

Abstract
Purpose: To perform preimplantation genetic diagnosis (PGD) of Leigh encephalopathy, we developed a rapid and reliable quantification assay for the percentage of T8993G mtDNA mutation and analyzed various specimens. Methods: We prepared the standard curve by measuring serial proportion of 8993T/G cloned plasmid DNA using real-time PCR, and measured (1) mutant DNA (known proportions by PCR-RFLP), (2) single lymphocytes from 46% mutant carrier, (3) 123 blastomeres from 20 abnormal embryos. Results: (1) These were within −5∼+6% error range, (2) mean 44.3%(11–70%), (3) Five embryos harbored T8993G mutation (4–22%). Embryos from same person indicated different degrees of heteroplasmy, and blastomeres from same embryo demonstrated limited dispersion of heteroplasmy (2–11%). Conclusions: (1) This method provides rapid and reliable PGD for Leigh encephalopathy. (2) The variable heteroplasmy with somatic mitosis was suggested. (3) T8993G mutation was existed in undeveloped embryo, and the bottleneck theory was supported. The limited heteroplasmy dispersion of blastomeres from same embryo also supported reliability of PGD for T8993G mutation.
Keywords: Mitochondrial DNA, Heteroplasmy, Real-time PCR. Preimplantation genetic diagnosis, Leigh encephalopathy
Introduction
The mitochondrial (mt) DNA is a circular double-stranded DNA molecule with 16.6 kilobases [1]. It presents in several copies in each mitochondrion. Human individual cells contain 100,000 to 200,000 copies of mtDNA [2]. The mtDNA has been recognized that it is maternally inherited and nearly all of the mtDNA is identical in normal individuals. Mutation of mtDNA is seriously concerned with mitochondrial disease. Heteroplasmy (the presence of both normal and mutant mtDNA at different levels within the same cell) accounts for the phenotypic variability of mitochondrial disease [3, 4]. From human maternal carrier of mitochondrial disease to pedigrees, rapid shifts in mtDNA mutant frequency have been observed. To explain this phenomenon, it has been hypothesized that the number of mtDNA within any one oocyte is reduced to as few as five or less during oogenesis, especially in primordial germ cell [5–9]. This is termed “bottleneck theory”. Thus oocytes develop extremely different heteroplasmy ratios of mtDNA mutation (a state termed “mutant load”).
To date, there have not been attempts generally for preimplantation genetic diagnosis (PGD) of mitochondrial disease caused by mtDNA mutation. This is due to three possible main reasons : (I) the mutant load measured in the blastomere will not be the same as the mutant load in the other fetal tissues, (II) the mutant load measured in the blastomere will change during embryogenesis or after birth, (III) the correlation between the mutant load and the disease severity is not obviously understood.
In the majority of mtDNA disorders, these features of mtDNA are fulfilled [10, 11] and prenatal diagnosis or PGD is difficult to perform. However in families with mutations at the mtDNA T8993G point mutation, the mutant load is usually similar in different tissues, indicating that there is no tissue-specific selection of age-related variation, and there is also a strong correlation between the mutant load and the disease severity [12, 13]. Thus PGD may be feasible for this mutation.
The T8993G point mutation in the ATPase6 gene was first described in an adult with neurogenic muscle weakness, ataxia and retinitis pigmentosa (NARP) [14], and is also a common cause of Leigh syndrome [15, 16]. The clinical severity is strongly associated with mutant load. The patient with Leigh syndrome has very high mutation load, typically > 90% mutation of mtDNA, and has severe clinical symptoms. NARP is related with intermediate mutant load of 60 to 80%, while mutant load less than 60% are commonly not associated with clinical symptoms [17, 18].
The only way to perform PGD for mitochondrial disease is analysis of the heteroplasmy ratio in a single blastomere of an early stage embryo (4–8 cells stage). Although the mutation ratio used to be quantified by PCR-RFLP and this method has high sensitivity with low standard deviation [19], it requires many procedures; PCR amplification, digestion with restriction enzyme, and electrophoresis in a genetic analyser. Consequently more simple procedure is desirable for safety and reliable PGD. In the present study, we developed a simple and precision quantification assay of the T8993G mtDNA point mutation using real-time single PCR for PGD of Leigh encephalopathy and analyzed various types of cells and DNAs about this mutation to verify this assay. We also analyzed the segregation patterns of mtDNA in preimplantation development by measuring heteroplasmy ratio of human blastomeres.
Materials and methods
Plasmid DNA and specimens preparation
DNA for the wild-type (8993T) and mutant (8993G) target sequences was generated from cloned plasmid DNA containing pCR® 2.1-TOPO® vector (Invitrogen, USA) and PCR products of primers mtF8838 and mtR9139, and to be sized about 4200bp. The copy numbers of the wild-type and mutant DNA sequences were calculated based on the size and molecular weight of the plasmid DNA. These were mixed to each mutant load, and diluted into 0.04 pg/μl (equivalent to mtDNA from a single blastomere) by TE buffer with 5 ng/μl salmon DNA (Sigma-Aldrich, USA) to avoid adsorption of small DNA to real-time PCR wells.
Four different heteroplasmy types of specimens (lymphocyte cell line or whole blood) from female carriers and patients affected by Leigh encephalopathy with T8993G mutation and whole blood samples from normal woman and MOLT4 cell line as controls were obtained. Four specimens have been analyzed to have 28%, 46%, 62%, 98% of T8993G mutant load by PCR-RFLP respectively. The PCR restriction analysis was undertaken as previously described [20]. MOLT4 cell line was established from lymphocyte of human acute lymphoblastic leukemia and considered to have no T8993G mutation. DNA was extracted from these six types of specimens by using SepaGene (Sanko Junyaku, Japan) and diluted into 10 pg/μl, amounts that equal mtDNA from a single blastomere (104 copies). When a fresh or cultured single lymphocyte with 46% mutation was analyzed, cells were handled in a clean bench with a mouth-controlled fine heat-polished glass micropipette in drops of PBS using an inverted microscope.
Twenty human embryos confirmed to contain three polar bodies (3PN) were donated to this research by thirteen unrelated normal couples. Embryos were considered to be abnormal inadequate embryo to transfer at 24 h following insemination and cultured up to 2–12 cleavage stage, and placed in an equilibrated medium (Sydney IVF Embryo Biopsy Medium: Cook, USA). Then Embryos were irradiated by a non-contact 1.48 μm diode laser system (OCTAX Laser Shot: MTG, Germany) for the piercing of the zona pellucida. Two or three short pulses (2.9 ms) were applied, and the biopsy pipette was inserted in the hole and a blastomere was removed by aspiration. Under an inverted microscope the blastomeres were rinsed in a drop of PBS, and transferred into individual PCR tubes containing 3 μl of cell lysis solution (0.2% sarcosyl + TE buffer with 10 mM EDTA). Whole blastomeres per embryo were examined.
All specimens were collected after receiving informed consent. The research procedure was approved by the Research Ethics Committee of Keio University School of Medicine and the Japan Society of Obstetrics and Gynecology.
Real-time PCR quantification of mtDNA heteroplasmy ratio
Real-time PCR primers and two fluorescent probes (TaqMan®MGB probe; Applied Biosystems, USA) corresponding to normal (8993T) and mutation (8993G) were prepared. The forward primer (5′-CGAAACCATCAGCCTACTCATTCAA-3′) spanned nt 8958 to nt 8982. The reverse primer (5′-CCTGCAGTAATGTTAGCGGTTAGG-3′) spanned nt 9026 to nt 9003, and total length of the amplified product was 69bp. The probe for normal (8993T; CCAATAGCCC[T]GGCCGT) had “VIC” fluorescent and the probe for mutation (8993G; AATAGCCC[G]GGCCGT) had “FAM” fluorescent. The source of the probes was obtained from Applied Biosystems. To confirm PCR product, PCR amplified DNA was sequenced using ABI PRISM 310 genetic analyzer.
Twentyfive μl PCR mixture was set up with final concentrations: 12.5 μl TaqMan Universal PCR Master Mix, No AmpErase UNG (2X), 0.625 μl 40X Assay Mix, 8.875 μl (6.875 μl) distilled water, 0.5 μl × 2 TaqMan MGB Probe, 1.0 μl × 2 PCR primer, 1.0 μl (3.0 μl) specimen. The reactions were performed as follows: initial denaturation at 50°C for 2 min and 95°C for 10 min, and 45 cycles at 92°C for 15 sec (denaturation), 60°C for 1 min (annealing and extension). The allelic discrimination assay using real-time PCR (ABI PRISM 7000) was used to measure each fluorescent, and proportion of these values (mutation [FAM]/normal [VIC]+ mutation [FAM]) was calculated and analyzed for the following studies.
Preparing the standard curve
(1) Two types of plasmid DNA (8993T and 8993G) were mixed and prepared to each mutation ratios (0, 1, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100%). The concentration of plasmid DNA was adjusted into 0.04 pg/μl as stated above. Measurements were performed in ten times, and the average value was applied for standard curves. (2) DNA extracted from 98% mutation carrier cell line and normal woman were mixed to each mutation ratios (0, 1, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 98%). The concentration of extracted DNA was adjusted into 10 pg as aforesaid. Measurements were performed in ten times.
Analysis of various specimens
(1) 28% mutation DNA (n = 20), (2) 46% mutation DNA (n = 20), (3) 62% mutation DNA (n = 20), (4) 98% mutation DNA (n = 20), (5) Single lymphocytes of fresh blood and established cell line derived from the same 46% mutation carrier (n = 40/40 respectively), (6) Single lymphocytes derived from MOLT4 cell line (n = 20), (7) Distilled water as negative control (n = 20), (8) 123 individual blastomeres from 20 cleavaged embryos as a simulation of the actual PGD.
Measured values were calculated and applied to standard curve of plasmid DNA, then converted to mutation percentage.
Results
Linear standard curves were obtained from the proportion of VIC and FAM fluorescence (Figs. 1 and 2). Values of correlation coefficient of VIC for normal sequence and of FAM for mutating sequence were 0.9697 and 0.9745 respectively. Standard curves from carrier DNA and plasmid DNA demonstrated an approximate curve, but carrier DNA showed slightly low values (0.0165∼0.0475; 0% ∼ −8%[mean − 3.6%]). A standard deviation of each curve was demonstrated in very low level (≤0.05).
Fig. 1.
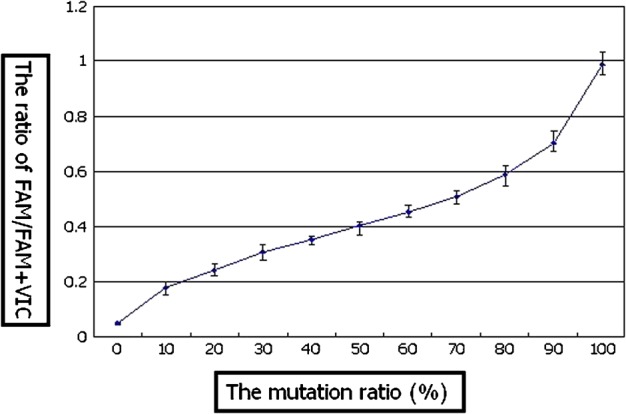
The standard curve of mutation ration analysis; two types of plasmid DNA (8993T/G)
Fig. 2.
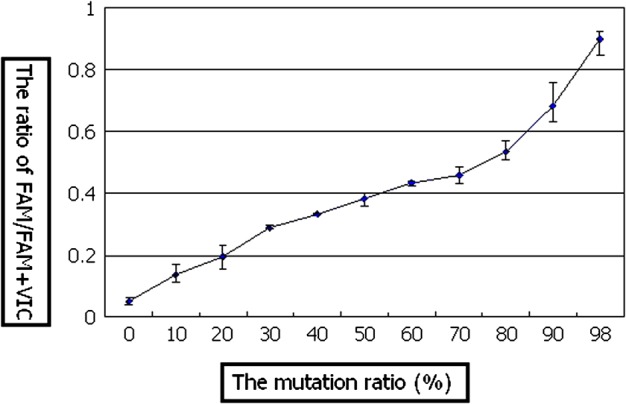
The standard curve of mutation ratio analysis; carrier DNA (98% mutation)
The sequencing result of PCR product showed identical arrangement with mtDNA nt 8958 to nt 9026 (Fig. 3).
Fig. 3.

The sequencing result of PCR product
The standard curve obtained from plasmid DNA was applied to measure mutant load of various specimens. Four different types of heteroplasmic DNA were consistent with standard curve within −5∼+6% error range (Fig. 4). Actual measurement data was as follows; (1) 28%DNA: upper extreme 34%/upper quartile 31%/lower quartile 29%/lower extreme 26%, (2) 46%DNA: 49/48/47/45%, (3) 62%DNA: 68/68/63/60%, (4) 98%DNA: 97/97/94/93%.
Fig. 4.

The mutation ration of each specimens applied to standard curve of Fig. 1
In the analysis of fresh blood single lymphocytes from 46% mutation carrier, a scattering was observed. An average mutant load of 40 lymphocytes was 44.3% with 11–70% wide range among individual cells (upper extreme 70%, upper quartile 52%, lower quartile 38%, lower extreme 11%), and this mean value was close to the result of PCR-RFLP (46%) (Fig. 4). While 80% lymphocytes (32/40) from fresh blood indicated 46% ± 10% range of mutant load, only 45% lymphocytes (18/40) from established cell line showed same range of mutant load (Fig. 5). Mutant load of MOLT4 lymphocytes and negative controls were both 0% (Figs. 4 and 6). While MOLT4 lymphocytes showed only normal fluorescent (VIC), negative controls indicated neither fluorescent.
Fig. 5.

The distribution of single lymphocytes from 46% mutation carrier fresh blood (F) and established cell line (E)
Fig. 6.

The variation of mutation ratio in blastomeres derived from normal women (3PN embryo → cleavage embryo)
T8993G mutation was detected in five embryos (25%; 5/20) with different degrees of heteroplasmy (4 ∼ 22%; embryo6,7,11,16,17) (Fig. 6). Blastomeres derived from same embryo indicated limited dispersion of heteroplasmy within 2 to 11% range (embryo6; 4 ∼ 15%, embryo7; 9 ∼ 20%, embryo11; 11 ∼ 22%, embryo16; 10 ∼ 12%, embyo17; 18 ∼ 21%). Embryos derived from same person indicated different degrees of heteroplasmy (embryo 5,6,7 [0 ∼ 8%,4 ∼ 15%,9 ∼ 20%]/embryo 16,17 [10 ∼ 12%,18 ∼ 21%]).
Discussion
A linearity of standard curve was secured with very low standard deviation and actual measurement data of heteroplasmic DNA was calculated within low error range. However in different DNA concentration, measurements showed a little change (data not shown). This standard curve was prepared under a condition of 104 mtDNA copies per blastomere, but several percentage differences were observed under 103 or 105 mtDNA concentration.
Studying single lymphocytes of the 46% carrier indicated wide range variation of mutant load (11 ∼ 70%[fresh]/0 ∼ 79%[cultured]). This data suggested the possibilities of the variable heteroplasmy at the cellular level by a mtDNA random distribution with somatic mitosis. Previously it has been pointed out that the mutant load of established cell lines might be changed as a result of cell selection during cell culture [21], therefore we expected the result to be different distribution between fresh and cultured lymphocytes. However, significant changes were not seen between them.
The analysis of blastomeres proved an existence of T8993G mtDNA mutation in 3PN embryo from women thought to have no mtDNA mutation. To the best of our knowledge, few existences have been reported so far, although in abnormal cleavage embryo. Mutant loads among embryos from same person were different (embryo 5,6,7 [0 ∼ 8%,4 ∼ 15%,9 ∼ 20%]/embryo 16,17 [10 ∼ 12%,18 ∼ 21%]). These differences were measured more than measurement error of present real-time PCR method. Therefore it suggested an existence of bottleneck theory during oogenesis. The ratio of the heteroplasmy was demonstrated to be a limited dispersion in the individual blastomeres of the same embryo. This result means that the mutant load of single blastomere is practically equal to that of the same embryo. These data are consistent with results from previous studies in mouse embryos [22] and human embryos [19], detecting the distribution of mtDNA polymorphisms in blastomeres. It also supports reliability of PGD for T8993G mutation.
In actual PGD of Leigh encephalopathy, considering various factors (measurement error, difference of first mtDNA amount, random distribution with somatic mitosis etc.), the cut-off value should be established to subtract 10 ∼ 15% from mutation ratio of carrier mother. However it is considered that the avoiding offspring of more severe mutant load will be not difficult, because previous data shows a much skewed segregation of the T8993G mutation in gametes of T8993G carriers. Most oocytes inherited either a very low or a very high proportion of mutant mtDNA [23].
There is a possibility of nuclear transplantation for mtDNA mutation carriers. However many problems exist in nuclear transplantation, principally on safety and the DNA originality of the individual human. At the moment, PGD by measuring mutant load is the only way which can provide an opinion to carrier mother and family for avoiding the transmission of high mutant mtDNA.
Conclusions
These results demonstrate that it is technically and logically possible to perform PGD for T8993G Leigh encephalopathy by measuring the mtDNA heteroplasmy ratio (even small percentages) by allelic discrimination assay using real-time single PCR from small amounts of DNA. This method requires only few hours to obtain results of mutant load, and needs only few procedures, therefore it is expected to avoid misdiagnosis by manipulations and to perform the rapid and accurate PGD.
The research of single lymphocytes and blastomeres suggested the variable heteroplasmy with somatic mitosis, the existence of T8993G mutation in abnormal embryo and the bottleneck theory during oogenesis
Acknowledgments
I appreciate my coworkers’ collaborations and advice on the study, especially Ms. Yoko Yasuda, Ms. Maya Higuchi, Ms. Satoko Moriya, and Ms. Yuko Matumoto, and also alongside the financial and institutional support from the Department of Obstetrics and Gynecology, Keio University School of Medicine.
References
- 1.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–65. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 2.Cummins J. Mitochondrial DNA in mammalian reproduction. J Reprod Fertil. 1998;3:172–82. doi: 10.1530/ror.0.0030172. [DOI] [PubMed] [Google Scholar]
- 3.DiMauro S. Mitochondrial encephalomyopathies. In: Rosenberg RN, Prusiner SB, DiMauro S, editors. The molecular and genetic basis of neurological disease. Boston: Butterworth-Heinemann; 1993. pp. 665–94. [Google Scholar]
- 4.Shoffner JM, Wallace DC. Oxidative phosphorylation diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle MD, editors. The metabolic and molecular bases of inherited disease, 7th ed. New York: McGraw Hill; 1995. pp. 1535–609. [Google Scholar]
- 5.Hauswirth WW, Laipis PJ. Mitochondrial DNA polymorphism in a maternal lineage of Holstein cows. Proc Natl Acad Sci USA. 1982;79:4686–90. doi: 10.1073/pnas.79.15.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hauswirth WW, Laipis PJ. Transmission genetics of mammalian mitochondria: a molecular model and experimental evidence. In: Quagliariello E, Slater EC, Palmieri F, eds. Achievements and perspectives of Mitochodrial Research. Amsterdam: Elsevier, 1985, Vol. II, pp. 49–59.
- 7.Laipis PJ, Van de Walle MJ, Hauswirth WW. Unequal partitioning of bovine mitochondrial genotypes among siblings. Proc Natl Acad Sci USA. 1988;85:8107–10. doi: 10.1073/pnas.85.21.8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashley MV, Laipid PJ, Hauswirth WW. Rapid segregation of heteroplasmic bovine mitochondria. Nucleic Acids Res. 1989;17:7325–31. doi: 10.1093/nar/17.18.7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koehler CM, Lindberg GL, Brown, Beitz DC, Freeman AE, Mayfield JE, Myers AM. Replacement of bovine mitochondrial DNA by a sequence variant within one generation. Genetics. 1991;129:247–55. doi: 10.1093/genetics/129.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. Molecular pathology of MELAS and MERRF: the relationship between mutation load and clinical phenotype. Brain. 1997;120:1713–21. doi: 10.1093/brain/120.10.1713. [DOI] [PubMed] [Google Scholar]
- 11.Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. MELAS and MERRF: the relationship between maternal mutation load and the frequency of clinically affected offspring. Brain. 1998;121:1889–94. doi: 10.1093/brain/121.10.1889. [DOI] [PubMed] [Google Scholar]
- 12.White SL, Shanske S, McGill JJ, Mountain H, Geraghty MT, DiMauro S, Dahl HH, Thorburn Mitochondrial DNA mutations at nucleotide 8993 show a lack of tissue- or age-related variation. J Inherit Metab Dis. 1999;22:899–914. doi: 10.1023/A:1005639407166. [DOI] [PubMed] [Google Scholar]
- 13.Dahl HH, Thorburn, White SL. Towards reliable prenatal diagnosis of mtDNA point mutations: studies of nt8993 mutations in oocytes, fetal tissues, children and adults. Hum Reprod. 2000;15:246–55. doi: 10.1093/humrep/15.suppl_2.246. [DOI] [PubMed] [Google Scholar]
- 14.Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428–33. [PMC free article] [PubMed] [Google Scholar]
- 15.Santorelli FM, Shanske S, Macaya A, DeVivo DC, DiMauro S. The mutation at nt 8993 of mitochondrial DNA is a common cause of Leith’s syndrome. Ann Neurol. 1993;34:827–34. doi: 10.1002/ana.410340612. [DOI] [PubMed] [Google Scholar]
- 16.Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–51. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- 17.Makela-Bengs P, Suomalainen A, Majander A, Rapola J, Kalimo H, Nuutila A, Pihko H. Correlation between the clinical symptoms and the proportion of mitochondrial DNA carrying the 8993 point mutation in the NARP syndrome. Pediatr Res. 1995;37:634–9. doi: 10.1203/00006450-199505000-00014. [DOI] [PubMed] [Google Scholar]
- 18.White SL, Collins VR, Wolfe R, Cleary MA, Shaske S, DiMauro S, Dahl HH, Thorburn Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet. 1999;65:474–82. doi: 10.1086/302488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steffann J, Frydman N, Gigarel N, Burlet P, Ray PF, Fanchin R, Feyereisen E, Kerbrat V, Tachdjian G, Bonnefont JP, Frydman R, Munnich A. Analysis of mtDNA variant segregation during early human embryonic development: a tool for successful NARP preimplantation diagnosis. J Med Genet. 2006;43:244–7. doi: 10.1136/jmg.2005.032326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makino M, Horai S, Goto Y, Nonaka I. Mitochondrial DNA mutations in Leigh syndrome and their phylogenetic implications. J Hum Genet. 2000;45:69–75. doi: 10.1007/s100380050014. [DOI] [PubMed] [Google Scholar]
- 21.Bourgeron T, Chretien D, Rotig A, Munnich A, Rustin P. Prenatal diagnosis of cytochrome c oxidase deficiency in cultured amniocytes is hazardous. Prenat Diagn. 1992;12:548–9. doi: 10.1002/pd.1970120614. [DOI] [PubMed] [Google Scholar]
- 22.Dean NL, Battersby BJ, Ao A, Gosden RG, LinTan SL, Shoubridge EA, Molnar MJ. Prospect of preimplantation genetic diagnosis for heritable mitochondrial DNA diseases. Mol Hum Rep. 2003;9:631–8. doi: 10.1093/molehr/gag077. [DOI] [PubMed] [Google Scholar]
- 23.Blok RB, Gook DA, Thorburn DR, Dahl HH. Skewed segregation of the mtDNA nt 8993(T→G) mutation in human oocytes. Am J Hum Genet. 1997;60:1495–501. doi: 10.1086/515453. [DOI] [PMC free article] [PubMed] [Google Scholar]