1671. [DOI] [PubMed] [Google Scholar]
- Kaletta T, Hengartner MO. Finding function in novel targets: C. elegans as a model organism. Nat Rev Drug Discov. 2006;5:387–98. doi: 10.1038/nrd2031. [DOI] [PubMed] [Google Scholar]
- Segalat L. Invertebrate animal models of diseases as screening tools in drug discovery. ACS Chem Biol. 2007;2:231–6. doi: 10.1021/cb700009m. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 2011;63:411–36. doi: 10.1124/pr.110.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier E, Bodmer R. Drosophila, an emerging model for cardiac disease. Gene. 2004;342:1–11. doi: 10.1016/j.gene.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Ocorr K, Perrin L, Lim HY, et al. Genetic control of heart function and aging in Drosophila. Trends Cardiovasc Med. 2007;17:177–82. doi: 10.1016/j.tcm.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter LT, Potocki L, Chien S, et al. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11:1114–25. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer R. Heart development in Drosophila and its relationship to vertebrate systems. Trends Cardiovasc Med. 1995;5:21–7. doi: 10.1016/1050-1738(94)00032-Q. [DOI] [PubMed] [Google Scholar]
- Rizki TM. The Circulatory System and Associated Cells and Tissues in the Genetics and Biology of Drosophila. New York: Academic Press; 1978. pp. 397–452. [Google Scholar]
- Cammarato A, Ahrens CH, Alayari NN, et al. A mighty small heart: the cardiac proteome of adult Drosophila melanogaster. PLoS ONE. 2011;6:e18497. doi: 10.1371/journal.pone.0018497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development. 1993;118:719–29. doi: 10.1242/dev.118.3.719. [DOI] [PubMed] [Google Scholar]
- Lyons I, Parsons LM, Hartley L, et al. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–66. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- Schott JJ, Benson DW, Basson CT, et al. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281:108–11. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- Satou Y, Satoh N. Gene regulatory networks for the development and evolution of the chordate heart. Genes Dev. 2006;20:2634–8. doi: 10.1101/gad.1485706. [DOI] [PubMed] [Google Scholar]
- Kim SK, Rulifson EJ. Conserved mechanisms of glucose sensing and regulation by Drosophila corpora cardiaca cells. Nature. 2004;431:316–20. doi: 10.1038/nature02897. [DOI] [PubMed] [Google Scholar]
- Baker KD, Thummel CS. Diabetic larvae and obese flies-emerging studies of metabolism in Drosophila. Cell Metab. 2007;6:257–66. doi: 10.1016/j.cmet.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton SJ, Piper MD, Ikeya T, et al. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc Natl Acad Sci USA. 2005;102:3105–10. doi: 10.1073/pnas.0405775102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gade G, Auerswald L. Mode of action of neuropeptides from the adipokinetic hormone family. Gen Comp Endocrinol. 2003;132:10–20. doi: 10.1016/s0016-6480(03)00159-x. [DOI] [PubMed] [Google Scholar]
- Noyes BE, Katz FN, Schaffer MH. Identification and expression of the Drosophila adipokinetic hormone gene. Mol Cell Endocrinol. 1995;109:133–41. doi: 10.1016/0303-7207(95)03492-p. [DOI] [PubMed] [Google Scholar]
- Lee G, Park JH. Hemolymph sugar homeostasis and starvation-induced hyperactivity affected by genetic manipulations of the adipokinetic hormone-encoding gene in Drosophila melanogaster. Genetics. 2004;167:311–23. doi: 10.1534/genetics.167.1.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabel G, Martin JR, Chidami S, et al. AKH-producing neuroendocrine cell ablation decreases trehalose and induces behavioral changes in Drosophila. Am J Physiol Regul Integr Comp Physiol. 2005;288:R531–8. doi: 10.1152/ajpregu.00158.2004. [DOI] [PubMed] [Google Scholar]
- Colombani J, Raisin S, Pantalacci S, et al. A nutrient sensor mechanism controls Drosophila growth. Cell. 2003;114:739–49. doi: 10.1016/s0092-8674(03)00713-x. [DOI] [PubMed] [Google Scholar]
- Birse R, Choi J, Reardon K, et al. High fat diet-induced obesity and heart dysfunction is regulated by the TOR pathway in the Drosophila model. Cell Metab. 2010;12:533–44. doi: 10.1016/j.cmet.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman LP, Fink JL, Narzinski K, et al. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis Model Mech. 2011;4:842–9. doi: 10.1242/dmm.007948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepaldi G, Maggi, S The metabolic syndrome: a historical context. Diabetes Voice. 2006;51:8–10. [Google Scholar]
- Fink M, Callol-Massot C, Chu A, et al. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. BioTechniques. 2009;46:101–13. doi: 10.2144/000113078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Budanov AV, Park EJ, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327:1223–8. doi: 10.1126/science.1182228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HY, Wang W, Wessells RJ, et al. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes Dev. 2011;25:189–200. doi: 10.1101/gad.1992411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunte AS, Matthews KA, Rawson RB. Fatty acid auxotrophy in Drosophila larvae lacking SREBP. Cell Metab. 2006;3:439–48. doi: 10.1016/j.cmet.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–20. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–39. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–76. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–35. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lustig Y, Ruas JL, Estall JL, et al. Separation of the gluconeogenic and mitochondrial functions of PGC-1{alpha} through S6 kinase. Genes Dev. 2011;25:1232–44. doi: 10.1101/gad.2054711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling J, Kelly DP. The PGC-1 cascade as a therapeutic target for heart failure. J Mol Cell Cardiol. 2011;51:578–83. doi: 10.1016/j.yjmcc.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, Leone TC, Zechner C, et al. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–61. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershman B, Puig O, Hang L, et al. High-resolution dynamics of the transcriptional response to nutrition in Drosophila: a key role for dFOXO. Physiol Genomics. 2007;29:24–34. doi: 10.1152/physiolgenomics.00061.2006. [DOI] [PubMed] [Google Scholar]
- Tiefenbock SK, Baltzer C, Egli NA, et al. The Drosophila PGC-1 homologue Spargel coordinates mitochondrial activity to insulin signalling. EMBO J. 2010;29:171–83. doi: 10.1038/emboj.2009.330. [DOI] [PMC free article] [PubMed] [Google Scholar]