

Abstract
The combination of synthetic stable branched DNA and sticky ended cohesion has led to the development of structural DNA nanotechnology over the past 30 years. The basis of this enterprise is that it is possible to construct novel DNA-based materials by combining these features in a self-assembly protocol. Thus, simple branched molecules lead directly to the construction of polyhedra whose edges consist of double helical DNA, and whose vertices correspond to the branch points. Stiffer branched motifs can be used to produce self-assembled two-dimensional and three-dimensional periodic lattices of DNA (crystals). DNA has also been used to make a variety of nanomechanical devices, including molecules that change their shapes, and molecules that can walk along a DNA sidewalk. Devices have been incorporated into two-dimensional DNA arrangements; sequence-dependent devices are driven by increases in nucleotide pairing at each step in their machine cycles.
Keywords: Branched DNA, Sticky-Ended Cohesion, DNA Objects, Designed DNA Lattices, DNA-Based Nanomechanical Devices, Chemical Organization by DNA
I. Introduction and Pre-requisites for Structural DNA Nanotechnology
The readers of this volume are all aware that DNA is the molecule that nature uses as genetic material. The iconic antiparallel double helical structure assumed by its two strands facilitates high fidelity recognition between the nucleotides of complementary molecules. Although every base can pair with every other base, including itself (1), the Watson-Crick pairing (2) of adenine (A) with thymine (T) and guanine (G) with cytosine (C) appears to be the favored type of interaction between polynucleotides if the sequences of the molecules permit it. Biology clearly exploits this form of interaction in the replication of genetic information and in its expression. Nevertheless, biology is no longer the only branch of science where DNA is finding a significant role: It is now possible to exploit DNA complementarity to control the structure of matter.
This article will review the history and current status of using DNA to construct novel nanomaterials and to control their structures over time. We shall see below that DNA is used today build specific objects, periodic lattices in two and three dimensions, and nanomechanical devices. The dimensions of DNA are inherently on the nanoscale: The diameter of the double helix is about 2 nm, and the helical pitch is about 3.5 nm; hence construction involving DNA is fundamentally an exercise in nanoscience and nanotechnology. Consequently, the area we are discussing is called ‘structural DNA nanotechnology’ and its goal is the finest possible level of control over the spatial and temporal structure of matter: Putting what you want where you want it in three dimensions (3D), when you want it there.
Structural DNA nanotechnology rests on three pillars: [1] Hybridization; [2] Stably branched DNA; and [3] Convenient synthesis of designed sequences.
[1] Hybridization. The self-association of complementary nucleic acid molecules or parts of molecules (3), is implicit in all aspects of structural DNA nanotechnology. Individual motifs are formed by the hybridization of strands designed to produce particular topological species. A key aspect of hybridization is the use of sticky ended cohesion to combine pieces of linear duplex DNA; this has been a fundamental component of genetic engineering for over 35 years (4). Sticky-ended cohesion is illustrated in Figure 1a, where two double helical molecules are shown to cohere by hydrogen bonding. Not only is hybridization critical to the formation of structure, but it is deeply involved in almost all the sequence-dependent nanomechanical devices that have been constructed, and it is central to many attempts to build structural motifs in a sequential fashion (5). Among the various types of cohesion known between biological molecules, sticky ended cohesion is very special: Not only do we know that two sticky ends will cohere with each other in a specific and programmable fashion (affinity), but we also know the structure that they form when they do cohere, a Watson-Crick double helix (6). This key point is illustrated in Figure 1b. Thus, in contrast to other biologically-based affinity interactions (e.g., an antigen and an antibody), we know on a predictive basis the local product structure formed when sticky ends cohere, without the need to do a crystal structure first to establish the relative orientation of the two components.
Figure 1. Sticky-Ended Cohesion. (a) Cohesion Between Two Molecular Overhangs.
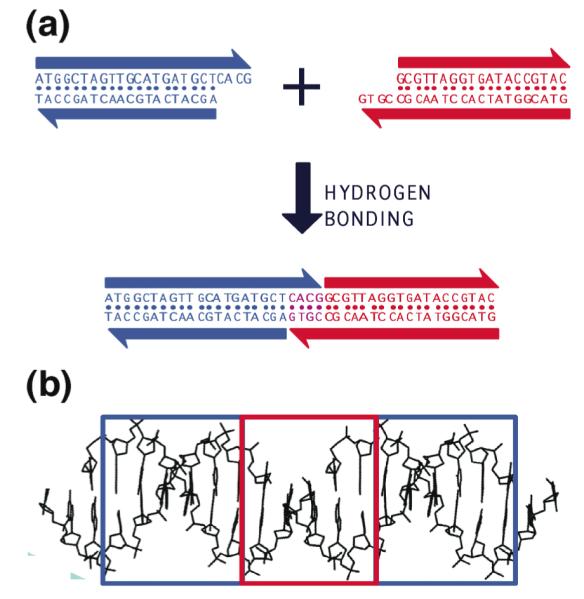
Two duplex molecules are shown, a red one and a blue one. Each has a single-stranded molecular overhang that is complementary to the overhang on the other molecule. When mixed, the two molecules can cohere in solution, as shown below. (b) Structural Features of Stick-Ended Cohesion. A crystal structure (6) is shown that contains DNA decamers whose cohesion in the direction of the helix axis is directed by dinucleotide sticky ends. This interaction is seen readily in the red box, where the continuity of the chains is interrupted by gaps. The two blue boxes contain B-form duplex DNA. It is a half-turn away from the DNA in the red box, so it is upside-down from it, but otherwise the structure is the same. Thus, sticky ends cohere to form B-DNA, and one can use this information in a predictive fashion to estimate the local structures of DNA constructs held together by sticky ends.
[2] Stably Branched DNA. Although the DNA double helix is certainly the best-known structure in biology, little biology would occur if the DNA molecule were locked tightly into that structure with an unbranched helix axis. For example, triply branched replication forks occur during semi-conservative replication (7), and four-arm branched Holliday junctions (8) are intermediates in genetic recombination. Likewise, branched DNA molecules are central to DNA nanotechnology. It is the combination of in vitro hybridization and synthetic branched DNA that leads to the ability to use DNA as a construction material (9). The fundamental notion behind DNA nanotechnology is illustrated in Figure 2a, which shows the cohesion of four copies of a 4-arm branched DNA molecule tailed in sticky ends associating to form a quadrilateral. In the example shown, only the inner sticky ends are used in forming the quadrilateral. Consequently, the structure can be extended to form an infinite lattice (9).
Figure 2. Self-Assembly of Branched DNA Molecules to Form Larger Arrangements.
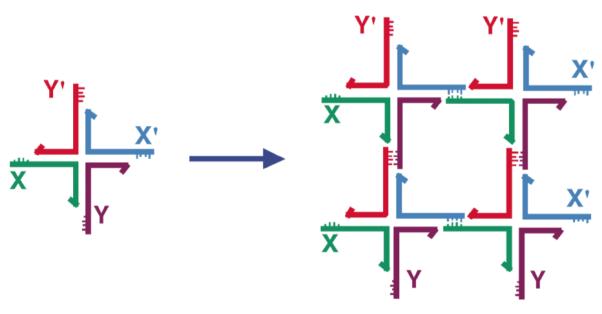
The image on the left shows a 4-arm branched junction made from four differently colored strands. Its double helical domains are tailed in 5′ sticky ends labeled (clockwise from the left) X, Y, X’ and Y’; the sticky ends are indicated by small extensions from the main strand (our convention is to represent 3′ ends by arrowheads or, as here, by half-arrowheads). The primed sticky ends complement the unprimed ones. The image on the right shows how four of these junctions can self-assemble through this complementarity to yield a quadrilateral. The sticky ends have come together in a complementary fashion. Note that this assembly does not use up all the available sticky ends, so that those that are left over could be used to generate a lattice in 2D, and, indeed, in 3D.
[3] Convenient Synthesis of Designed Sequences. Biologically derived branched DNA molecules, such as Holliday junctions, are inherently unstable, because they exhibit sequence symmetry; i.e., the four strands actually consist of two pairs of strands with the same sequence. This symmetry enables an isomerization known as branch migration that allows the branch point to relocate (10). Branch migration can be eliminated if one chooses sequences that lack symmetry in the vicinity of the branch point. We will discuss below different approaches to the use of symmetry in DNA nanotechnology, but the first approaches to DNA nanotechnology entailed sequence design that attempted to minimize sequence symmetry in every way possible. Such sequences are not readily obtained from natural sources, which leads us to the third pillar supporting DNA nanotechnology, the synthesis of DNA molecules of arbitrary sequence (11). Fortunately, this is a capability that has existed for about as long as needed by this enterprise: Synthesis within laboratories or centralized facilities has been around since the 1980s; today it is possible to order all the DNA components needed for DNA nanotechnology, so long as they lack complex modifications, i.e., so-called ‘vanilla’ DNA. In addition, the biotechnology enterprise has generated demand for many variants on the theme of DNA (e.g., biotinylated molecules), and these molecules are also readily synthesized or purchased.
II. Initial Steps in the Process
[1] Motif Design
There are two fundamental steps needed for to perform projects in structural DNA nanotechnology: Motif design and sequence design. Motif design relies on the operation of reciprocal exchange, the switching of the connections between DNA strands in two double different double helices to produce a new connectivity. This notion is illustrated in Figure 3a, where a red and a blue strand undergo reciprocal exchange to produce red-blue and blue-red strands. It is important to recognize that this is not an operation performed in the laboratory; it is done on paper or in the computer, and then the strands corresponding to the results of the operation are synthesized. Owing to the polar nature of DNA backbones, the operation can be performed between strands of the same polarity or between strands of opposite polarity. If only a single reciprocal exchange is performed, there is no difference, because the two products are just conformers of each other; however, if two or more operations are performed different topologies result. Often a different ease of formation accompanies the two different topologies; empirically, the best-behaved molecules are those in which exchange takes place between strands of opposite polarity.
Figure 3. Motif Generation by Reciprocal Exchange.

(a) The Fundamental Operation. The basic operation of reciprocal exchange is shown: A red stand and a blue strand become a red-blue and a blue-red strand following the operation. (b) Motifs that can result from Reciprocal Exchange of DNA Molecules. At the top of the panel are shown the DX motif and the DX+J motif. The DX motif results from two reciprocal exchanges between double helical motifs. The DX+J motif contains another DNA domain. Usually this domain is oriented perpendicular to the plane of the two helix axes in the DX part of the motif. When this orientation is achieved, the extra domain can behave as a topographic marker for 2D arrays containing the DX+J motif. The bottom row of the panel shows the TX motif at left, wherein a third domain has been added; again the exchanges take place between strands of opposite polarity. In the center and right are the PX motif and its topoisomer, the JX2 motif. The PX molecule is formed by exchanges between strands of identical polarity at every possible position. The JX2 molecule lacks two of these exchanges.
A number of important motifs generated this way are illustrated in Figure 3b. The DX motif (12) with exchanges between strands of opposite polarity is shown at the upper left of that panel. This motif has been well characterized, and it is known that its persistence length is about twice that of conventional linear duplex DNA (13). The DX+J motif is shown at the upper right of Figure 3b. In this motif the extra domain is usually oriented so as to be nearly perpendicular to the plane of the two helix axes; this orientation enables the domain to act as a topographic marker in the atomic force microscope (AFM). The motif shown at the bottom left of Figure 3b is the three-domain TX molecule. Below, we shall see that by joining these motifs in a 1-3 fashion (the top helical domain of one molecule joins with the bottom domain of another molecule), 2D arrays can be created that contain useful cavities (14, 15).
By contrast with the DX and TX motifs shown, the PX motif and its topoisomer, the JX2 motif (bottom right of Figure 3b), result from reciprocal exchange between strands of the same polarity. In the case of the PX molecule, this happens everywhere that two double helices can be juxtaposed; two exchanges are missing in the JX2 molecule. These motifs are central to many of the robust nanomechanical devices that have been constructed (16).
[2] Sequence Design and Symmetry Minimization
The design of DNA sequences that do not adhere to the strict linear duplex DNA paradigm is likely to result in molecules that correspond to excited states of some sort. Of course, the vast bulk of our knowledge about DNA structure and thermodynamics is predicated on ‘ground state’ linear duplex, rather than various excited states. It is evident that the goal of sequence design would naturally be to get the molecules to form the excited states that we seek to make. The free energy cost of introducing a 4-arm branch into a DNA molecule is not very high (+1.1 (±0.4) kcal mol−1 at 18 °C in the presence of 10 mM Mg2+) (17). However, before this was established, an effective method based on sequence symmetry minimization was worked out that has served well for the design of branched molecules (9, 18). This method is used for assigning small DNA motif sequences in many laboratories.
The basic approach to sequence symmetry minimization is illustrated in Figure 4a, using the example of a 4-arm branched junction built from four strands that each contain 16 nucleotides. Each strand has been broken up into a series of 13 overlapping tetramers, so there are 52 tetramers in the entire molecule. The first two tetramers in strand one have been boxed, CGCA and GCAA. Sequence symmetry minimization would insist that each tetramer be unique. Furthermore, to ensure that the molecule cannot form linear duplex DNA at any point around the designed branch point, the linear complements to each of the twelve tetramers flanking the branch point are also forbidden. For example, the complement to the boxed CTGA (i.e., TCAG) is nowhere to be found in this molecule. Using these criteria, competition with the target structure (the four octamer duplex components of the branch) will only come from trimers; thus, the boxed ATG segments could, in principle, be in the wrong place, but the free energy differences between octamers and trimers win out and the target is obtained without detectable impurities. In the case of 4-arm branched molecules, branch migration may also be a problem (10), so the junction-flanking nucleotides are forbidden from having a two-fold symmetric relationship among themselves. Other tricks used to avoid getting the wrong associations include: the prevention of long stretches of G’s that could form other structures (particularly near crossover points), avoiding homopolymer tracts, polypurine or polypyrimidine tracts, alternating purine-pyrimidine tracts, and anything else that looks to the designer like it might be symmetric, in the broadest sense of the word. Such precautions are clearly less important in the middle of a large stretch of linear duplex than they are near branch points.
Figure 4.

(a) Sequence Design. The four-arm junction shown contains four 16-mers that are each broken up into 13 overlapping tetramers. Insisting that each tetramer be unique and that no tetramer complement the those that flank the branch point leads to the formation of a stable branch, particularly if the twofold symmetry that enables branch migration is forbidden. Tetramers provide a ‘vocabulary’ of 256 (less 16 self-complementary units) possible sequences to use, leading to competition from trimers. It would be difficult to design this molecule to have 56 unique trimers. Larger units clearly are to be used with larger constructs. (b) A Twelve-Arm Junction. It is not possible to eliminate symmetry around the center of this junction, so identical nucleotide pairs were spaced at four-step intervals around the junction.
In recent years, some issues involving symmetry minimization have been raised, explicitly or implicitly. For example, in a 12-arm junction (Figure 4b), it is not possible to flank the branch point with different base pairs, as it was with the 4-arm junction. An entropic argument was used to design the junction in Figure 4b, and it seems to have been successful (19). Mao has used physical restraints (e.g., strand length) and concentration, while maximizing symmetry, to obtain large arrangements of molecules designed to have as few as one strand (20). Both of these approaches consider nucleic acids as physical entities whose properties can be exploited; they represent advances from original considerations of nucleic acids sequences as arbitrary mathematical constructs. Arguably the most dramatic example of ignoring sequence symmetry and just designing structures is Rothemund’s DNA origami (21). We will discuss DNA origami in more detail below, but basically a long viral single strand, the sequence accepted as a given, is used to scaffold about 250 shorter strands to produce a 2D (21) or 3D shape, either with straight (22) or bent (23) features. Recently, Shih has demonstrated that two long complementary DNA molecules can be folded successfully into two different shapes before they re-hybridize to form a linear duplex (24).
III. Individual Constructs
[1] Unscaffolded Targets
The earliest DNA constructs were best described as topological species, rather than geometrical species. This feature is owing to the fact that the earliest DNA motifs, i.e., branched junctions, were not robust, but could be described as floppy if they were ligated (25, 26). Thus, both the linking and branching topologies of these molecules were well-defined, because these features could be established by gels, but their detailed structures were not fixed. Idealized pictures of the first two structures, a cube (27) and a truncated octahedron (28), each with two double helical turns between vertices are shown in Figure 5. All nicks in these molecules were sealed, and they were characterized topologically by denaturing gel electrophoresis. One of the issues with these structures is that they were not deltahedra, polyhedra whose faces are all triangles; deltahedra, such as tetrahedra (29), octahedra (30), and icosahedra (22), have all been produced during the current decade (22, 31). In addition, a protein has been encapsulated within a tetrahedron (32). The larger species have been characterized by electron microscopy. Other polyhedra, such as DNA buckyballs (truncated icosahedra) have been produced by carefully exploiting the interplay between junction flexibility and edge rigidity (31).
Figure 5. Early Topological Constructs Built from DNA.
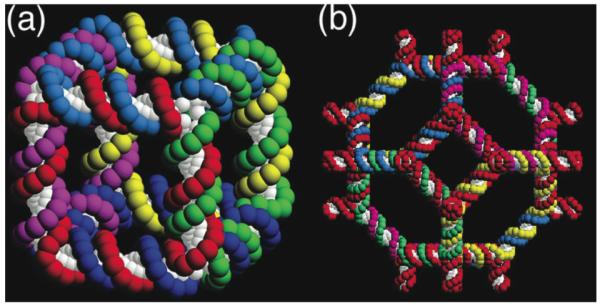
(a) A Cube-Like Molecule. This molecule is a hexacatenane; each edge corresponds to two double helical turns of DNA. Each backbone strand is drawn in a different color, and each one corresponds to a given face of the cube. Each is linked twice to each of the four strands that flank it, owing to the two-turn lengths of the edges. (b) A DNA Truncated Octahedron. This molecule is a 14-catenane, again with each edge consisting of two turns of double helical DNA. Although the truncated octahedron has 3 edges flanking each vertex (i.e., it is ‘3- connected’), it has been built using 4-arm junctions.
The topological features of the early constructs also led to the development of single-stranded DNA topology. A crossover in a knot or a catenane can be regarded as being equivalent to a half-turn of DNA, which can be exploited accordingly (34). Thus, it has proved fairly simple to produce a variety of knotted molecules from single-stranded DNA (35), as well as a number of specifically linked catenanes. Some of these constructs have been used to characterize the topology of Holliday junctions (36). By using left-handed Z-DNA, it is possible to produce nodes of both signs in topological products. This aspect of DNA topology was exploited to produce the first Borromean rings from DNA (37). An RNA knot was used to discover that E. coli DNA topoisomerase III (but not topoisomerase I) can act as an RNA topoisomerase (38).
[2] DNA Origami
DNA origami entails the use of a scaffolding strand to which a series of smaller ‘staple’ strands or ‘helper’ strands are added. The staple strands are used to fold the scaffolding strand into a well-defined shape. The first example of a scaffolding strand in structural DNA nanotechnology was reported by Yan et al. (39), who used a scaffolding strand to make a 1D ‘barcode array. This application of scaffolding was followed quickly by Shih et al. (30), who used a long strand of DNA with five short helper strands to build an octahedron held together by PX cohesion (40). However, neither of these two advances had the dramatic impact of Rothemund’s 2006 publication (21). He demonstrated that he was able to take single-stranded M13 viral DNA and get it to fold into a variety of shapes, including a smiley face (Figure 6a). One of the great advantages of this achievement is that it creates an addressable surface area roughly 100 nm square. One can use the DX+J motif (Figure 3b) developed for patterning 2D arrays (41) (see below) to place patterns on DNA origami constructs. An example is seen in Figure 6b, which shows a map of the western hemisphere. DNA origami has become widely used since it was introduced, and has been employed for embedding nanomechanical devices (42), for making long 6-helix bundles (43) for use as an aid to NMR structure determination (44), and for building 3D objects (22), including a box that can be locked and unlocked, with potential uses in therapeutic delivery (33).
Figure 6. Atomic Force Micrographs of DNA Origami Constructs.

(a) A Smiley Face. The scaffold strand, bound to helper strands, zigzags back and forth from left to right, yielding the structure seen. (b) A Map of the Western Hemisphere. This image demonstrates dramatically the addressability of the DNA array. Each of the white pixels is made by a small DNA double helical domain, similar to the DX+J tile (Figure 3).
IV. Crystalline Arrays
[1] 2D Crystals
The original goal of structural DNA nanotechnology was to produce designed periodic matter (9). To do so, it was necessary to have robust motifs that did not bend and flex readily; otherwise, a repeating pattern could fold up to form a cycle, poisoning the growth of the array. The DX molecule (see Figure 3b) was the first molecule shown to be sufficiently rigid for this purpose (13, 45). The molecule was quickly exploited to produce periodic matter in two dimensions (41). The DX+J motif was used to impose patterns on these arrays. When DX+J molecules were included specifically in the pattern, deliberately striped features could be seen in the atomic force microscope (AFM), as shown in Figure 7. DNA motifs that form 2D periodic (or aperiodic) arrays are often called ‘tiles,’ because they can tile the plane. There are numerous tiles that have been developed to tile the 2D plane, including the 3-domain TX tile (14), the 6-helix hexagonal bundle (43), and the DX triangle (46). For reasons that are not well understood, 2D crystals are relatively small, typically no more than a few microns in either dimension.
Figure 7. Two-Dimensional Arrays of DX and DX+J Molecules.
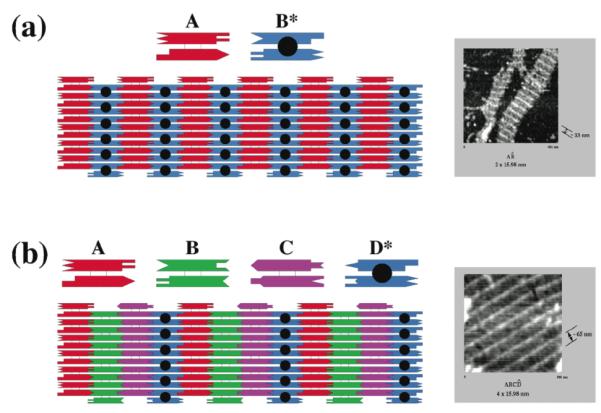
(a) An Alternating Array of DX and DX+J Molecules. Two tiles are shown, a DX tile labeled A and a DX+J tile labeled B*. The black dot represents the extra domain of the DX+J. Sticky ends are shown as geometrically complementary shapes. The array that would be formed by these two tiles is shown below them, including a stripe-like feature formed by the extra domain of the DX+J tile. The horizontal direction of each tile is 16 nm. The atomic force micrograph at right shows stripes separated by 32 nm. (b) An Array of Three DX and One DX+J Tiles. The A, B and C tiles are DX molecules and the D* tile is a DX+J tile. All tiles have the same dimensions as in (a). This should lead to a 64 nm separation of stripes, as seen in the atomic force micrograph at the right.
2D crystals have proved to be an extremely good way to introduce students and other new investigators to structural DNA nanotechnology. The preparation of the AB* or ABCD* array (Figure 7), for example, is extremely simple: All the strands are mixed stoichiometrically, heated to 90-95° and then cooled over 40 hours in a styrofoam container (47); they are readily observed by atomic force microscopy when deposited on mica (47). Rotating graduate students, undergraduates and high school students are usually successful at producing beautiful atomic force micrographs on the first pass. There are some experiments that can discourage new investigators in structural DNA nanotechnology, but making 2D periodic arrays is not among them.
[2] 3D Crystals
Control of the structure of matter would be incomplete without the ability to produce 3D crystals of high quality. Although 2D crystals are examined by AFM, the way to characterize the molecular structure of 3D crystals is by X-ray crystallography. Under exceptionally good circumstances, one can resolve two double helices separated by 7 nm (two turns of DNA), but such resolution is not usually available to AFM (e.g., ref. 48). By contrast, X-ray crystallography is capable of resolutions ~1 Å, the limitations being largely a function of crystal quality. All the early attempts to obtain high quality crystals of DNA motifs resulted in crystals diffracting to no better than 10 Å resolution. If there are issues of molecular boundaries within the crystal to be determined, such a resolution is unlikely to provide an unambiguous answer (49). Recently, however, a tensegrity triangle (50) with two double helical turns per edge, has been crystallized to 4 Å resolution and its structure has been determined by the single anomalous dispersion method (51). The tensegrity triangle has three double helical domains that point in three independent directions, so as to define a 3D structure. An individual tensegrity triangle and its six neighbors are shown in stereoscopic projection in Figure 8a. The three independent directions of the tensegrity triangle lead to a rhombohedral structure that flanks a cavity. This cavity, along with seven of the eight tensegrity triangles that flank it, is shown in stereoscopic projection in Figure 8b; the volume of the cavity is ~100 nm3.
Figure 8. The 3D Lattice Formed by the Tensegrity Triangles.

(a) The Surroundings of an Individual Triangle. This stereoscopic simplified image distinguishes the three independent directions by the colors (red, green and yellow) of their base pairs. Thus, the central triangle is shown flanked by three other pairs of triangles in the three differently colored directions. (b) The Rhombohedral Cavity Formed by the Tensegrity Triangles. This stereoscopic projection shows seven of the eight tensegrity triangles that comprise the corners of the rhombohedron. The outline of the cavity is shown in white. The red triangle at the back connects through one edge each to the three yellow triangles that lie in a plane somewhat closer to the viewer. The yellow triangles are connected through two edges each to two different green triangles that are in a plane even nearer the viewer. A final triangle that would cap the structure has been omitted for clarity. That triangle would be directly above the red triangle, and would be even closer to the viewer than the green triangles.
It is evident from Figure 8a that the structure is held together in the directions of the helix axes. This is not happenstance. Nine other crystals have been characterized that are also based on the tensegrity triangle, and are held together by two-nucleotide sticky ends (51). Some of the crystals (like the one shown in Figure 8) are 3-fold rotationally averaged by sticky-end selection, and some of the tensegrity triangles have longer edges, either three turns or four turns. The resolution of the crystals is inversely related to edge length, ~6.5 Å for 3 turns and ~10-11 Å for 4 turns. It is unclear as to why this is the case, but it may be related to the fact that these are stick-like structures, rather than the ball-like structures usually examined by macromolecular crystallography. Whereas there are no molecular boundaries to establish for these structures, the resolution is not a key factor, so one of each of the larger structures has been determined by molecular replacement. The volume of the largest cavity is around 1100 nm3 (roughly a zeptoliter). In contrast to 2D crystals, the crystal sizes are macroscopic, ranging from 0.25 mm to 1 mm in linear dimensions. One of the strengths of the notion of holding molecules together by sticky ends is that one can design the number of molecules per crystallographic repeat. A crystal with two distinct two-turn motifs has been designed, and its structure determined by molecular replacement (R. Sha, T. Wang, J. Zheng, J.J. Birktoft, C. Mao and N.C. Seeman, in preparation).
V. Other Types of DNA-Based Nanomaterials
[1] Organization of Other Species
The use of DNA to organize other molecular species has been a central component of structural DNA nanotechnology since its inception (9). Originally, the notion was that 3D crystals could organize biological macromolecules that were resistant to crystallization by traditional methods. It is clear from the last section that some progress is needed in resolution before such a goal can be realized. Nevertheless, structural DNA nanotechnology has done other things with proteins. Yan and his colleagues have attached biotin groups to DNA arrays and used them to bind streptavidin (52). The same group has used aptamers on the surface of DNA origami to establish the cooperativity of protein-protein interactions (53).
Another target for structural DNA nanotechnology is the organization of nanoelectronic or nanophotonic components (54). In this regard, a number of studies have been devoted to organizing nanoparticles using DNA, sometimes within individual objects (55, 56), and sometimes within 1D (57) or 2D arrays containing multiple components (58, 59). Figure 9 illustrates how multiple components, each carrying a different cargo, can be used to organize complex arrangements of metallic nanoparticles (59). The motif used is the 3D-DX motif, based on the tensegrity triangle, but containing 8-turn DX components in each direction of crystal propagation, rather than single helices. A 5 nm gold nanoparticle has been placed on the end of one of the propagation directions in one of the 3D-DX molecules, and a 10 nm gold nanoparticle has been put in the same site in the other 3D-DX molecule (Figure 9a). Thus, one propagation direction is eliminated, but 2D arrays can be formed. Figure 9b shows the 3D-DX motif and its lattice in greater detail, and Figure 9c shows a TEM image of the checkerboard pattern formed by the nanoparticles.
Figure 9. Organizing Gold Nanoparticles with a 2D Motif.
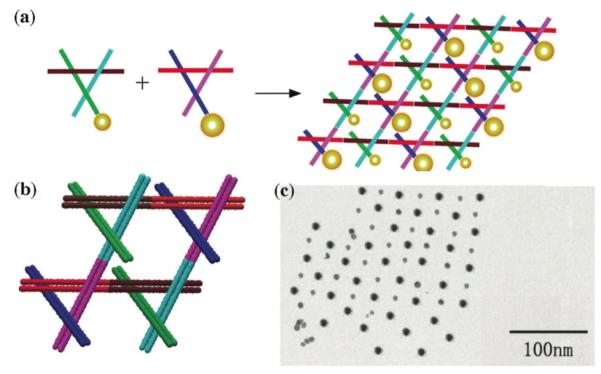
(a) Two different 3D-DX motifs, containing 5 nm or 10 nm particles on one end of a propagation direction yield a checkerboard nanoparticle array. (b) The two 3D-DX motifs in greater detail assembled to form a 2D array. (c) A TEM image showing a checkerboard array of gold nanoparticles.
[2] Algorithmic Assembly
The crystallographic background of the author has led to an emphasis on periodic matter in this article. However, it is also possible to organize aperiodic matter. Winfree and his colleagues have focused on this aspect of assembly. The advantages of algorithmic assembly are that complex algorithmic patterns can be generated using only a few tiles. For example, Pascal’s triangle modulo 2, in which all the even numbers are replaced by 0 and the odd numbers by 1, is known as a Sierpinski triangle. This fractal pattern can be generated by four tiles that correspond to the logical operations of exclusive OR (XOR), as well as three tiles for the vertex and sides of the triangle. The XOR gate yields a 0 if the two inputs are the same, and a 1 if they are different. Currently, the main issue in algorithmic assembly is its extreme sensitivity to errors relative to periodic assembly. This problem derives from the fact that correct tiles in periodic assembly are competing only with incorrect tiles for a slot in the growing array, whereas in algorithmic assembly correct tiles are competing with half-correct tiles (42, 60). Winfree and his colleagues have built Sierpinski triangles (61), most successfully when using an approach that ‘chaperones’ the area of the aperiodic array (62).
The same group has also built a set of tiles that can count in binary (63). The great promise of counting is that 2D crystals, and perhaps 3D crystals, might be built to precise dimensions.
[3] DNA Analogs
It is a natural question as to whether other nucleic acids are capable of being used as the basis of precise self-assembly. Jaeger’s group has mined the loop-loop tertiary interactions in a variety of RNA crystal structures, and has exploited them as the basis for self-assembled arrangements. He has taken robust motifs, such as tRNA molecules, and assembled them to form square-like species and small arrays (64, 65). Nobody seems to have tried to use RNA in the exclusively sticky-ended fashion that structural DNA nanotechnology has usually employed. Chaput and his colleagues have built a branched junction from glycerol nucleic acid (66). Combining different types of nucleic acid molecules in the same construct is tricky, because constructs like the DX molecule are very sensitive to double helical twist. 2D arrays mixing DNA and PNA were used to establish the helical repeat of the DNA/PNA double helix (67).
[4] Semi-Natural Constructs
In addition to DNA analogs, it is possible to combine other chemical species with DNA in nanoconstructs. Sleiman’s laboratory has performed a number of studies in this vein. For example, they have placed metallo-organic complexes in junction sites (68), yielding different properties in the presence or absence of the metal; they have also placed metals at the corners of metal-nucleic acid cages (69). In a related development, Kieltyka et al. have shown that it is possible to stimulate G-tetrad formation by use of an organometallic molecule that is square-shaped (70). The same group has shown that DNA can pair with a molecule containing a conjugated backbone (71). Although DNA is readily recognized as a special molecule, it is extremely valuable for the future of DNA-based materials to be able to combine it with the other molecular species that can be synthesized.
VI. DNA-Based Nanomechanical Devices
[1] Devices Based on Structural Transitions
The description above has emphasized structure and the placement of atoms in space. It is natural to ask whether it is also possible to change the arrangement with time, by exploiting some sort of transition that can be induced. There are a number of structural transitions of DNA that can be exploited to produce nanomechanical motion. The first such device was based of the transition from right-handed B-DNA to left-handed Z-DNA (72). There are two conditions necessary for the B-Z transition, a sequence that is capable of undergoing it readily, and solution conditions that promote it (73). One can use the first requirement to limit how much of the DNA undergoes the transition (the ‘proto-Z DNA’), and the second requirement to drive the transition one way or the other. The device consists of two DX components flanking a stretch of proto-Z DNA. Figure 10a shows how the two domains of the DX portions switch position when the device undergoes the transition. Other devices have been built that exploit the transition of oligo-dC sequences to the I-form of DNA (74). The pH-dependence of this device has been used to measure intracellular pH by combining it with fluorescent labels. The propensity of oligo-dG sequences to form tetraplex structures has also been used to build potassium-responsive nanomechanical DNA-based devices (75).
Figure 10. DNA-Based Nanomechanical Devices.

(a) A DNA Nanomechanical Device Based on the B-Z Transition. The device consists of two DX molecules connected by a shaft containing 20 nucleotide pairs (yellow) capable of undergoing the B-Z transition. Under B-promoting conditions the short domains are on the same side of the shaft, but under Z-promoting conditions (added Co(NH ) 3+ 3 6 ) they are on opposite sides of the shaft. The pink and green FRET pair are used to monitor this change. (b) The Machine Cycle of a PX-JX2 Device. Starting with the PX device on the left, the green set strands are removed by their complements (Process I) to leave an unstructured frame. The addition of the yellow set strands (process II) converts the frame to the JX2 structure, in which the top and bottom domains are rotated a half turn relative to their arrangement in the PX conformation. Processes III and IV reverse this process to return to the PX structure. (c) AFM Demonstration of the Operation of the Device. A series of DNA trapezoids are connected by devices. In the PX state, the trapezoids are in a parallel arrangement, but when the system is converted to the JX2 state, they are in a zigzag arrangement. (d) Insertion of a Device Cassette into a 2D Array. The eight TX tiles that form the array are shown in differently colored outlined tiles. For clarity the cohesive ends are shown to be the same geometrical shape, although they all contain different sequences. The domain connecting the cassette to the lattice is not shown. The cassette and reporter helix are shown as red filled components; the marker tile is labeled ‘M’ and is shown with a black filled rectangle representing the domain of the tile that protrudes from the rest of the array. Both the cassette and the marker tile are rotated about 103° from the other components of the array (three nucleotides rotation). The PX arrangement is shown at the left and the JX2 arrangement is on the right. Note that the reporter hairpin points towards the marker tile in the PX state, but points away from it in the JX2 state.
[2] Sequence Dependent Devices
As intriguing and useful as DNA devices may be when based on structural transitions, the true power of using DNA is its programmability; single-trigger structural transitions can lead to only a few variants, even with nuanced DNA chemistry (35). The most effective way to construct multiple devices is to make them sequence dependent. Although the rate of response is limited by strand diffusion times, this seems to be the most robust way of achieving different responses from multiple devices in the same solution. The first sequence-dependent device was a molecular tweezers constructed by Yurke and his colleagues (76). The method that they used to change states was to add an 8-nucleotide ‘toehold’ to a controlling strand in the system. This toehold is designed to be unpaired when added to the device. It is removed by the addition of the complete complement to the strand containing the toehold. The complement binds to the toehold and removes the rest of the strand through branch migration. The reaction is downhill, because more base pairs are formed by removing the toehold-containing stand than existed when it was bound to the device. Virtually every sequence-specific device utilizes this toehold principle.
A nanomechanical device is termed ‘robust’ if it behaves like a macroscopic device, neither multimerizing nor dissociating when going through its machine cycle. The machine cycle of a robust sequence-dependent device controlled by ‘set’ strands (16) is shown in Figure 10b. This device has two states, called PX and JX2, which differ from each other by a half-turn rotation between their tops and bottoms. Starting in the PX state at left, addition of the full complement to the green set strands (tailed in a biotin, represented by a black dot) removes the green strands, leaving a naked frame. Addition of the yellow set strands switches the device to the JX2 state. The cycle is completed when the complements of the yellow strands are added to strip them from the frame and the green set strands are added. Figure 10c shows in schematic that changing states can affect the structure of reporter trapezoids connected by the device. This panel also shows AFM images of these state changes.
The notion of combining devices and lattices enables one not only to place atoms at desired locations in a fixed fashion, but to vary their positions with time in a programmed fashion. This goal has been achieved in 2D (15, 42) by developing a cassette with three components: One component is the PX-JX2 device, the second is a domain that attaches the cassette to a 2D array, and the third is a reporter arm whose motion can be detected by AFM. This system is illustrated in Figure 10d, where the device has been incorporated into an eight-tile TX lattice. The PX state is shown on the left and the JX2 state is shown on the right. The reporter arm is seen to change its orientation relative to a marker tile (black) when the device state is switched.
The notion of exploiting toeholds that lead to systems with more base pairs dominates many activities in structural DNA nanotechnology. Clocked DNA walkers (77, 78) (devices that walk on sidewalks, where each step require intervention) have been developed using this principle. Recently, the same notion has been used to produce an autonomous walker that can move without intervention, once initiated (79). Pierce and his colleagues have recently suggested using this approach as a route to programming structural assembly (5). Likewise, Winfree and his colleagues have used the same approach to control molecular circuitry (80). No purely-DNA device based on any other reaction has been reported.
VII. Further Considerations
[1] Other Systems
DNA is a convenient material to use to build specific structural species. Space does not permit us to discuss the use of loosely organized DNA combined with proteins, a system that has great utility and has been pioneered by Niemeyer (81). The use of unstructured DNA as a ‘smart glue’ has been utilized by Mirkin and his colleagues (82) when combined with gold nanoparticles. The difference between nanoparticles free in solution and those bound together by the presence of a DNA molecule is readily detected by a color change. Gang’s (83) and Mirkin’s (84) groups have also produced 3D nanoparticle microcrystals using unstructured DNA.
[2] Symmetry
We emphasized earlier that minimizing symmetry leads to greater control over any system, although Mao has indicated that the maximization of symmetry also leads to minimization of purification. This can lead to components of greater purity and possibly greater extent. For example, he and his colleagues have made large 2D arrays using a single strand (20). In another take on symmetry, Yan has made finite symmetric arrangements of DNA with specific symmetries (85). As a consequence of the symmetry, those investigators are able to use a relatively small number of DNA tiles for each of their constructs.
[3] Cloning
As soon as one hears that DNA is being used to build materials, the question immediately presents itself: Can this material be reproduced biologically, rather than by complex laboratory procedures? The first suggestion for this approach is general, but has not been attempted (86). Shih et al. (30) were able to clone a strand that folded into an octahedron, along with the aid of five helper strands. In 2007, Yan and his colleagues reproduced a PX structure by in vitro rolling circle methods, a key initial step in the process (87). More recently, this group has managed to perform the same task in vivo for both a branched junction and a PX structure (88). However, it is important to point out that both of these cloned structures are topologically trivial: They are both equivalent to circles, and an open challenge is to produce a complex topology by cloning.
VIII. Conclusion and Prospects
[1] A Toolbox Provided by Nature
The complementarity of DNA (2) and the ability of complementary strands to hybridize (3) facilitate the programmability of intermolecular affinity (4). In addition, the structural predictability of both sticky ended cohesion products (6) and of specially designed branched DNA motifs (12, 14, 43, 45, 50) enable the programmability of structure. In this article, I have focused only on the aspects of DNA nanotechnology that derive from both of these features, emphasizing the capability of placing matter in particular spatial positions and the ability to change those structures at particular times through programmed molecular motion (16,73,76,77-79). There are many approaches to new materials, including new materials based on DNA [e.g., references 81 and 82], that are valuable and do not require this level of precision; however, the prejudices of this author and the limitations of space have prevented coverage of this aspect of DNA materials. The highest known precision of DNA constructs in 3D (250 microns in extent) is now 4 Å resolution, but other crystals (up to 1 mm in dimension) are readily available to 10 Å resolution (51). DNA origami has been estimated to provide a pixel size of about 60 Å (21). The quality of 2D crystals (typical dimensions ~1 micron) is unclear, because it is rare for the primary method of observing them, AFM, to give resolutions better than ~7 nm). It is clear that nature has provided a molecular basis for synthesizing molecules whose structures can be controlled with high precision outside the biological context. The properties of DNA may or may not be ideal for particular materials applications. However, there are large numbers of analogs (e.g., refs. 89, 90) whose features may be more suitable and might be used instead.
[2] A Growing Rapidly Field
The most exciting development in nucleic acid nanotechnology involving in the last decade is the growth of the field. Around the turn of the millennium, the effort in this field was basically limited to the originating laboratory (9, 91). Today, 40- 50 laboratories are involved in the effort. This article has a tilt that emphasizes my personal interests. Unfortunately, length restrictions have prevented me from mentioning much of the exciting work done by numerous investigators; I apologize to them for these omissions. The expansion of the field is arguably the most powerful development of the last decade: Different people have different perspectives on solutions to the problems that exist, leading to greater likelihoods of success. Furthermore, more investigators lead to more innovative approaches, exemplified by the developments of Rothemund’s origami (21), Shih’s NMR system (44), Gothelf et al.’s DNA box (33), Sleiman’s use of coordination chemistry (69), Yan’s cloning (88), Jaeger (64) and Paukstelis’s (92) uses of tertiary interactions and Mao’s autonomous device (93). The increasing recognition of the power of directing molecular interactions by internally programmed molecular information is leading to an extremely bright future for this field. It is impossible to predict where this field is going, but with so many new and imaginative investigators involved, it will undoubtedly lead to increasingly exciting applications.
Summary Points List.
The development of structural DNA nanotechnology relies on the control of DNA hybridization, on the ready availability of synthetic DNA and on the ability to design and self-assemble stable branched DNA molecules.
Sequence design and motif design are key elements of this field.
It is possible to form molecules with the connectivities of various DNA polyhedra by joining branched species via sticky ends; the shapes of molecules with only triangular faces will be correctly formed.
Stiff motifs are needed for periodic arrangements of DNA motifs. Using them, it is possible to self-assemble two-dimensional and three-dimensional crystalline arrangements. More complex arrangements, known as algorithmic assemblies, can also be self-organized within the context of a chaperoning border.
It is possible to use DNA to build nanomechanical devices. Some are based on DNA structural transitions, but others are sequence-dependent, availing themselves of the full power of DNA programmability.
DNA can be used to organize other species in space, including proteins and nanoelectronic components.
DNA origami has resulted in facile construction of both two dimensional and three dimensional patterns and motifs. These range from a ‘smiley face’ in two dimensions, to a box with a keyed lock in three dimensions.
During the past decade the field has expanded from a single laboratory to nearly 50 investigators, signaling increasing interest in finding applications for programming the information in DNA beyond its genetic role, for structural and dynamic purposes.
Key Future Issues.
Can the resolution of 3D DNA crystals be improved from 4 Å to 1-2 Å? If so, it will be possible to use this system as the basis for crystallizing biological macromolecules.
Can the sizes of 2D DNA arrays be increased from ~1 micron to large areas? This would enable the use of DNA to organize nanoelectronics on a practical scale.
Can DNA origami tiles be self-assembled in the same way as small motifs? This would greatly increase the addressability of DNA-based surfaces.
Can nanoparticles and other nanoelectronic components be fit into 3D DNA arrays? This would usher in a new era in the ability to organize nanoelectronics and nanophotonics from the bottom up, with an entirely new paradigm.
Can error-free algorithmic assembly be increased in extent and can it be extended to 3D? This will enable the construction of 3D crystals of designated size.
Can nucleic acid analogs be used as effectively as DNA for nanoconstruction? If so, applications will not be limited to those compatible with an extensive polyanion with the structural characteristics of DNA.
Can some source of energy other than increased base pairing be employed for DNA-based nanoedevices? If so, more sophisticated systems can be developed.
Can DNA-based nanodevices by made sufficiently sophisticated, autonomous and inert to be used within a biological context? If so, cellular-level robotic therapy might be possible.
ACKNOWLEDGEMENTS
I wish to thank all of my students, postdocs, collaborators and colleagues, who brought this field to its current state. I also wish to thank Dr. Paul W.K. Rothemund for the use of Figure 6. This work has been supported by grants GM-29544 from the National Institute of General Medical Sciences, CTS-0608889 and CCF-0726378 from the National Science Foundation, 48681-EL and W911NF-07-1-0439 from the Army Research Office, N000140910181 from the Office of Naval Research and a grant from the W.M. Keck Foundation.
Footnotes
Glossary. No terms not obvious to any educated biochemist that I can tell.
Acronyms. None that are not in common usage.
LITERATURE CITED
- 1.Voet D, Rich A. The crystal structures of purines, pyrimidines and their intermolecular complexes. Prog. Nucl. Acid. Res. & Mol. Biol. 1970;10:183–265. doi: 10.1016/s0079-6603(08)60565-6. [DOI] [PubMed] [Google Scholar]
- 2.Watson JD, Crick FHC. Molecular structure of nucleic acids - a structure for deoxyribose nucleic acid. Nature. 1953;171:737–8. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 3.Rich A, Davies DR. A new, 2-stranded helical structure, polyadenylic acid and polyuridylic acid. J. Am. Chem. Soc. 1956;78:3548–9. [Google Scholar]
- 4.Cohen SN, Chang ACY, Boyer HW, Helling RB. Construction of biologically functional bacterial plasmids in vitro. Proc. Nat. Acad. Sci. (USA) 1973;70:3340–4. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin P, Choi HMT, Calvert CR, Pierce NA. Programming biomolecular self-assembly pathways. Nature. 2008;451:318–23. doi: 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- 6.Qiu H, Dewan JD, Seeman NC. A DNA Decamer with a Sticky End: The Crystal Structure of d-CGACGATCGT. J. Mol. Biol. 1997;267:881–98. doi: 10.1006/jmbi.1997.0918. [DOI] [PubMed] [Google Scholar]
- 7.Watson JD, Crick FHC. Genetical implications of the structure of deoxyribonucleic acid. Nature. 1953;171:964–67. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- 8.Holliday R. Mechanism for gene conversion in fungi. Genet. Res. 1964;5:82–304. doi: 10.1017/S0016672308009476. [DOI] [PubMed] [Google Scholar]
- 9.Seeman NC. Nucleic acid junctions and lattices. J. Theor. Biol. 1982;99:237–47. doi: 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- 10.Hsieh P, Panyutin IG. DNA branch migration. In: Eckstein F, Lilley DMJ, editors. Nucleic Acids & Mol. Biol. Vol. 9. Springer-Verlag; Berlin: 1995. pp. 42–65. [Google Scholar]
- 11.Caruthers MH. Gene synthesis machines: DNA chemistry and its uses. Science. 1985;230:281–5. doi: 10.1126/science.3863253. [DOI] [PubMed] [Google Scholar]
- 12.Fu T-J, Seeman NC. DNA double crossover structures. Biochem. 1993;33:3311–20. doi: 10.1021/bi00064a003. [DOI] [PubMed] [Google Scholar]
- 13.Sa-Ardyen P, Vologodskii AV, Seeman NC. The flexibility of DNA double crossover molecules. Biophys. J. 2003;84:3829–37. doi: 10.1016/S0006-3495(03)75110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LaBean TH, Yan H, Kopatsch J, Liu F, Winfree E, Reif JH, Seeman NC. The construction, analysis, ligation and self-assembly of DNA triple crossover complexes. J. Am. Chem. Soc. 2000;122:1848–60. [Google Scholar]
- 15.Ding B, Seeman NC. Operation of a DNA robot arm inserted into a 2D DNA crystalline substrate. Science. 2006;31:1583–85. doi: 10.1126/science.1131372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan H, Zhang X, Shen Z, Seeman NC. A robust DNA mechanical device controlled by hybridization topology. Nature. 2002;415:62–65. doi: 10.1038/415062a. [DOI] [PubMed] [Google Scholar]
- 17.Lu M, Guo Q, Marky LA, Seeman NC, Kallenbach NR. Thermodynamics of DNA chain branching. J. Mol. Biol. 1992;223:781–89. doi: 10.1016/0022-2836(92)90989-w. [DOI] [PubMed] [Google Scholar]
- 18.Seeman NC. De novo design of sequences for nucleic acid structure engineering. J. Biomol. Struct. Dyns. 1990;8:573–81. doi: 10.1080/07391102.1990.10507829. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Seeman NC. The assembly and characterization of 8-arm and 12-arm DNA branched junctions. J. Am. Chem. Soc. 2007;129:8169–76. doi: 10.1021/ja0693441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, Chen Y, He Y, Ribbe AE, Mao C. Approaching the limit: Can one oligonucleotide assemble into large nanostructures? Angew. Chemie Int. Ed. 2006;45:1942–45. doi: 10.1002/anie.200504022. [DOI] [PubMed] [Google Scholar]
- 21.Rothemund PWK. Scaffolded DNA origami for nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 22.Douglas SM, Dietz H, Liedl T, Högborg B, Graf F, Shih WM. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature. 2009;459:414–18. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dietz H, Douglas SM, Shih WM. Folding DNA into twisted and curved nanoscale shapes. Science. 2009;325:725–30. doi: 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Högborg B, Liedl T, Shih WM. Folding DNA origami from a double-stranded source of scaffold. J. Am. Chem. Soc. 2009;131:9154–55. doi: 10.1021/ja902569x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma R-I, Kallenbach NR, Sheardy RD, Petrillo ML, Seeman NC. 3-Arm nucleic acid junctions Are flexible. Nucl. Acids Res. 1986;14:9745–53. doi: 10.1093/nar/14.24.9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petrillo ML, Newton CJ, Cunningham RP, Ma R-I, Kallenbach NR, Seeman NC. The ligation and flexibility of 4-arm DNA junctions. Biopolymers. 1988;27:1337–52. doi: 10.1002/bip.360270902. [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Seeman NC. The synthesis from DNA of a molecule with the connectivity of a cube. Nature. 1991;350:631–33. doi: 10.1038/350631a0. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Seeman NC. The construction of a DNA truncated octahedron. J. Am. Chem. Soc. 1994;116:1661–69. [Google Scholar]
- 29.Goodman RP, Schaap IAT, Tardin CF, Erben CM, Berry RM, Schmidt CF, Turberfield AJ. Rapid chiral assembly of rigid DNA building blocks for molecular nanofabrication. Science. 2005;310:1661–65. doi: 10.1126/science.1120367. [DOI] [PubMed] [Google Scholar]
- 30.Shih WM, Quispe JD, Joyce GF. DNA that folds into a nanoscale octahedron. Nature. 2004;427:618–21. doi: 10.1038/nature02307. [DOI] [PubMed] [Google Scholar]
- 31.He Y, Ye T, Su M, Zhang C, Ribbe AE, Jiang W, Mao C. Hierarchical self-assembly of DNA into symmetric supramolecular polyhedra. Nature. 2008;452:198–201. doi: 10.1038/nature06597. [DOI] [PubMed] [Google Scholar]
- 32.Erben CM, Goodman RP, Turberfield AJ. Single molecule protein encapsulation in a rigid DNA cage. Angew. Chemie Int. Ed. 2006;45:7414–17. doi: 10.1002/anie.200603392. [DOI] [PubMed] [Google Scholar]
- 33.Andersen ES, Dong M, Nielsen MM, Jahn K, Subramani R, Mamdouh W, Golas MM, Sander B, Stark H, Oliviera CLP, Pedersen JS, Birkedal V, Besenbacher F, Gothelf KV, Kjems J. Self-assembly of a nanoscale box with a controllable lid. Nature. 2009;459:p73–76. doi: 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- 34.Seeman NC. The design of single-stranded nucleic acid knots. Molec. Eng. 1992;2:297–307. [Google Scholar]
- 35.Du SM, Stollar BD, Seeman NC. A synthetic DNA molecule in three knotted topologies. J. Am. Chem. Soc. 1995;117:1194–1200. [Google Scholar]
- 36.Fu T-J, Tse-Dinh Y-C, Seeman NC. Holliday junction crossover topology. J. Mol. Biol. 1994;236:91–105. doi: 10.1006/jmbi.1994.1121. [DOI] [PubMed] [Google Scholar]
- 37.Mao C, Sun W, Seeman NC. Assembly of Borromean rings from DNA. Nature. 1997;386:137–38. doi: 10.1038/386137b0. 1997. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Di Gate RJ, Seeman NC. An RNA Topoisomerase. Pro. Nat. Acad. Sci. (USA) 1996;93:9477–82. doi: 10.1073/pnas.93.18.9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan H, LaBean TH, Feng L, Reif JH. Directed nucleation assembly of DNA tile complexes for barcode-patterned lattices. Proc. Nat. Acad. Sci. 2003;100:8103–08. doi: 10.1073/pnas.1032954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Yan H, Shen Z, Seeman NC. Paranemic cohesion of topologically-closed DNA molecules. J Am. Chem. Soc. 2002;124:12940–41. doi: 10.1021/ja026973b. [DOI] [PubMed] [Google Scholar]
- 41.Winfree E, Liu F, Wenzler LA, Seeman NC. Design and self-assembly of two-dimensional DNA crystals. Nature. 1998;394:539–44. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 42.Gu H, Chao J, Xiao SJ, Seeman NC. Dynamic patterning programmed by DNA tiles captured on a DNA origami substrate. Nature Nanotech. 2009;4:245–49. doi: 10.1038/nnano.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathieu F, Liao S, Mao C, Kopatsch J, Wang T, Seeman NC. Six-helix bundles designed from DNA. NanoLett. 2005;5:661–65. doi: 10.1021/nl050084f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Douglas SM, Chou JJ, Shih WM. DNA-nanotube-induced alignment of membrane proteins for NMR structure determination. Proc. Nat. Acad. Sci. (USA) 2007;104:6644–48. doi: 10.1073/pnas.0700930104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, Yang X, Qi J, Seeman NC. Antiparallel DNA double crossover molecules as components for nanoconstruction. J. Am. Chem. Soc. 1996;118:6131–40. [Google Scholar]
- 46.Ding B, Sha R, Seeman NC. Pseudohexagonal 2D DNA crystals from double crossover cohesion. J. Am. Chem. Soc. 2004;126:10230–31. doi: 10.1021/ja047486u. [DOI] [PubMed] [Google Scholar]
- 47.Seeman NC. Curr. Protocols Nucl. Acid Chem. John Wiley & Sons; New York: 2002. Key experimental approaches in DNA nanotechnology. Unit 12.1. [DOI] [PubMed] [Google Scholar]
- 48.Mao C, Sun W, Seeman NC. Designed two-dimensional DNA Holliday junction arrays visualized by atomic force microscopy. J. Am. Chem. Soc. 1999;121:5437–43. [Google Scholar]
- 49.Kim SH, Quigley GJ, Suddath FL, McPherson A, Sneden D, Kim JJ, Weinzierl J, Blattman P, Rich A. 3 Dimensional structure of yeast phenylalanine transfer-RNA -- shape of the molecule at 5.5 Å resolution. Proc. Nat. Acad. Sci. 1972;69:3746–50. doi: 10.1073/pnas.69.12.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu D, Wang W, Deng Z, Walulu R, Mao C. Tensegrity: Construction of rigid DNA triangles with flexible four-arm junctions. J. Am. Chem. Soc. 2004;126:2324–25. doi: 10.1021/ja031754r. [DOI] [PubMed] [Google Scholar]
- 51.Zheng J, Birktoft JJ, Chen Y, Wang T, Sha R, Constantinou PE, Ginell SL, Mao C, Seeman NC. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature. 2009 doi: 10.1038/nature08274. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park SH, Yin P, Liu Y, Reif JH, LaBean TH, Yan H. Programmable DNA self-assemblies for nanoscale organization of ligands and proteins. NanoLett. 2005;5:729–33. doi: 10.1021/nl050175c. [DOI] [PubMed] [Google Scholar]
- 53.Rinker S, Ke YG, Liu Y, Chhabra R, Yan H. Self-assembled DNA nanostructures for distance-dependent multivalent ligand-protein binding. Nature Nanotech. 2008;7:418–422. doi: 10.1038/nnano.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson BH, Seeman NC. The design of a biochip: A self-assembling molecular-scale memory device. Protein Eng. 1987;1:295–300. doi: 10.1093/protein/1.4.295. [DOI] [PubMed] [Google Scholar]
- 55.Alivisatos AP, Johnson KP, Peng XG, Wilson TE, Loweth CJ, Bruchez MP, Schultz PG. Organization of ‘nanocrystal molecules’ using DNA. Nature. 1996;382:609–11. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]
- 56.Mastroianni AJ, Claridge SA, Alivisatos AP. Pyramidal and chiral groupings of gold nanocrystals assembled using DNA scaffolds. J. Am. Chem. Soc. 2009;131:8455–59. doi: 10.1021/ja808570g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li HY, Park SH, Reif JH, LaBean TH, Yan H. DNA-templated self-assembly of protein and nanoparticle linear arrays. J. Am. Chem. Soc. 2004;126:418–419. doi: 10.1021/ja0383367. [DOI] [PubMed] [Google Scholar]
- 58.Pinto YY, Le JD, Seeman NC, Musier-Forsyth K, Taton TA, Kiehl RA. Sequence-encoded self-assembly of multiple-nanocomponent arrays by 2D DNA scaffolding. NanoLett. 2005;5:2399–2402. doi: 10.1021/nl0515495. [DOI] [PubMed] [Google Scholar]
- 59.Zheng J, Constantinou PE, Micheel C, Alivisatos AP, Kiehl RA, Seeman NC. 2D nanoparticle arrays show the organizational power of robust DNA motifs. NanoLett. 2006;6:1502–04. doi: 10.1021/nl060994c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mao C, LaBean TH, Reif JH, Seeman NC. Logical computation using algorithmic self-assembly of DNA triple crossover molecules. Nature. 2000;407:493–96. doi: 10.1038/35035038. [DOI] [PubMed] [Google Scholar]
- 61.Rothemund PWK, Papadakis N, Winfree E. Algorithmic self-assembly of DNA Sierpinski triangles. PLOS Biol. 2004;2:2041–52. doi: 10.1371/journal.pbio.0020424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fujibayashi K, Hariadi R, Park SH, Winfree E, Murata S. A fixed-width cellular automaton pattern. NanoLett. 2008;8:1791–97. doi: 10.1021/nl0722830. [DOI] [PubMed] [Google Scholar]
- 63.Barish RD, Rothemund PWK, Winfree E. Two computational primitives for algorithmic self-assembly: copying and counting. NanoLett. 2005;5:2586–92. doi: 10.1021/nl052038l. [DOI] [PubMed] [Google Scholar]
- 64.Chworos A, Severcan I, Koyfman AY, Weinkam P, Oroudjev E, Hansma HG, Jaeger L. Building programmable jigsaw puzzles with RNA. Science. 2004;306:2068–72. doi: 10.1126/science.1104686. [DOI] [PubMed] [Google Scholar]
- 65.Severcan I, Geary C, Venzemnieks A, Jaeger L. Square-shaped RNA particles from different RNA folds. NanoLett. 2009;9:1270–77. doi: 10.1021/nl900261h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang RS, McCullum EO, Chaput JC. Synthesis of two mirror-image 4-helix junctions derived from glycerol nucleic acid. J. Am. Chem. Soc. 2008;130:5846–47. doi: 10.1021/ja800079j. [DOI] [PubMed] [Google Scholar]
- 67.Lukeman PS, Mittal A, Seeman NC. Two dimensional PNA/DNA arrays: Estimating the helicity of unusual nucleic acid polymers. Chem. Comm. 2004;2004:1694–95. doi: 10.1039/b401103a. [DOI] [PubMed] [Google Scholar]
- 68.Yang H, Sleiman HF. Templated synthesis of highly stable, electroactive and dynamic metal-DNA branched junctions. Angew. Chemie Int. Ed. 2008;47:2443–46. doi: 10.1002/anie.200703741. [DOI] [PubMed] [Google Scholar]
- 69.Yang H, McLaughlin CK, Aldaye FA, Hamblin GD, Rys AZ, Rouiller I, Sleiman HF. Metal-nucleic acid cages. Nature Chem. 2009;1:390–96. doi: 10.1038/nchem.290. [DOI] [PubMed] [Google Scholar]
- 70.Kieltyka R, Engelbienne P, Fakhoury J, Autexier C, Moitessier N, Sleiman HF. A platinum supramolecular square as an effective G-quadruplex binder and telomerase inhibitor. J. Am. Chem. Soc. 2008;130:10040–41. doi: 10.1021/ja8014023. [DOI] [PubMed] [Google Scholar]
- 71.Lo PK, Sleiman HF. Synthesis and molecular recognition of conjugated polymer with DNA-mimetic properties. Macromolecs. 2008;41:5590–5603. [Google Scholar]
- 72.Rich A, Nordheim A, Wang AHJ. The chemistry and biology of left-handed Z-DNA. Ann. Rev. Biochem. 1984;53:791–846. doi: 10.1146/annurev.bi.53.070184.004043. [DOI] [PubMed] [Google Scholar]
- 73.Mao C, Sun W, Shen Z, Seeman NC. A DNA nanomechanical device based on the B-Z transition. Nature. 1999;397:144–46. doi: 10.1038/16437. [DOI] [PubMed] [Google Scholar]
- 74.Modi S, Swetha MG, Goswami D, Gupta G, Mayor S, Krishnan Y. A DNA nanomachine maps spatiotemporal pH changes in living cells. Nature Nanotech. 2009 doi: 10.1038/nnano.2009.83. in press. [DOI] [PubMed] [Google Scholar]
- 75.Hou X, Guo W, Xia F, Nie FQ, Dong H, Tian Y, Wen LP, Wang L, Cao LX, Yang Y, Xue JM, Song YL, Wang YG, Liu DS, Jiang L. A biomimetic potassium-responsive nanochannel: G-quadruplex conformational switching in a synthetic nanopore. J. Am. Chem. Soc. 2009;131:7800–05. doi: 10.1021/ja901574c. [DOI] [PubMed] [Google Scholar]
- 76.Yurke B, Turberfield AJ, Mills AP, Jr., Simmel FC, Newmann JL. A DNA-fuelled molecular machine made of DNA. Nature. 2000;406:605–08. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- 77.Sherman WB, Seeman NC. A precisely controlled DNA bipedal walking device. NanoLett. 2004;4:1203–07. [Google Scholar]
- 78.Shin JS, Pierce NA. A synthetic DNA walker for molecular transport. J. Am. Chem. Soc. 2004;126:10834–35. doi: 10.1021/ja047543j. [DOI] [PubMed] [Google Scholar]
- 79.Omabegho T, Sha R, Seeman NC. A bipedal DNA Brownian motor with coordinated legs. Science. 2009;324:67–71. doi: 10.1126/science.1170336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang DY, Turberfield AJ, Yurke B, Winfree E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 2007;318:1121–25. doi: 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]
- 81.Niemeyer CM. Functional devices from DNA and proteins. Nano Today. 2007;2:42–52. [Google Scholar]
- 82.Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Science. 1996;382:607–09. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 83.Nykypanchuk D, Maye MM, van der Lelie D, Gang O. DNA-guided crystallization of colloidal nanoparticles. Nature. 2008;451:549–52. doi: 10.1038/nature06560. [DOI] [PubMed] [Google Scholar]
- 84.Park SY, Lytton-Jean AKR, Lee B, Weigand S, Schatz GC, Mirkin CA. DNA-programmable nanoparticle crystallization. Nature. 2008;451:553–56. doi: 10.1038/nature06508. [DOI] [PubMed] [Google Scholar]
- 85.Liu Y, Ke Y, Yan H. Self-assembly of symmetric finite-size DNA arrays. J. Am. Chem. Soc. 2005;127:17140–41. doi: 10.1021/ja055614o. [DOI] [PubMed] [Google Scholar]
- 86.Seeman NC. The construction of 3-D stick figures from branched DNA. DNA & Cell Biol. 1991;10:475–86. doi: 10.1089/dna.1991.10.475. [DOI] [PubMed] [Google Scholar]
- 87.Lin C, Wang X, Liu Y, Seeman NC, Yan H. Rolling circle enzymatic replication of a complex multi-crossover DNA nanostructure. J. Am. Chem. Soc. 2007;129:14475–81. doi: 10.1021/ja0760980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin C, Rinker S, Wang X, Liu Y, Seeman NC, Yan H. In vivo cloning of artificial DNA nanostructures. Proc. Nat. Acad. Sci. (USA) 2008;105:17626–31. doi: 10.1073/pnas.0805416105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Freier SM, Altmann KH. The ups and downs of nucleic acid stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucl. Acids Res. 1997;25:4429–43. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Egholm M, Buchardt O, Nielsen PE, Berg RH. Peptide nucleic-acids (PNA) - oligonucleotide analogs with an achiral peptide backbone. J. Am. Chem. Soc. 1992;114:1895–97. [Google Scholar]
- 91.Seeman NC. DNA nanotechnology: novel DNA constructions. Ann. Rev. Biophys. Biomol. Struct. 1998;27:225–48. doi: 10.1146/annurev.biophys.27.1.225. [DOI] [PubMed] [Google Scholar]
- 92.Paukstelis P, Nowakowski J, Birktoft JJ, Seeman NC. The crystal structure of a continuous three-dimensional DNA lattice. Chem. Biol. 2004;11:1119–26. doi: 10.1016/j.chembiol.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 93.Chen Y, Mao C. Putting a brake on an autonomous DNA nanomotor. J. Am. Chem. Soc. 2004;126:8626–27. doi: 10.1021/ja047991r. [DOI] [PubMed] [Google Scholar]