

Abstract
Physiologic wound healing is highly dependent on the coordinated functions of vascular and non-vascular cells. Resolution of tissue injury involves coagulation, inflammation, formation of granulation tissue, remodeling and scarring. Angiogenesis, the growth of microvessels the size of capillaries, is crucial for these processes, delivering blood-borne cells, nutrients and oxygen to actively remodeling areas. Central to angiogenic induction and regulation is microvascular remodeling, which is dependent upon capillary endothelial cell and pericyte interactions. Despite our growing knowledge of pericyte-endothelial cell crosstalk, it is unclear how the interplay among pericytes, inflammatory cells, glia and connective tissue elements shape microvascular injury response. Here, we consider the relationships that pericytes form with the cellular effectors of healing in normal and diabetic environments, including repair following injury and vascular complications of diabetes, such as diabetic macular edema and proliferative diabetic retinopathy. In addition, pericytes and stem cells possessing “pericyte-like” characteristics are gaining considerable attention in experimental and clinical efforts aimed at promoting healing or eradicating ocular vascular proliferative disorders. As the origin, identification and characterization of microvascular pericyte progenitor populations remains somewhat ambiguous, the molecular markers, structural and functional characteristics of pericytes will be briefly reviewed.
Keywords: Adipose-derived stem cell, Angiogenesis, Capillary, Diabetes, Extracellular matrix, Fibroblast, Glia, Inflammatory Cells, Injury, Mesenchymal stem cell
1. Introduction
Physiologic wound healing progresses through coagulation, inflammation, formation of granulation tissue, and remodeling events (Falanga, 2005), all of which rely upon the active involvement and contributions derived from the microvasculature and its associated cellular and metabolic components. As the epidermis and all epithelia are avascular, dermal angiogenesis is an essential response following injury. Importantly, post-injury capillary expansion is controlled by microvascular cell-cell interactions. Microvascular cellular responses are further shaped by cues from the extracellular matrix (ECM), local non-vascular resident and immigrant cells, and immune surveillance cells present within the wound microenvironment. As have been well studied, platelets, inflammatory cells, and connective tissue components foster neovascularization through regulated secretion of soluble factors and ECM reorganization (Schultz et al., 2011). How these cell-based and cell-extracellular signals provide meaningful wound healing-associated “crosstalk” to control microvascular remodeling in response to injury is of particular interest. Early studies identified a role for pericyte-endothelial cell (EC) interactions in wound healing, but its scope was limited to the vasculature (Crocker et al., 1970). In this review, we examine the molecular and cellular interactions governing wound healing responses, revealing what might be under-appreciated or emergent roles for pericytes in microvascular remodeling and wound repair.
Non-healing wounds, such as those seen in diabetic patients suffering from the ulceration of their extremity-associated wounds (plantar ulcers) or patients presenting with proliferative diabetic retinopathy (DR) remain a problem in the clinic. Diverse in nature, yet reflective of the important role that the local microenvironment plays in shaping the spectrum of pathologic microvascular wound healing responses, DR results in a marked formation of neovessels (Crawford et al., 2009); whereas, diabetic dermal wounds are deficient in angiogenic processes (Brem and Tomic-Canic, 2007). As changes to pericytes have been proposed to mediate DR (Hammes, 2005) and foot ulceration (Tilton et al., 1985), we will review the current literature on pericyte dysfunction in diabetic wound healing. Since it is hypothesized that DR embodies a wound healing response (Gariano and Gardner, 2005), we aim to highlight convergent, divergent and overlapping signaling pathways that may help to reveal the basic molecular and cellular mechanisms controlling pericyte-dependent microvascular remodeling observed during healing or proliferative DR.
Use of mesenchymal stem cells (MSC) to promote wound healing is an expanding field of therapeutic interest (Brower et al., 2011). Recent studies describe pericytes as a potential perivascular pool for MSCs, giving rise to skeletal myofibers, bone, cartilage, and adipocytes (Crisan et al., 2008a, 2008b). MSCs and adipose-derived stem cells (AdSC) are being deployed in experimental models of wound healing (Huang et al., 2012; Natesan 2011b), and MSCs (Sasaki et al., 2008) and AdSCs (Natesan et al., 2011b) have been described as possessing pericyte-specific properties; these attributes may be linked to enhanced healing. Herein, we propose a more uniform classification system for these stem cells or presumed pericyte progenitors, offering an overview of key molecular markers expressed, structural elements presented, and functional properties possessed by microvascular pericytes.
2. Pericytes, Microvascular Endothelial Cells and Non-vascular Cells Control Wound Repair
Microvascular pericytes act as a crucial cellular interface between blood-borne and connective tissue signals. In normal vascular physiology, pericytes are closely associated with a stable endothelium. However, in response to injury, pericytes are not only contacted with ECs, but also encounter infiltrating inflammatory and connective tissue cells (Figure 1). How pericytes interact with the microenvironment and cellular players of wound healing will be reviewed below.
Figure 1. Schematic Diagram of Pericyte Interactions with Cellular Effectors of Wound Healing.

Pericytes form relationships with vascular, immune, connective tissue and glial cells during injury repair. This diagram demonstrates that pericytes may regulate multiple points of the wound healing cascade. Pericyte functions are not confined to the microvasculature, and range from modulation of immune cell infiltration and activation to glial scar formation. Hypothesized interactions are indicated by (?).
2.1. Vascular Cells
2.1.1. Endothelial Cells
ECs serve as the fundamental unit of the vasculature. Composing the inner lining of vessels, they act as a conduit for oxygen, nutrients and blood-borne cells. Endothelial tubes possess two surfaces: adluminal and abluminal. Adluminally, ECs are immunologically inactive, which prohibits spontaneous clot formation and immune cell activation. Low levels of adhesion receptors line inactive endothelial monolayers, allowing monocytes to roll along the vessel wall (Cook-Mills and Deem, 2005). Abluminally, microvascular ECs are ensheathed by a shared basement membrane (BM) with pericytes; the abluminal surface is interspersed with EC-pericyte and EC-matrix contact points (Hirschi and D’Amore, 1996).
In injury, the endothlium acts to home infiltrating inflammatory cells, and provide nutrients to actively remodeling tissues. Chemokines and cytokines from the wound bed change the repertoire of luminal surface receptors on ECs. For example, P-selectin and E-selectin, glycoprotein receptors responsible for leukocyte rolling and adhesion, are virtually absent from normal EC surfaces. Yet, inflammatory factors induce translocation of P-selectin from Weibel-Palade bodies, and increase expression of E-selectin in the endothelium (Weller et al., 1992). P- and E-selectins appear to function concertedly as double deficient mice for P-/E-selectin display decreased leukocyte transmigration and wound closure (Subramaniam et al., 1997). Furthermore, tumor necrosis factor-alpha (TNF-α) and interleukin-1 (IL-1) upregulate vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) in ECs, which cause arrest of leukocyte movement (Muller et al., 1993). Studies with membrane-bound ICAM-1 knockout mice and knockout mice for all isoforms of ICAM-1 demonstrate attenuated wound healing responses (Gay et al., 2011; Nagaoka et al., 2000). Since transmigration of leukocytes through the endothelial barrier is mediated by the binding of platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) on leukocytes and ECs (Muller et al., 1993), decreased leukocyte accumulation and angiogenesis is observed when endothelial PECAM-1 expression is disrupted. Results using this PECAM-deficient murine model suggest that wound healing may be negatively affected, as leukocyte extravasation into the connective tissue or wound bed is necessary to foster injury resolution (Solowiej et al., 2003). Taken together, leukocyte homing to injured tissue is highly dependent on EC expression of adhesion receptors, and these receptors are crucial for microvascular-immune cell interactions needed for the progression of the healing cascade.
As angiogenesis provides oxygen, nutrients and blood-borne cells to the site of tissue injury, microvascular expansion is a tightly regulated process influenced by local cell-matrix interactions and soluble mediators released or produced within the wound bed (Schultz et al., 2011). For example, formation of fibrin clots and release of platelet factors such as vascular endothelial growth factor (VEGF) (Möhle et al., 1997) stimulates EC migration and capillary morphogenesis. As healing progresses, matrix remodeling influences EC integrin-matrix interactions, which stimulates angiogenesis. During dermal wound healing, clot-derived fibronectin and vitronectin as well as EC production of laminins may foster increased angiogenesis. For example, fibronectin and vitronectin facilitate EC adhesion and migration (Tonnesen et al., 2000). Further, laminin 8 increases capillary tube formation in vitro, and laminin 10 is strongly expressed by microvessels in dermal granulation tissue (Li J et al., 2003). It is currently unknown how endothelial-pericyte interactions modulate laminin expression and whether this impacts matrix remodeling or the dynamic reciprocity driving healing (Shultz et al., 2011). Together, the microenvironment is an important factor in modulation of the endothelium, and during injury repair, induces a pro-angiogenic phenotype in ECs.
Pericyte crosstalk with the endothelium may regulate the extent of neovascularization in wound healing. Early wound healing studies observed gradual increases in pericyte coverage of ECs as the healing cascade progressed (Crocker et al., 1970), suggesting pericyte contact may mediate vessel stabilization in wound repair. Indeed, two independent pathways of pericyte regulation of endothelial growth state have been identified. Firstly, soluble transforming growth factor-beta (TGF-β) promotes endothelial quiescence. When pericytes and ECs are cocultured, latent TGF-β is converted to its active form, which inhibits EC growth (Antonelli-Orlidge et al., 1989). In addition, an alternative pathway of contact-dependent regulation of EC proliferative phenotype exists. Work by Lee et al. (2010) provides evidence that pericyte mechanical stiffness may control the tension of the underlying BM and endothelium. Furthermore, work from our group reveals adenoviral infection of pericytes with dominant negative or active Rho GTPase alters pericyte contractile state, which in turn, modulates EC proliferation (Kutcher et al., 2007). These results suggest that perturbations in pericyte contractility may serve as an angiogenic switch in wound healing or pathologic ocular angiogenesis seen in diabetes. Moreover, tissue inhibitor of metalloproteinase-2 (TIMP-2) and TIMP-3 expressed by ECs and pericytes respectively, enhance capillary stability (Saunders et al., 2006). Thus, pericyte regulation of EC proliferation is multiform, and modulation of these signaling pathways may play important roles in neovascularization, including the maturation of the capillary plexus associated with the wound bed.
2.1.2. Vascular Progenitor Cells
Unlike angiogenesis, which arises from a pre-existing microvascular plexus, vasculogenesis is driven by the in situ differentiation of endothelial (EPC) or vascular progenitor cells. The embryonic and postnatal vasculature is formed from multiple sources including mesodermal tissue, bone marrow and local stem cell reservoirs (Bautch, 2011). Mesoderm-derived angioblasts produce ECs of the major vessels, and angioblast migration is VEGF-dependent (Cleaver et al., 1997). Moreover, chimera studies demonstrate angioblasts also contribute to the vessels of the trunk and limbs, as well as perineural vessels (Ambler et al., 2001). Hemangioblasts, bipotential progenitors, are an additional source of endothelium in the developing vasculature. Fate map studies revealed that hemangioblasts give rise to erythrocytes and ECs (Vogeli et al., 2006). Newly formed vessels must be stabilized, which is fostered by mural cell associations. Mural progenitor cell recruitment and differentiation into smooth muscle cells (SMC)/pericytes are mediated by EC contact (Hirschi et al., 1998). Furthermore, during postnatal vasculogenic expansion, EPCs and native endothelium stimulate differentiation of vascular stem cells into pericytes by JAGGED-1 contact-dependent signals (Boscolo et al., 2011). Thus, the formation and maturation of nascent vascular networks relies on interactions among vascular progenitor cells, ECs and mural cells, and this process may be important at wound sites where areas of actively growing and remodeling microvessels are present.
EPCs produce functional vascular networks in cutaneous wounds and ischemic tissues (Asahara et al., 1999). Isolation and in vitro culture of putative EPCs, mononuclear blood cells expressing CD34, demonstrated that these cells possess EC lineage markers and form tube-like structures. Furthermore, in vivo hind limb ischemia studies reveal that CD34+ EPCs can be observed as they incorporate into the endothelium of neovessels (Asahara et al., 1997). These results highlight the blood-borne nature of this progenitor pool, which is capable of EC differentiation in wounded dermal compartments recovering from injury.
Peripheral vascular trauma places hypoxic stress on the surrounding tissue, as is the case in wound microenvironments (Knighton et al., 1983). Gill et al. (2001) examined the peripheral blood of burn patients for mobilization of EPCs, and observed significant increases in bone marrow derived EPCs concomitant with augmented VEGF plasma levels. Moreover, EPCs were shown to contribute to neovascularization in ischemic tissue, and this EPC-driven neovascularization was enhanced by cytokine pre-treatment (Takahashi et al., 1999). Interestingly, during wound healing, chemokine signaling through the CCL5/CCR5 pathway appears to contribute to EPC homing since CCR5 null mice display decreased EPC accumulation and wound closure (Ishida et al., 2012). In addition, this study revealed that EPCs not only participate in wound neovascularization, but also secrete growth factors such as TGF-β and VEGF. Conversely, Bluff et al. (2007) demonstrated that dermal wound healing increases EPC accumulation 5–14 days after injury, but that EPCs do not significantly add to neovascularization as angiogenesis was implicated as the prevailing mechanism of neovascularization in the healing of surgical incisions. Together, these results suggest that soluble factors and low oxygen concentrations from the wound bed stimulate EPC mobilization and accumulation to foster increased vascularization. Furthermore, EPC-driven neovascularization may only function in wounds inflicted by significant trauma where the wound area is large and requires a more robust vascular response.
Pericytes may foster EPC differentiation while stabilizing neovessel formation during vasculogenesis. Yet, direct evidence revealing such a functional linkage is lacking. Interplay between pericytes and EPCs could be important in controlling microvascular morphogenesis, and results derived from in vitro experimentation support this notion. Matrigel-derived cocultures containing defined ratios of EPCs and pericytes reveal that EPCs can form capillary-like networks, and that pericytes become closely associated with these structures (Bagley et al., 2005). Further, pericyte extensions appeared to connect groups of EPCs; therefore, it was hypothesized that pericytes might guide EPC-derived vascular structures (Bagley et al., 2005). This observation could be important in wound healing, as pericytes may direct EPC migration and tube formation into the wound area. Moreover, shear stress or vascular smooth muscle cell (vSMC) contact have been shown to induce EPC differentiation into mature endothelium (Ye et al., 2008), suggesting that local mechanical or mural cell-based signals shape EPC differentiation in situ. However, whether pericyte-EPC contact acts as an initiator or merely helps to strengthen an already differentiating EPC remains equivocal. Clearly, more work will be needed to reveal whether pericyte and EPC interactions foster wound healing by promoting capillary EC differentiation and capillary network formation.
2.2. Inflammatory Cells
2.2.1. Platelets
Platelets, the primary cellular responders to injury, represent a plasma reservoir of pro-inflammatory, pro-coagulatory and pro-angiogenic factors that contribute to the healing cascade. During vessel injury, platelets adhere to exposed collagen fibers mediating the formation of fibrin clots (Brass, 2003); upon activation, platelets release a milieu of soluble factors including TGF-β, platelet derived growth factor (PDGF), basic-fibroblast growth factor (b-FGF), VEGF, ATP (Blair and Flaumenhaft, 2009) and sphingosine 1-phosphate (S1P) (English et al., 2000). And, with local control of coagulation critical for the initiation of the myriad of acute wound healing responses that ensue, platelets function to integrate healing effector pathways ranging from coagulation to angiogenesis and re-epithelialization.
At the onset of injury, cessation of hemorrhagic events requires formation of a fibrin clot. Interestingly, fibrin and fibrinogen, key players in the coagulation cascade, have been shown to bind VEGF and b-FGF. These growth factor-laden molecules induce angiogenesis, fibroblast migration and proliferation, and interact with infiltrating inflammatory cells (Laurens et al., 2006). Moreover, the use of platelet-rich fibrin matrices has been shown to induce wound neovascularization, and accelerate wound closure through the release of VEGF, TGF-β and PDGF-BB (Roy S et al., 2011). Because platelets and their associated soluble factors demonstrate pro-healing effects, it is not surprising that platelet-rich plasma has been employed to foster enhanced healing in clinical settings. Indeed, application of platelet rich plasma to dermal lesions increases EC proliferation and migration, angiogenesis and injury resolution (Park HB et al., 2011; Yang et al., 2011). Furthermore, work in our laboratory and wound healing center indicate that synthetic peptides “mimicking” and augmenting the naturally occurring plasma- and/or platelet-derived healing activities can significantly promote wound healing in vitro and in vivo (Demidova-Rice et al., 2012). These bioactive wound healing peptides, which we engineered based on key platelet rich plasma fragments have a marked effect when added to capillary EC cultures, significantly augmenting cell proliferation and tube formation. Importantly, the wound healing peptides also markedly enhance epithelial cell migration in vitro and in vivo. Thus, platelet functionality ensures initiation of the healing responses to injury, while fostering wound closure through enhanced epithelializiation
No direct interactions between platelets and pericytes have been identified, yet pericytes may cooperate with platelets to control vascular hemorrhage. Bouchard et al. (1997) demonstrated that brain pericytes possess the ability to modulate the extrinsic clotting cascade. Brain pericytes express tissue factor, and their surface permits the assembly of the prothrombinase complex. Further, dermal pericytes were also shown to express tissue factor (McDonald et al., 2008), although the clotting efficacy of dermal pericytes was not investigated. When pericyte-EC cocultures were embedded in fibrin clots, only EC sprouts were observed to emanate from the clot (Nehls et al., 1994), suggesting pericytes may remain within the fibrin matrix to mediate additional coagulation. Alternatively, fibrin may sequester pericytes in the clot to permit expansion of the capillary plexus into the wound bed. To test this, pericyte-EC cocultures on several matrix components such as collagen, laminin and fibrin could be assessed for endothelial S-phase entry and tube formation. Fibrin cocultures might exhibit significant angiogenic activity and capillary-like structures devoid of pericytes, indicating fibrin may switch pericyte physiology to a state that is permissive to endothelial activation, allowing revascularization of the wound bed.
2.2.2. Neutrophils
During the cellular response to injury, neutrophil transmigration is coordinated by pericytes. Feng et al. (1998) observed that neutrophil movement through the endothelium was followed by a transcellular migration path across cutaneous pericytes. Further, electron microscopy revealed pauses in neutrophil migration when contacted with pericytes. Indeed, studies on cremaster muscle venules have identified BM and possibly pericyte-produced matrix microdomains through which neutrophil extravasation is facilitated. Such matrix microdomains are heralded by decreased expression of laminin 10, collagen IV, and nidogen-2, i.e. low expression regions (LERs) (Wang et al., 2006). Strikingly, nearly 100% of LERs were associated with gaps between adjacent venular pericytes. Further, Voisin et al. (2010) demonstrated that LERs are not confined to venules of cremaster muscles, but are ubiquitous. Taken together, these results identify a critical role for pericytes in the construction of the BM as well as in shaping whether or to what extent immigrant cells can transit from the blood into the connective tissues of healing wounds. Furthermore, the close proximity of LERs to pericytes and preferential migration of neutrophils through these channels suggests pericytes and neutrophils may be in direct communication during inflammatory responses.
Through chemotactic signals neutrophils are able to home to sites of inflammation, transmigrate through the vasculature, and enter the wound bed (Woodfin et al., 2010). Neutrophils produce early inflammatory responses, as their entry into the wound area is strongest within 24 hours of injury. Investigation of neutrophil infiltration kinetics revealed that compared to saline treated wounds, wounds inoculated with Staphylococcus aureus exhibited higher numbers of neutrophils, but did not significantly alter wound closure (Kim MH et al., 2008). This result confirmed an early study by Simpson and Ross (1972), which reported control and neutropenic wounds had comparable healing rates. However, a recent study implicated neutrophil derived-TNF-α as a promoter of neovascularization and re-epithelialization in Pseudomonas aeruginosa infected lesions (Kanno et al., 2011). Moreover, neutrophils were observed to secrete VEGF (Gaudry et al., 1997) and TIMP-free matrix metalloproteinase (MMP)-9 (Ardi et al., 2007), which stimulate angiogenesis in vitro. These results indicate neutrophil contribution to injury repair may be dictated by the presence or absence of infection, as well as the type of bacterial infection. While several studies indicate neutrophils possess pro-angiogenic molecules, they have yet to be tested in vivo. Therefore, further investigation is required to gain a fuller understanding of the extent of neutrophil participation in wound closure.
2.2.3. Monocytes/Macrophages
Pericytes and macrophages possess overlapping functions, and may coordinate wound neovascularization. Evidence for phagocytic properties of pericytes has been shown in brain-derived pericytes in vivo (Jeynes, 1985) and in vitro (Balabanov et al., 1996). Furthermore, injurious stimuli may convert brain pericytes to macrophages or upregulate their macrophage-like activities (Thomas, 1999). Therefore, pericytes could aid to clear wound debris in dermal lesions; although, the ability of dermal pericytes to phagocytose particles remains to be demonstrated.
Macrophage and pericyte interactions may foster vascular expansion and stability in the wound bed. In vitro and in vivo experiments revealed that macrophages and monocytes produce tube-like structures in Matrigel (Anghelina et al., 2004). These tubes were posited to influence neovessel distribution as erythrocytes and cells with EPC markers co-localized with these structures. Moreover, recruitment of bone marrow derived-pericytes and macrophages were shown to establish immature vascular networks in dermal Matrigel plugs via FGF-2 dependent mechanisms (Tigges et al., 2008). Macrophage migration may also influence neovascularization. Nucleoside triphosphate diphosphohydrolase-1 (CD39) mediates the breakdown of extracellular nucleoside di- and triphosphate molecules such as ATP and ADP. In nucleotide migration studies, CD39-null macrophage chemotaxis was altered (Goepfert et al., 2001). When CD39-null mice were used to investigate Matrigel plug capillary ingrowth, CD39-null mice exhibited a stratified cell infiltrate of macrophages, pericytes and ECs devoid of neovessel formation, suggesting macrophage migration is crucial to wound neovascularization (Goepfert et al., 2001). Further, macrophage-pericyte interactions may not be limited to early vessel formation as population and subsequent stabilization of the tumor vasculature by pericytes appears to be macrophage MMP-9 dependent (Chantrain et al., 2004).
Several subtypes of macrophages exist, including resident tissue, M1 and M2 macrophages; however, during wound healing, the distinction between M1 and M2 macrophages becomes blurred, and are commonly referred to as “wound associated macrophages” (Rodero and Khosrotehrani, 2010). Wound-associated macrophages function as phagocytic cells capable of interacting with vascular and non-vascular cells through an array of soluble factors. Indeed, through time-specific ablation of macrophages, Lucas et al. (2010) revealed that macrophages differentially regulate wound healing. For example, elimination of macrophages in the early stages of injury results in decreased neovascularization and re-epithelialization, whereas depletion of macrophages in the later stages of healing appears to have no significant effect on tissue repair (Lucas et al., 2010). Moreover, decreased macrophage infiltration mitigates angiogenesis and secretion of pro-angiogenic factors (Goren et al., 2009; Mirza et al., 2009). There is also evidence that macrophages may control the soluble milieu of the wound microenvironment through phenotypic switching. Wound macrophages produce high concentrations of cytokines such as TNF-α and IL-6 early, but secrete higher levels of TGF-β in the later stages of healing (Daley et al., 2010). As TGF-β mediates EC quiescence in pericyte-EC cocultures (Antonelli-Orlidge et al., 1989), macrophage-derived TGF-β in concert with pericyte investment of nascent capillaries may further enhance vessel maturation. Thus, macrophages may be an additional regulator of angiogenic potential during wound repair, and contribute to vessel guidance and stabilization with pericytes.
2.2.4. Pericyte Regulation of Lymphocyte Function: A Role in Modulating Wound Healing
Pericytes may regulate T cell activation, and recruit T and B-lymphocytes to areas of tissue injury. Under inflammatory conditions, brain pericytes gain the ability to present antigens to T cells, which leads to lymphocyte activation (Balabanov et al., 1999). In contrast, retinal pericytes inhibit T lymphocyte activity (Tu et al., 2011). Hyperglycemia reduces pericyte inactivation of T cells, suggesting pericyte modulation of T lymphocytes is highly dependent on cues from the microenvironment. Therefore, pericytes may influence T cell activity during wound healing, and cues from the wound microenvironment may dictate whether these pericyte signals are stimulatory or inhibitory.
Pericyte recruitment of lymphocytes to inflamed tissue could be mediated by several cytokines. Immunohistochemical analysis of stromal-cell derived factor (SDF-1) and CXCR4 in the dermal microvasculature revealed a potential role for pericyte-derived SDF-1 in lymphocyte recruitment (Pablos et al., 1999). Moreover, secretion of CXCL9 and CXCL12 by tumor microvessel-associated pericytes enhances B and T cell tissue infiltration (Venetz et al., 2010). Further, dermal pericytes have been shown to express CXCL12 (Avniel et al., 2006), which signals through CXCR4 to mediate T cell chemotaxis (Prasad et al., 2007); therefore, the CXCL12/CXCR4 axis may represent an additional pathway for pericyte-lymphocyte communication.
Pericytes modulate adhesion receptor expression and lymphocyte infiltration. Stellate cells, pericyte-like cells of the liver, regulate adhesion molecule expression in response to injury and inflammation. In vitro application of inflammatory cytokines to stellate cells increased levels of ICAM-1 and VCAM-1 compared to control cultures. These results were recapitulated in vivo as acute liver injury resulted in augmented levels of ICAM-1 and VCAM-1 mRNA (Knittel et al., 1999). Further, brain pericyte exposure to TNF-α mediated T cell infiltration via very late antigen-4 (VLA-4)/VCAM-1 binding (Verbeek et al., 1995). Pericytes possess soluble and contact-dependent means of regulating lymphocyte infiltration to inflamed tissue; therefore, these pathways could importantly influence tissue repair.
2.3. Connective Tissue
2.3.1. Fibroblasts/Myofibroblasts
Fibroblasts are a major cellular component of connective tissue, and are crucial for maintenance of the ECM in normal and injured tissue. In the dermis, subpopulations of fibroblast exist, which exhibit differences in ECM deposition, and highlight fibroblast heterogeneity (Sorrell and Caplan, 2004). Characteristic of wounds, myofibroblasts represent specialized fibroblasts that express α-smooth muscle actin (α-SMA) (Darby et al., 1990). The concerted functions of these cells facilitate injury repair through matrix remodeling, growth factor secretion and contractile forces.
Release of PDGF from platelets initiates fibroblast migration and proliferation into the wound bed (Rajkumar et al., 2006). Fibroblasts secrete numerous growth factors including epidermal growth factor (EGF), FGF-2, TGF-β, PDGF and VEGF (Barrientos et al., 2008), which promote epithelialization, vascularization and granulation tissue formation. Activation of fibroblasts causes differentiation to contractile myofibroblasts (Li B and Wang, 2011). Fibroblast-myofibroblast transitions require both mechanical signals and TGF-β signaling (Serini et al., 1998), and myofibroblasts can liberate latent TGF-β to stimulate further fibroblast differentiation (Wipff et al., 2007). Collagen network stability is mediated by myofibroblast contractions. Tightening of previously secreted collagen fibers allows adjacent myofibroblasts to secrete more collagen, thereby increasing the strength of wound granulation tissue (Tomasek et al., 2002). This cyclical process of contraction and ECM deposition may also facilitate wound closure in a TGF-β dependent manner (Grinnell and Ho, 2002). A recent study by Kilarski et al. (2009) illuminates an additional role for fibroblasts and myofibroblasts: non-angiogenic neovascularization. Myofibroblasts generate forces that are capable of translocating capillaries into the wound area, suggesting sprouting angiogenesis follows this contraction-mediated vascular growth. These studies illuminate critical roles for fibroblast and myofibroblast force generation; not only do these forces influence wound closure, but may also provide a blood supply to expanding granulation tissue.
The extent of fibroblast interactions with pericytes remains unclear. As fibroblasts are known to secrete numerous soluble factors including FGF-2, which induces phenotypic changes to pericytes (Papetti et al., 2003), it is conceivable that fibroblast secretions may increase pericyte numbers during the proliferative phase of wound healing. With the formation of immature capillary networks during repair, an enhanced pool of pericytes would be advantageous for stabilization and remodeling of these neovessels.
There is emerging evidence that pericytes may act as a source of myofibroblasts. In vivo dermal healing revealed overlapping immunologic markers between myofibroblasts and pericytes (Juniantito et al., 2012), and pericyte IL-33 induction caused differentiation to myofibroblasts (Sponheim et al., 2010). However, Sponheim et al. (2010) suggested these IL-33 positive myofibroblasts could have originated from pre-existing dermal fibroblasts as well. Furthermore, pericyte to myofibroblast differentiation appears to not be skin specific, but can also occur in kidney fibrosis through PDGF-dependent mechanisms (Chen et al., 2011). These studies offer an intriguing novel role for pericytes as a potential source of myofibroblasts in wound healing. However, whether these pericyte-derived myofibroblasts are bona fide myofibroblasts remains to be determined. An alternative hypothesis is that these myofibroblasts within the wound bed could be contractile, ECM depositing pericytes that have become dissociated from microvessels (Figure 2). Fate map studies of microvascular pericytes during wound healing could elucidate the origin of myofibroblast-like cells found in wound tissue. Thus, pericyte to myofibroblast differentiation may reflect a migration and phenotypic switching event, whereby pericytes derived from a vascular niche are activated to migrate into the wound ECM and possess the functional roles of a myofibroblast.
Figure 2. Myofibroblast Emergence: A Perivascular Origin?

In normal tissue, the microvasculature is stable and covered by pericytes, and inactivated fibroblasts occupy the surrounding connective tissue. During wound healing, activated fibroblasts may differentiate into myofibroblasts or alternatively, pericytes may differentiate into myofibroblasts. However, pericytes may migrate from the vasculature, and enter the wound bed. Signals and matrix from the wound microenvironment may cause pericytes to assume a more contractile and matrix depositing phenotype, similar to that of a myofibroblast. Further validation will reveal the essence of the different contractile cell types that occupy the wound bed. Hypothesized interactions are indicated by (?).
2.4. Neuronal Support Cells
2.4.1. Müller Cells/Astrocytes
Association of mural cells with non-vascular, resident parenchymal cells has long been thought to influence microvascular dynamics during development and disease. For example, Müller cell (MC) and astrocytes are glial support cells that act as a critical interface between the vasculature and nervous system in what has been described as “the neurovascular unit” (Fisher, 2009). Confined to the retina, MCs function to provide lactate and pyruvate to neurons, and maintain the blood-retina barrier (Bringmann et al., 2006; Tout et al., 1993). Also, MCs secrete growth factors that promote retinal cell survival (Saint-Geneiz et al., 2008). Similarly, astrocytes are glia of the spinal cord and brain. Central nervous system (CNS) astrocytes interact with the vasculature and neurons, regulating blood flow dynamics, ion transport, and metabolism (Ransom and Ransom, 2012).
During injury, glial cells maintain neurovascular homeostasis through gliosis (Streit et al., 1999), which is characterized by an upregulation of glial fibrillary acidic protein (GFAP) expression (Pekny and Nilsson, 2005). Gliosis represents a spectrum of astrocyte responses aimed at maintaining neuronal homeostasis (Sofroniew, 2009). When reactive gliosis is prolonged in MCs and astrocytes, the result is the formation of a glial scar. Glial scars impede axon regeneration (Silver and Miller, 2004), and in the retina, produce abnormal neuron growth and neovascularization (Bringmann et al., 2006). A recent study by Göritz et al. (2011) illuminates a subpopulation of CNS pericytes that participates in scar formation during spinal cord injury, suggesting glial scar formation may no longer be a glial-specific phenomenon, but include pericytes as well. Therefore, development of future therapeutics may benefit from examining glial and mural cell interactions in CNS lesions.
MCs and astrocytes are in close contact with capillaries of the CNS and retina, where astrocytic end-feet are observed to make contacts with pericytes (Mathiisen et al., 2010). Functionally, glia and pericyte interactions have been shown to maintain the blood-retina and blood-brain barriers (Al Ahmad et al., 2011; Kim JH et al., 2009). This linkage may also be important in response to injury in DR. Rescue of MC apoptosis in the diabetic retina appears to prevent capillary cell death, including pericyte loss. These results suggest that pathology to neuronal and glial cells might directly affect the health of vascular cells (Hammes et al., 1995). When pericytes and astrocytes are cultured with ECs in vitro both cell types migrate to capillary-like structures; furthermore, pericytes and astrocytes become invested in these endothelial structures as well (Itoh et al., 2011; Minakawa et al., 1991). Astrocyte-EC cocultures reveal that astrocytes inhibit EC proliferation through contact-dependent means (Garcia et al., 2004). Therefore, pericyte and astrocyte investment of microvessels may cooperate to maintain a quiescent endothelium, and regulate the angiogenic potential of the CNS microvasculature in response to injury.
3. Wound Healing in Diabetes: A Focus on Pericytes
Diabetes presents a significant burden on society, affecting 25.8 million Americans and costing $174 billion dollars annually (American Diabetes Association, 2011), with a projected 64% increase in patients and $514 billion in health care delivery costs by 2025 (Rowley and Bezold, 2012). Diabetic complications affect multiple organ systems, increasing the risk for myocardial infarction, stroke and atherosclerosis (Beckman et al., 2002). In addition, diabetic vascular complications plague macro- and microvascular structures causing stroke, atherosclerosis, impaired healing, retinopathy, neuropathy and nephropathy (Reddy and Natarajan, 2011). The cellular and molecular changes associated with DR encompass a misregulated healing response (Gariano and Gardner, 2005); therefore, we will frame the progression of DR within the events of a normal wound healing cascade (Figure 3). Here, we will also examine the pathogenesis of impaired injury repair in the context of pericyte dysfunction.
Figure 3. Microvascular Dynamics During Normal Wound Healing, Diabetic Retinopathic and Chronic Wound-Associated Healing.

(A) Normal tissue repair progresses through coagulation, inflammation, proliferation and remodeling phases without complication resulting in wound closure. (B) DR exhibits abnormal coagulation, inflammation and proliferation phases. Abnormalities in these phases of wound healing result in pathologic neovascularization and vascular leakage. (C) Chronic wounds possess normal coagulation, but display perturbed inflammation and proliferation phases. Without proper immune and vascular cell responses, wound closure is impaired or prevented.
3.1. Diabetic Retinopathy
Recent successes with anti-angiogenic therapies in age-related macular degeneration (AMD) have turned attention to seeking advances in combating other ocular diseases such as DR (Durham and Herman, 2011). Development of innovative treatments is imperative, as diabetes-induced retinopathy causes the majority of new cases of adult blindness, and from 2005–2008 it was estimated that 28.5% of diabetics were afflicted with DR (American Diabetes Association, 2011). While the initial stimuli driving retinal neovascularization remain unknown, hyperglycemia produces a chaotic biochemical and cellular environment, a combined result of oxidative stresses and advanced glycation end products. Through altered signaling pathways and cellular interactions, an aberrant wound healing response is initiated, and if left unchecked, results in pathologic angiogenesis and retinal detachment.
Similar to coagulation events in physiologic wound healing, stasis of retinal blood flow (Frank, 2004) and abnormal platelet function produce microthrombi, which release vasoactive substances that may alter capillary integrity, permitting egress of platelet fragments and hyperglycemic plasma into perivascular spaces (Dobbie et al., 1974). While platelets do not adhere to uninjured endothelium in normal physiology, hyperglycemic plasma induces platelet hyper-reactivity through increased adhesion receptor and platelet activation molecule expression (Ferreiro et al., 2010). Moreover, hypoxia downstream of microthrombosis and concomitant leukostasis may also contribute to retinal microvascular leakage and angiogenic induction. Indeed, fibrinogen and fibrin, potent inflammatory cell and EC activators, were found in diabetic retinal exudates (Murata et al., 1992). Thus, platelets may play an important role in the early progression of DR by facilitating augmented clot formation and vascular leakage.
Prolonged exposure to hyperglycemia perturbs vascular cell physiology, and creates a scenario reminiscent of the inflammatory and proliferative phases of tissue repair. In vitro culture of ECs and pericytes provide evidence that the diabetic microenvironment may upregulate inflammatory gene expression (Kowluru et al., 2010; Perrone et al., 2009) and matrix deposition (Roy S et al., 1996). In vivo mouse studies reveal a critical role for inflammation in DR progression. Diabetic CD18 knockout and ICAM knockout mice both exhibited decreased leukostasis, vessel leakage and vascular lesions compared to wild type diabetic mice. Moreover, pericyte loss was attenuated in diabetic knockout mice (Joussen et al., 2004). When cultured in hyperglycemic conditions, retinal pericytes exhibit attenuated inhibition of T lymphocyte activity (Tu et al., 2011). Taken together, retinal pericytes are sensitive to inflammation, and their inactivation of lymphocytes is essential to the stability of the microcirculation.
Retinal endothelial dysfunction and inflammatory responses produce vascular permeability that permits increased amounts of glucose to invade the mural cell microenvironment. Elevated levels of glucose have a deleterious effect on pericyte physiology, and have been shown to induce pericyte apoptosis (Geraldes et al., 2009), a histopathologic hallmark of DR (Hammes, 2005). Loss of pericytes may facilitate a permissive environment for neovascularization, as a 50% reduction in pericyte density was shown to lead to the re-initiation of EC proliferation (Enge et al., 2002). Ultimately, pathologic angiogenesis ensues, producing retinal capillaries that become acellular entities devoid of pericytes and ECs, enclosed by a thickened BM (Lorenzi and Gerhardinger, 2001).
3.2. Chronic Wounds in Diabetes
As retinal pericyte dysfunction and degeneration promotes pathologic angiogenesis, perturbations in dermal pericyte physiology result in decreased neovascularization and healing responses. These observations highlight the heterogeneity of microvascular function, and reflect differences between pericytes of the retina and dermis. Impaired wound healing is a significant complication in diabetes, which often results in foot ulcers. Diabetic foot ulcers have a lifetime incidence upwards of 25%, and are found in 70–84% of lower limb amputees (Evans et al., 2011). The exact mechanisms of chronic wound healing are unclear, but evidence suggests pathology to the microvasculature is essential to the pathogenesis of diabetic wound healing (Pham et al., 1998). Because pericytes mediate interactions with numerous cellular effectors of wound healing, their loss may be crucial to the pathogenesis of non-healing wounds in diabetes.
Pericyte loss from dermal and muscle capillary networks of diabetics has been documented (Tilton 1981, 1985); yet, little connection has been made linking pericyte absence with alterations to inflammatory responses, microvascular stability, wound closure and matrix deposition. Chronic wounds possess impaired inflammatory cell infiltration and function (Fahey et al., 1991; Nolan et al., 1978). Pericyte dysfunction may affect neutrophil and macrophage accumulation in chronic wounds, as pericyte-derived matrix components and localization to LERs is crucial for inflammatory cell infiltration. In addition, pericytes modulate lymphocyte activity, suggesting alterations to pericyte physiology could prolong or shorten the inflammatory response.
The microenvironment of chronic wounds is hostile, characterized by markedly elevated levels of proteases and glycated matrix components. Punch biopsies of diabetic foot ulcers reveal increased concentrations of MMP-1, MMP-2, MMP-8, and MMP-9, but decreased concentrations of TIMP-2 (Lobmann et al., 2002). Imbalances in proteases and their inhibitors would begin to cannibalize growth factors and growth factor receptors, prohibiting capillary expansion. Moreover, glycated matrices possess altered elasticity and rigidity. When fibroblasts were challenged to proliferate and contract on glycated matrix, they were prohibited (Liao et al., 2009). These results could have implications for pericyte-EC signaling, as changes to the mechanical properties of the BM may impair chemo-mechanical regulation of endothelial growth state by pericytes. Thus, abnormal protease activity and glycation of ECM components render the wound bed to a state of angiogenic incompetence, thereby inhibiting neovascularization of granulation tissue.
Impaired wound healing illuminates the complexity of molecular and cellular organization required for proper healing. Wound healing events are interconnected, and as pericytes coordinate with multiple cell types, understanding pericyte pathology in diabetic ulcers may drive the discovery of future wound healing therapeutics.
4. Experimental Use of Pericyte-like Stem Cells: Implications for Cell-Based Wound Healing Therapeutics
4.1. Adipose-derived Stem Cells
AdSCs represent a source of multipotent stem cells that are typically isolated from the stromal vascular fraction derived from adipose (Lin et al., 2010). Traktuev et al. (2008) first identified CD34+ AdSCs as pericyte-like cells. These AdSCs express “pericyte” or mural cell markers, NG2 and α-SMA, as well as the mesenchymal marker, CD90. On Matrigel, AdSCs make abluminal associations with EC tube networks (Traktuev et al., 2008). In addition, a recent in vitro study confirmed the pericyte-like properties of AdSCs, and identified soluble and contact-dependent interactions are responsible for the production of EC tube morphogenesis and increased vessel stability (Merfeld-Clauss et al., 2010). However, AdSC differentiation appears to not be limited to pericytes, as a study using CD90+/CD34− AdSCs observed that AdSCs not only differentiate into pericytes, but also into EC lineages (Natesan et al., 2011b). Thus, while there is increasing evidence that AdSCs are capable of pericyte differentiation, questions still remain as to what microenvironmental cues direct AdSCs to mural cell lineages, and what subpopulations are capable of this transition.
While the endothelium and mural cells appear to be unsuitable for the toxic microenvironment of diabetic ulcers, AdSCs may represent a more resilient cell type. Because neovascularization is absent in diabetic lesions, the wound bed becomes extremely hypoxic. Evidence suggests that CD34+/NG2+/α-SMA+ AdSCs, cells with pericyte-like properties, increase secretion of pro-angiogenic molecules, and maintain vascularization in inflamed tissue (Amos 2008, 2011). Further, application of conditioned media derived from CD90+/CD34− AdSCs cultured in hypoxia augmented wound area closure (Lee EY et al., 2009). Moreover, AdSCs have been shown to enhance wound healing in diabetic mice through secretion of growth factors and neovascularization (Kim EK et al., 2011; Nambu et al., 2009). However, one study using diabetic rats observed increased healing, but no significant difference in wound vascularization when treated with AdSCs (Maharlooei et al., 2011). Thus, the mechanisms underlying AdSC mediated healing requires further characterization, and more studies are needed to determine if AdSC to pericyte transitions are necessary to foster increased healing.
Taken together, AdSCs promote wound healing in diabetic and hypoxic conditions. Because AdSCs are an abundant and easily isolated population of adult stem cells in both normal (Yoshimura et al., 2006) and injured tissues (Natesan et al., 2011a), they may be an excellent tool for future pericyte replacement and chronic wound therapies.
4.2. Mesenchymal Stem Cells
Controversy seems to surround MSCs and pericytes. Both cell types have been noted to share several mesenchymal markers including CD44, CD90, CD105 and CD73 (Corselli et al., 2010), and evidence suggests that perivascular MSCs express the pericyte markers NG2 and PDGFR-β (Crisan et al., 2008b). Furthermore, pericytes have been purported to be stem cells themselves, capable of differentiation into osteogenic, adipogenic and chondrogenic lineages (Dar et al., 2012). This multipotency has been observed in CNS pericytes as well (Dore-Duffy et al., 2006). Early in vivo studies add more complexity, as MSCs were unable to differentiate into pericytes, although, these MSCs expressed the pericyte marker α-SMA; furthermore, these α-SMA+ cells were not found in perivascular locations (Anjos-Afonso et al., 2004; Silva et al., 2005). As therapeutic use of MSCs increases, and the delineation between MSCs and pericytes becomes further blurred, we suggest that pericytes and MSCs are two separate cells types. Pericytes may undergo phenotypic switching upon leaving the vascular niche, but do not undergo differentiation. In contrast, MSCs may reside in perivascular locations, but may not possess functional similarities to pericytes. Additional experimentation is required to determine whether MSCs regulate vascular stability and tone as well as endothelial quiescence.
MSCs may represent a potential source of pericytes in dermal wound healing. Sasaki et al. (2008) identified capillary associated α-SMA+/CD31− pericytes in dermal lesions using GFP labeled MSCs (Sasaki et al., 2008); it should be noted that these GFP+ cells composed only 33% of wound bed α-SMA+ cells. This study suggests bone marrow MSCs may participate in wound healing by differentiating into a variety of cells including pericytes; although, it appears that a majority of pericytes arise from local vascular sources.
Innovative delivery systems are being deployed to enhance MSC-dependent healing. For example, MSC-containing hydrogels are “pro-angiogenic,” augment cytokine secretion, and accelerate wound closure in vivo (Rustad et al., 2012). Further, approximately 39% of implanted MSCs were NG2 positive. Another study provides evidence that microspheres loaded with MSCs could increase healing, as wounds treated with microspheres had significantly greater wound closure compared to control tissues (Huang et al., 2012). In addition to experimental models of healing, MSCs are being used clinically to alleviate limb ischemia. A recent Phase II trial suggests administration of EPCs and MSCs may elevate limb perfusion through increased angiogenesis or vascular remodeling (Lasala et al., 2011).
These studies illuminate a potential role for MSCs as progenitors of wound bed pericytes. Furthermore, animal studies demonstrate MSCs possess potent wound healing effects. These healing properties potentially hold therapeutic value in human wounds as MSCs were shown to be effective in treating limb ischemia. To enhance the pericytic properties of MSCs, more accurate sorting and pre-conditioning of MSCs could be used. Moreover, treating MSCs with molecules such as TGF-β, which induce pericyte-like phenotypes (Hirschi et al., 1998), before implantation might better ensure that stem cells produce a pericyte outcome in diseased tissues.
5. Pericyte Markers: A Perivascular Identity Crisis?
Pericytes are an extremely heterogeneous population of mural cells that accompany microvascular structures of the body. Enveloped by BM in the abluminal perivascular space of microvessels (Sims, 1986), proper identification is dependent on a mixture of molecular markers and location cues. While a growing list of pericyte markers exists, a bona fide marker remains to be discovered. As reviewed by Armulik et al. (2011), molecular markers of pericytes are promiscuous. For example, the glycoprotein 3G5 (Nayak et al., 1988), proteoglycan NG2 (Ozerdem et al., 2001) and PDGFR-β (Hellstrom et al., 1999) react with other cell types including renal medullary cells, vSMC and myofibroblasts, respectively. Moreover, several pericyte markers may be useful in areas of actively remodeling vessels only. These markers include an intermediate filament protein desmin (Nehls et al., 1992), high molecular weight melanoma-associated antigen (Schlingemann et al., 1991) and aminopeptidase A (Schlingemann et al., 1996). Therefore, with such ambiguity concerning the characterization of pericytes, it warrants the following line of questioning: “What defines a pericyte based on the current molecular markers, and can we develop methods to better identify pericytes?”
5.1. The Pericyte Actin Network: An Old Story with a New Twist
Original studies by Herman and D’Amore revealed actin networks as potential pericyte markers. Immunofluorescence analyses of actin networks could be used to distinguish pericytes from vSMCs and ECs in vitro and in vivo (Herman and D’Amore, 1985). Pericyte stress fibers were shown to strongly react with antibodies to both muscle and non-muscle actin, whereas stress fibers of vSMCs and ECs reacted only with muscle or non-muscle actin isoform-specific antibodies, respectively. Moreover, anti-muscle actin antibodies illuminated qualitative differences between pericyte and vSMC cytoskeletons. Pericytes and vSMCs possess robust muscle actin enriched stress fibers, yet only pericytes exhibit a granular meshwork of muscle actin in motile cytoplasmic segments (Figure 4). Non-vascular cells such as dermal fibroblasts also express α-SMA as do myofibroblasts, stellate cells and mesangial cells (Johnson et al., 1991; Rubbia-Brandt et al., 1197; Skalli et al., 1986). Recent work from our lab suggests that the actin cytoskeleton not only acts as a biomarker, but could be used as a functional marker for pericytes as well. Kolyada et al. (2003) revealed alteration to Rho GTPase signaling affects pericyte contractile phenotype through actin cytoskeletal rearrangements. Building on these results, Kutcher et al. (2007) demonstrated that perturbations to pericyte contraction influences pericyte regulation of endothelial proliferation. Therefore, comparing EC growth inhibition in AdSC/MSC-EC cocultures with pericyte-EC cocultures could determine whether these stem cells are functionally equivalent to pericytes. Further, in vitro contractility assays paired with investigation of the non-muscle and muscle actin networks could provide evidence that MSCs and AdSCs are capable of mechanical regulation of capillary physiology. Taken together, pericytes are dynamic regulators of capillary stability and flow (Kutcher and Herman, 2009); therefore, functional assays may add another level of resolution for distinguishing pericytes from other pericyte-like stem cells.
Figure 4. The Pericyte Actin Network Regulates Cell Shape, Contractility and Endothelial Cell Quiescence.
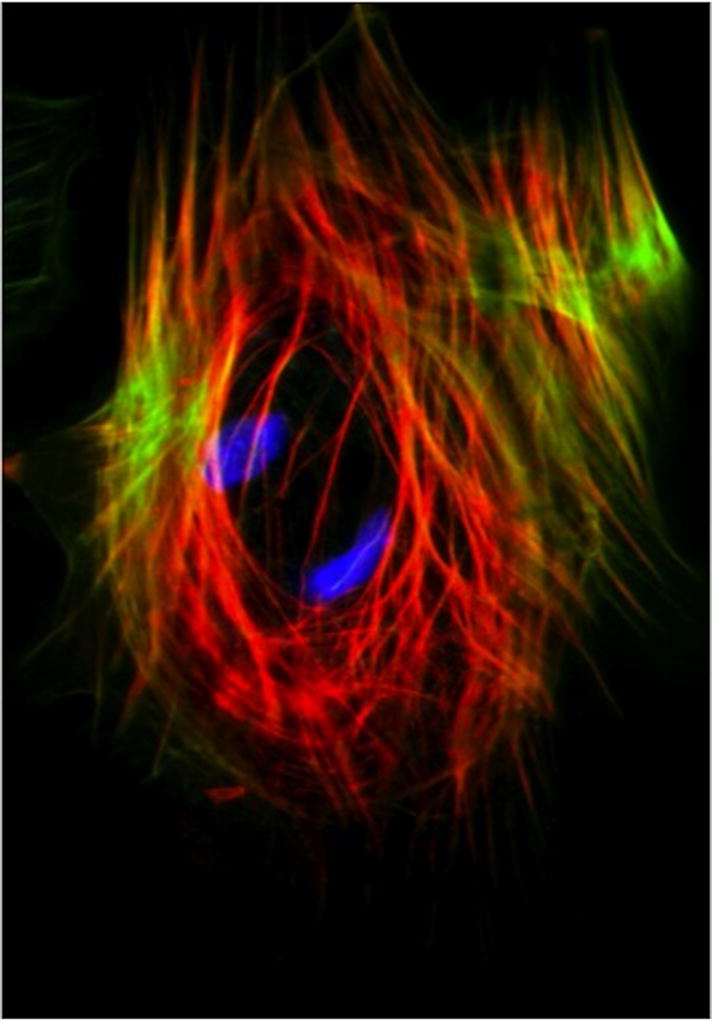
Immunofluorescence imaging of the smooth muscle and cytoskeletal containing compartments within a Bovine Retinal Pericyte. Phalloidin staining (green) and anti-α-SMA antibody (red) reveals F-actin and muscle actin networks in pericytes. α-SMA may be an important molecular and functional marker of pericytes and pericyte-like stem cells.
6. Conclusion
Orchestration of cellular and molecular events is crucial for wound repair in normal physiology. Pericyte interactions with inflammatory cells, glia and connective tissue highlight the diverse roles pericytes play in mediating injury repair. These interactions may extend to other pathologies such as tumor progression, where pericyte investment may not only stabilize the tumor microvasculature, but also aid the tumor in recruiting stromal and immune components. Moreover, induction of wound healing pathways may not be limited to tissue injuries, as events in DR exhibit many similarities with the phases of physiologic wound repair. DR and chronic wounds represent reciprocal wound healing responses; DR possesses enhanced angiogenic, inflammatory and proliferative processes, while diabetic ulcers are lacking, yet both exhibit pericyte loss. Therefore, elucidating the signaling pathways of either disease, and targeting pericytes may be beneficial to treatment of the other. Further, it is advantageous that a more uniform classification system be developed, as pericyte identification remains unclear and pericyte-like stem cells are emerging as therapeutic options in wound healing. Molecular markers and an association with the perivascular space may no longer be sufficient to identify pericytes. Pericytes are not static cells, but dynamic regulators of microvascular physiology. Thus, while “pericytic stem cells” are enticing candidates for pro-wound healing treatments, they must be rigorously validated before they can be truly called pericytes.
Abbreviations
- AdSC
adipose-derived stem cell
- BM
basement membrane
- DR
diabetic retinopathy
- EC
endothelial cell
- ECM
extracellular matrix
- EPC
endothelial progenitor cell
- LER
low expression region
- MC
Müller cell
- MSC
mesenchymal stem cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al Ahmad A, Taboada CB, Gassmann M, Ogunshola OO. Astrocytes and pericytes differentially modulate blood–brain barrier characteristics during development and hypoxic insult. J Cereb Blood Flow Metab. 2011;31(2):693–705. doi: 10.1038/jcbfm.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambler CA, Nowicki JL, Burke AC, Bautch VL. Assembly of trunk and limb blood vessels involves extensive migration and vasculogenesis of somite-derived angioblasts. Dev Biol. 2001;234(2):352–64. doi: 10.1006/dbio.2001.0267. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. 2011 Jan 26; http://www.diabetes.org/diabetes-basics/diabetes-statistics/?loc=DropDownDB-stats.
- Amos PJ, Shang H, Bailey AM, Taylor A, Katz AJ, Peirce SM. IFATS collection: The role of human adipose-derived cells in inflammatory microvascular remodeling and evidence of a perivascular phenotype. Stem Cell. 2008;26(10):2682–90. doi: 10.1634/stemcells.2008-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos PJ, Mulvey CL, Seaman SA, Walpole J, Degen KE, Shang H, et al. Hypoxic culture and in vivo inflammatory environments affect the assumption of pericyte characteristics by human adipose and bone marrow progenitor cells. Am J Physiol Cell Physiol. 2011;301(6):C1378–88. doi: 10.1152/ajpcell.00460.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anghelina M, Krishnan P, Moldovan L, Moldovan NI. Monocytes and macrophages form branched cell columns in matrigel: implications for a role in neovascularization. Stem Cells Dev. 2004;13(6):665–76. doi: 10.1089/scd.2004.13.665. [DOI] [PubMed] [Google Scholar]
- Anjos-Afonso F, Siapati EK, Bonnet D. In vivo contribution of murine mesenchymal stem cells into multiple cell-types under minimal damage conditions. J Cell Sci. 2004;117:5655–64. doi: 10.1242/jcs.01488. [DOI] [PubMed] [Google Scholar]
- Antonelli-Orlidge A, Saunders KB, Smith SR, D’Amore PA. An activated form of transforming growth factor β is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci U S A. 1989;86(12):4544–8. doi: 10.1073/pnas.86.12.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104(51):20262–7. doi: 10.1073/pnas.0706438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85(3):221–8. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- Avniel S, Arik Z, Maly A, Sagie A, Basst HB, Yahana MD, et al. Involvement of the CXCL12/CXCR4 pathway in the recovery of skin following burns. J Invest Dermatol. 2006;126(2):468–76. doi: 10.1038/sj.jid.5700069. [DOI] [PubMed] [Google Scholar]
- Bagley RG, Weber W, Rouleau C, Teicher BA. Pericytes and endothelial precursor cells: cellular interactions and contributions to malignancy. Cancer Res. 2005;65(21):9741–50. doi: 10.1158/0008-5472.CAN-04-4337. [DOI] [PubMed] [Google Scholar]
- Balabanov R, Washington R, Wagnerova J, Dore-Duffy P. CNS microvascular pericytes express macrophage-like function, cell surface integrin aM and macrophage marker ED-2. Microvasc Res. 1996;52:127–142. doi: 10.1006/mvre.1996.0049. [DOI] [PubMed] [Google Scholar]
- Balabanov R, Beaumont T, Dore-Duffy P. Role of central nervous system microvascular pericytes in activation of antigen-primed splenic T-lymphocytes. J Neurosci Res. 1999;55(5):578–87. doi: 10.1002/(SICI)1097-4547(19990301)55:5<578::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16(5):585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- Bautch VL. Stem cells and the vasculature. Nat Med. 2011;17(11):1437–43. doi: 10.1038/nm.2539. [DOI] [PubMed] [Google Scholar]
- Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA. 2002;287(19):2570–81. doi: 10.1001/jama.287.19.2570. [DOI] [PubMed] [Google Scholar]
- Blair P, Flaumenhaft R. Platelet α-granules: basic biology and clinical correlates. Blood Rev. 2009;23(4):177–89. doi: 10.1016/j.blre.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluff JE, Ferguson MW, O’Kane S, Ireland G. Bone marrow–derived endothelial progenitor cells do not contribute significantly to new vessels during incisional wound healing. Exp Hematol. 2007;35(3):500–6. doi: 10.1016/j.exphem.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Boscolo E, Stewart CL, Greenberger S, Wu JK, Durham JT, Herman IM, et al. JAGGED1 signaling regulates hemangioma stem cell-to-pericyte/vascular smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol. 2011;31(10):2181–92. doi: 10.1161/ATVBAHA.111.232934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard BA, Shatos MA, Tracy PB. Human brain pericytes differentially regulate expression of procoagulant enzyme complexes comprising the extrinsic pathway of blood coagulation. Arterioscler Thromb Vasc Biol. 1997;17(1):1–9. doi: 10.1161/01.atv.17.1.1. [DOI] [PubMed] [Google Scholar]
- Brass LF. Thrombin and platelet activation. Chest. 2003;124(3 Suppl):18S–25S. doi: 10.1378/chest.124.3_suppl.18s. [DOI] [PubMed] [Google Scholar]
- Brem H, Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes. J Clin Invest. 2007;117(5):1219–22. doi: 10.1172/JCI32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, et al. Müller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25(4):397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Brower J, Blumberg S, Carroll E, Pastar I, Brem H, Chen W. Mesenchymal stem cell therapy and delivery systems in nonhealing wounds. Adv Skin Wound Care. 2011 Nov;24(11):524–32. doi: 10.1097/01.ASW.0000407648.89961.a6. [DOI] [PubMed] [Google Scholar]
- Chantrain CF, Shinmada H, Jodele S, Groshen S, Ye W, Shalinsky DR, et al. Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res. 2004;64(5):1675–86. doi: 10.1158/0008-5472.can-03-0160. [DOI] [PubMed] [Google Scholar]
- Chen Y, Chang F, Wu C, Chou Y, Hsu H, Chiang W, et al. Platelet-derived growth factor receptor signaling activates pericyte–myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80(11):1170–81. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- Cleaver O, Tonissen KF, Saha MS, Krieg PA. Neovascularization of the xenopus embryo. Dev Dyn. 1997;210(1):66–77. doi: 10.1002/(SICI)1097-0177(199709)210:1<66::AID-AJA7>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Cook-Mills JM, Deem TL. Active participation of endothelial cells in inflammation. J Leukoc Biol. 2005;77(4):487–95. doi: 10.1189/jlb.0904554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corselli M, Chen CW, Crisan M, Lazzari L, Péault B. Perivascular ancestors of adult multipotent stem cells. Arterioscler Thromb Vasc Biol. 2010;30(6):1104–9. doi: 10.1161/ATVBAHA.109.191643. [DOI] [PubMed] [Google Scholar]
- Crawford TN, Alfaro DV, Kerrison JB, Jablon EP. Diabetic retinopathy and angiogenesis. Curr Diabetes Rev. 2009;5(1):8–13. doi: 10.2174/157339909787314149. [DOI] [PubMed] [Google Scholar]
- Crisan M, Deasy B, Gavina M, Zheng B, Huard J, Lazzari L, Péault B. Purification and long-term culture of multipotent progenitor cells affiliated with the walls of human blood vessels: myoendothelial cells and pericytes. Methods Cell Biol. 2008a;86:295–309. doi: 10.1016/S0091-679X(08)00013-7. [DOI] [PubMed] [Google Scholar]
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008b;3(3):301–13. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Crocker DJ, Murad TM, Geer JC. Role of the pericyte in wound healing an ultrastructural study. Exp Mol Pathol. 1970;13(1):51–65. doi: 10.1016/0014-4800(70)90084-5. [DOI] [PubMed] [Google Scholar]
- Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. J Leukoc Biol. 2010;87(1):59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar A, Domev H, Ben-Yosef O, Tzukerman M, Zeevi-Levin N, Novak A, et al. Multipotent vasculogenic pericytes from human pluripotent stem cells promote recovery of murine ischemic limb. Circulation. 2012;125(1):87–99. doi: 10.1161/CIRCULATIONAHA.111.048264. [DOI] [PubMed] [Google Scholar]
- Darby I, Skalli O, Gabbiani G. Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest. 1990;63(1):21–9. [PubMed] [Google Scholar]
- Demidova-Rice TN, Wolf L, Deckenback J, Hamblin MR, Herman IM. Human platelet-rich plasma- and extracellular matrix- derived peptides promote impaired cutaneous wound healing in vivo. PLoS One. 2012;7(2):e32146. doi: 10.1371/journal.pone.0032146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbie JG, Kwaan HC, Colwell J, Suwanwela N. Role of platelets in pathogenesis of diabetic retinopathy. Arch Ophthalmol. 1974;91(2):107–9. doi: 10.1001/archopht.1974.03900060113005. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P, Katychev A, Wang X, Van Buren E. CNS microvascular pericytes exhibit multipotential stem cell activity. J Cereb Blood Flow Metab. 2006;26(5):613–24. doi: 10.1038/sj.jcbfm.9600272. [DOI] [PubMed] [Google Scholar]
- Durham JT, Herman IM. Microvascular modifications in diabetic retinopathy. Curr Diab Rep. 2011;11(4):253–64. doi: 10.1007/s11892-011-0204-0. [DOI] [PubMed] [Google Scholar]
- Enge M, Bjarnegård M, Gerhardt H, Gustafsson E, Kalén M, Asker N, et al. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 2002;21(16):4307–16. doi: 10.1093/emboj/cdf418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English D, Welch Z, Kovala AT, Harvey K, Volpert OV, Brindley DN, et al. Sphingosine 1-phosphate released from platelets during clotting accounts for the potent endothelial cell chemotactic activity of blood serum and provides a novel link between hemostasis and angiogenesis. FASEB J. 2000;14(14):2255–65. doi: 10.1096/fj.00-0134com. [DOI] [PubMed] [Google Scholar]
- Evans KK, Attinger CE, Al-Attar A, Salgado C, Chu CK, Mardini S, et al. The importance of limb preservation in the diabetic population. J Diabetes Complications. 2011;25(4):227–31. doi: 10.1016/j.jdiacomp.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Fahey TJ, 3rd, Sadaty A, Jones WG, 2nd, Barber A, Smoller B, Shires GT. Diabetes impairs the late inflammatory response to wound healing. J Surg Res. 1991;50(4):308–13. doi: 10.1016/0022-4804(91)90196-s. [DOI] [PubMed] [Google Scholar]
- Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;12;366(9498):1736–43. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Neutrophils emigrate from venules by a transendothelial cell pathway in response to FMLP. J Exp Med. 1998;187(6):903–15. doi: 10.1084/jem.187.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro JL, Gómez-Hospital JA, Angiolillo DJ. Platelet abnormalities in diabetes mellitus. Diab Vasc Dis Res. 2010;7(4):251–9. doi: 10.1177/1479164110383994. [DOI] [PubMed] [Google Scholar]
- Fisher M. Pericyte signaling in the neurovascular unit. Stroke. 2009;40(3 Suppl):S13–5. doi: 10.1161/STROKEAHA.108.533117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350(1):48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- Garcia CM, Darland DC, Massingham LJ, D’Amore PA. Endothelial cell–astrocyte interactions and TGFβ are required for induction of blood–neural barrier properties. Brain Res Dev Brain Res. 2004;152(1):25–38. doi: 10.1016/j.devbrainres.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005;438(7070):960–6. doi: 10.1038/nature04482. [DOI] [PubMed] [Google Scholar]
- Gaudry M, Brégerie O, Andrieu V, El Benna J, Pocidalo MA, Hakim J. Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood. 1997;90(10):4153–61. [PubMed] [Google Scholar]
- Gay AN, Mushin OP, Lazar DA, Naik-Mathuria BJ, Yu L, Gobin A, et al. Wound healing characteristics of ICAM-1 null mice devoid of all isoforms of ICAM-1. J Surg Res. 2011;171(1):e1–7. doi: 10.1016/j.jss.2011.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, et al. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15(11):1298–306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill M, Dias S, Hattori K, Rivera ML, Hicklin D, Witte L, et al. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88(2):167–74. doi: 10.1161/01.res.88.2.167. [DOI] [PubMed] [Google Scholar]
- Goepfert C, Sundberg C, Sévigny J, Enjyoji K, Hoshi T, Csizmadia E, et al. Disordered cellular migration and angiogenesis in cd39-null mice. Circulation. 2001;104(25):3109–15. doi: 10.1161/hc5001.100663. [DOI] [PubMed] [Google Scholar]
- Goren I, Allmann N, Yogev N, Schürmann C, Linke A, Holdener M, et al. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol. 2009;175(1):132–47. doi: 10.2353/ajpath.2009.081002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisén J. A pericyte origin of spinal cord scar tissue. Science. 2011;333(6039):238–42. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- Grinnell F, Ho CH. Transforming growth factor beta stimulates fibroblast-collagen matrix contraction by different mechanisms in mechanically loaded and unloaded matrices. Exp Cell Res. 2002;273(2):248–55. doi: 10.1006/excr.2001.5445. [DOI] [PubMed] [Google Scholar]
- Hammes HP, Federoff HJ, Brownlee M. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol Med. 1995;1(5):527–34. [PMC free article] [PubMed] [Google Scholar]
- Hammes HP. Pericytes and the pathogenesis of diabetic retinopathy. Horm Metab Res. 2005;37 (Suppl 1):39–43. doi: 10.1055/s-2005-861361. [DOI] [PubMed] [Google Scholar]
- Hellström M, Kalén M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126(14):3047–55. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- Herman IM, D’Amore PA. Microvascular pericytes contain muscle and nonmuscle actins. J Cell Biol. 1985;101(1):43–52. doi: 10.1083/jcb.101.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschi KK, D’Amore PA. Pericytes in the microvasculature. Cardiovasc Res. 1996;32(4):687–98. [PubMed] [Google Scholar]
- Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141(3):805–14. doi: 10.1083/jcb.141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Lu G, Wu Y, Jirigala E, Xu Y, Ma K, et al. Mesenchymal stem cells delivered in a microsphere-based engineered skin contribute to cutaneous wound healing and sweat gland repair. J Dermatol Sci. 2012;66(1):29–36. doi: 10.1016/j.jdermsci.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Kimura A, Kuninaka Y, Inui M, Matsushima K, Mukaida N, et al. Pivotal role of the CCL5/CCR5 interaction for recruitment of endothelial progenitor cells in mouse wound healing. J Clin Invest. 2012;122(2):711–21. doi: 10.1172/JCI43027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Toriumi H, Yamada S, Hoshino H, Suzuki N. Astrocytes and pericytes cooperatively maintain a capillary-like structure composed of endothelial cells on gel matrix. Brain Res. 2011;1406:74–83. doi: 10.1016/j.brainres.2011.06.039. [DOI] [PubMed] [Google Scholar]
- Jeynes B. Reactions of granular pericytes in a rabbit cerebrovascular ischemia model. Stroke. 1985;16(1):121–5. doi: 10.1161/01.str.16.1.121. [DOI] [PubMed] [Google Scholar]
- Johnson RJ, Iida H, Alpers CE, Majesky MW, Schwartz SM, Pritzi P, et al. Expression of smooth muscle cell phenotype by rat mesangial cells in immune complex nephritis. Alpha-smooth muscle actin is a marker of mesangial cell proliferation. J Clin Invest. 1991;87(3):847–58. doi: 10.1172/JCI115089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18(12):1450–2. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- Juniantito V, Izawa T, Yuasa T, Ichikawa C, Yamamoto E, Kuwamura M, et al. Immunophenotypical analyses of myofibroblasts in rat excisional wound healing: possible transdifferentiation of blood vessel pericytes and perifollicular dermal sheath cells into myofibroblasts. Histol Histopathol. 2012;27(4):515–27. doi: 10.14670/HH-27.515. [DOI] [PubMed] [Google Scholar]
- Kanno E, Kawakami K, Ritsu M, Ishii K, Tanno H, Toriyabe S, et al. Wound healing in skin promoted by inoculation with Pseudomonas aeruginosa PAO1: The critical role of tumor necrosis factor-α secreted from infiltrating neutrophils. Wound Repair Regen. 2011;19(5):608–21. doi: 10.1111/j.1524-475X.2011.00721.x. [DOI] [PubMed] [Google Scholar]
- Kilarski WW, Samolov B, Petersson L, Kvanta A, Gerwins P. Biomechanical regulation of blood vessel growth during tissue vascularization. Nat Med. 2009;15(6):657–64. doi: 10.1038/nm.1985. [DOI] [PubMed] [Google Scholar]
- Kim EK, Li G, Lee TJ, Hong JP. The effect of human adipose-derived stem cells on healing of ischemic wounds in a diabetic nude mouse model. Plast Reconstr Surg. 2011;128(2):387–94. doi: 10.1097/PRS.0b013e31821e6de2. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim JH, Yu YS, Kim DH, Kim KW. Recruitment of pericytes and astrocytes is closely related to the formation of tight junction in developing retinal vessels. J Neurosci Res. 2009;87(3):653–9. doi: 10.1002/jnr.21884. [DOI] [PubMed] [Google Scholar]
- Kim MH, Liu W, Borjesson DL, Curry FR, Miller LS, Cheung AL, et al. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J Invest Dermatol. 2008;128(7):1812–20. doi: 10.1038/sj.jid.5701223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knighton DR, Hunt TK, Scheuenstuhl H, Halliday BJ, Werb Z, Banda MJ. Oxygen tension regulates the expression of angiogenesis factor by macrophages. Science. 1983;221(4617):1283–5. doi: 10.1126/science.6612342. [DOI] [PubMed] [Google Scholar]
- Knittel T, Dinter C, Kobold D, Neubauer K, Mehde M, Eichhorst S, et al. Expression and regulation of cell adhesion molecules by hepatic stellate cells (HSC) of rat liver: involvement of HSC in recruitment of inflammatory cells during hepatic tissue repair. Am J Pathol. 1999;154(1):153–67. doi: 10.1016/s0002-9440(10)65262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolyada AY, Riley KN, Herman IM. Rho GTPase signaling modulates cell shape and contractile phenotype in an isoactin-specific manner. Am J Physiol Cell Physiol. 2003;285(5):C1116–21. doi: 10.1152/ajpcell.00177.2003. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Zhong Q, Kanwar M. Metabolic memory and diabetic retinopathy: role of inflammatory mediators in retinal pericytes. Exp Eye Res. 2010;90(5):617–23. doi: 10.1016/j.exer.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutcher ME, Kolyada AY, Surks HK, Herman IM. Pericyte Rho GTPase mediates both pericyte contractile phenotype and capillary endothelial growth state. Am J Pathol. 2007;171(2):693–701. doi: 10.2353/ajpath.2007.070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutcher ME, Herman IM. The pericyte: cellular regulator of microvascular blood flow. Microvasc Res. 2009;77(3):235–46. doi: 10.1016/j.mvr.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasala GP, Silva JA, Minguell JJ. Therapeutic angiogenesis in patients with severe limb ischemia by transplantation of a combination stem cell product. J Thorac Cardiovasc Surg. 2011 Nov 12; doi: 10.1016/j.jtcvs.2011.08.053. Epub. [DOI] [PubMed] [Google Scholar]
- Laurens N, Koolwijk P, de Maat MP. Fibrin structure and wound healing. J Thromb Haemost. 2006;4(5):932–9. doi: 10.1111/j.1538-7836.2006.01861.x. [DOI] [PubMed] [Google Scholar]
- Lee EY, Xia Y, Kim WS, Kim MH, Kim TH, Kim KJ, et al. Hypoxia-enhanced wound-healing function of adipose-derived stem cells: increase in stem cell proliferation and up-regulation of VEGF and bFGF. Wound Repair Regen. 2009;17(4):540–7. doi: 10.1111/j.1524-475X.2009.00499.x. [DOI] [PubMed] [Google Scholar]
- Lee S, Zeiger A, Maloney JM, Kotecki M, Van Vliet KJ, Herman IM. Pericyte actomyosin-mediated contraction at the cell-material interface can modulate the microvascular niche. J Phys Condens Matter. 2010;22(19):194115. doi: 10.1088/0953-8984/22/19/194115. [DOI] [PubMed] [Google Scholar]
- Li B, Wang JH. Fibroblasts and myofibroblasts in wound healing: force generation and measurement. J Tissue Viability. 2011;20(4):108–20. doi: 10.1016/j.jtv.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang YP, Kirsner RS. Angiogenesis in wound repair: angiogenic growth factors and the extracellular matrix. Microsc Res Tech. 2003;60(1):107–14. doi: 10.1002/jemt.10249. [DOI] [PubMed] [Google Scholar]
- Liao H, Zakhaleva J, Chen W. Cells and tissue interactions with glycated collagen and their relevance to delayed diabetic wound healing. Biomaterials. 2009;30(9):1689–96. doi: 10.1016/j.biomaterials.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CS, Xin ZC, Deng CH, Ning H, Lin G, Lue TF. Defining adipose tissue-derived stem cells in tissue and in culture. Histol Histopathol. 2010;25(6):807–15. doi: 10.14670/HH-25.807. [DOI] [PubMed] [Google Scholar]
- Lobmann R, Ambrosch A, Schultz G, Waldmann K, Schiweck S, Lehnert H. Expression of matrix-metalloproteinases and their inhibitors in the wounds of diabetic and non-diabetic patients. Diabetologia. 2002;45(7):1011–6. doi: 10.1007/s00125-002-0868-8. [DOI] [PubMed] [Google Scholar]
- Lorenzi M, Gerhardinger C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia. 2001;44(7):791–804. doi: 10.1007/s001250100544. [DOI] [PubMed] [Google Scholar]
- Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Müller W, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184(7):3964–77. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- Maharlooei MK, Bagheri M, Solhjou Z, Jahromi BM, Akrami M, Rohani L, et al. Adipose tissue derived mesenchymal stem cell (AD-MSC) promotes skin wound healing in diabetic rats. Diabetes Res Clin Pract. 2011;93(2):228–34. doi: 10.1016/j.diabres.2011.04.018. [DOI] [PubMed] [Google Scholar]
- Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. 2010;58(9):1094–103. doi: 10.1002/glia.20990. [DOI] [PubMed] [Google Scholar]
- McDonald AG, Yang K, Roberts HR, Monroe DM, Hoffman M. Perivascular tissue factor is down-regulated following cutaneous wounding: implications for bleeding in hemophilia. Blood. 2008;111(4):2046–8. doi: 10.1182/blood-2007-05-092916. [DOI] [PubMed] [Google Scholar]
- Merfeld-Clauss S, Gollahalli N, March KL, Traktuev DO. Adipose tissue progenitor cells directly interact with endothelial cells to induce vascular network formation. Tissue Eng Part A. 2010;16(9):2953–66. doi: 10.1089/ten.tea.2009.0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakawa T, Bready J, Berliner J, Fisher M, Cancilla PA. In vitro interaction of astrocytes and pericytes with capillary-like structures of brain microvessel endothelium. Lab Invest. 1991 Jul;65(1):32–40. [PubMed] [Google Scholar]
- Mirza R, DiPietro LA, Koh TJ. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol. 2009;175(6):2454–62. doi: 10.2353/ajpath.2009.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möhle R, Green D, Moore MA, Nachman RL, Rafii S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci U S A. 1997;94(2):663–8. doi: 10.1073/pnas.94.2.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178(2):449–60. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Ishibashi T, Inomata H. Immunohistochemical detection of extravasated fibrinogen (fibrin) in human diabetic retina. Graefes Arch Clin Exp Ophthalmol. 1992;230(5):428–31. doi: 10.1007/BF00175927. [DOI] [PubMed] [Google Scholar]
- Nagaoka T, Kaburagi Y, Hamaguchi Y, Hasegawa M, Takehara K, Steeber DA, et al. Delayed wound healing in the absence of intercellular adhesion molecule-1 or L-selectin expression. Am J Pathol. 2000;157(1):237–47. doi: 10.1016/S0002-9440(10)64534-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu M, Kishimoto S, Nakamura S, Mizuno H, Yanagibayashi S, Yamamoto N, et al. Accelerated wound healing in healing-impaired db/db mice by autologous adipose tissue-derived stromal cells combined with atelocollagen matrix. Ann Plast Surg. 2009;62(3):317–21. doi: 10.1097/SAP.0b013e31817f01b6. [DOI] [PubMed] [Google Scholar]
- Natesan S, Wrice NL, Baer DG, Christy RJ. Debrided skin as a source of autologous stem cells for wound repair. Stem Cells. 2011a;29(8):1219–30. doi: 10.1002/stem.677. [DOI] [PubMed] [Google Scholar]
- Natesan S, Zhang G, Baer DG, Walters TJ, Christy RJ, Suggs LJ. A bilayer construct controls adipose-derived stem cell differentiation into endothelial cells and pericytes without growth factor stimulation. Tissue Eng Part A. 2011b;17(7–8):941–53. doi: 10.1089/ten.tea.2010.0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak RC, Berman AB, George KL, Eisenbarth GS, King GL. A monoclonal antibody (3G5)-defined ganglioside antigen is expressed on the cell surface of microvascular pericytes. J Exp Med. 1988;167(3):1003–15. doi: 10.1084/jem.167.3.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehls V, Denzer K, Drenckhahn D. Pericyte involvement in capillary sprouting during angiogenesis in situ. Cell Tissue Res. 1992;270(3):469–74. doi: 10.1007/BF00645048. [DOI] [PubMed] [Google Scholar]
- Nehls V, Schuchardt E, Drenckhahn D. The effect of fibroblasts, vascular smooth muscle cells, and pericytes on sprout formation of endothelial cells in a fibrin gel angiogenesis system. Microvasc Res. 1994;48(3):349–63. doi: 10.1006/mvre.1994.1061. [DOI] [PubMed] [Google Scholar]
- Nolan CM, Beaty HN, Bagdade JD. Further characterization of the impaired bactericidal function of granulocytes in patients with poorly controlled diabetes. Diabetes. 1978;27(9):889–94. doi: 10.2337/diab.27.9.889. [DOI] [PubMed] [Google Scholar]
- Ozerdem U, Grako KA, Dahlin-Huppe K, Monosov E, Stallcup WB. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn. 2001;222(2):218–27. doi: 10.1002/dvdy.1200. [DOI] [PubMed] [Google Scholar]
- Pablos JL, Amara A, Bouloc A, Santiago B, Caruz A, Galindo M, et al. Stromal-cell derived factor is expressed by dendritic cells and endothelium in human skin. Am J Pathol. 1999;155(5):1577–86. doi: 10.1016/S0002-9440(10)65474-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papetti M, Shujath J, Riley KN, Herman IM. FGF-2 antagonizes the TGF-beta1-mediated induction of pericyte alpha-smooth muscle actin expression: a role for myf-5 and Smad-mediated signaling pathways. Invest Ophthalmol Vis Sci. 2003;44(11):4994–5005. doi: 10.1167/iovs.03-0291. [DOI] [PubMed] [Google Scholar]
- Park HB, Yang JH, Chung KH. Characterization of the cytokine profile of platelet rich plasma (PRP) and PRP-induced cell proliferation and migration: Upregulation of matrix metalloproteinase-1 and -9 in HaCaT cells. Korean J Hematol. 2011;46(4):265–73. doi: 10.5045/kjh.2011.46.4.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50(4):427–34. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Perrone L, Devi TS, Hosoya K, Terasaki T, Singh LP. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol. 2009;221(1):262–72. doi: 10.1002/jcp.21852. [DOI] [PubMed] [Google Scholar]
- Pham HT, Economides PA, Veves A. The role of endothelial function on the foot. Microcirculation and wound healing in patients with diabetes. Clin Podiatr Med Surg. 1998;15(1):85–93. [PubMed] [Google Scholar]
- Prasad A, Qamri Z, Wu J, Ganju RK. Slit-2/Robo-1 modulates the CXCL12/CXCR4-induced chemotaxis of T cells. J Leukoc Biol. 2007;82(3):465–76. doi: 10.1189/jlb.1106678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar VS, Shiwen X, Bostrom M, Leoni P, Muddle J, Ivarsson M, et al. Platelet-derived growth factor-beta receptor activation is essential for fibroblast and pericyte recruitment during cutaneous wound healing. Am J Pathol. 2006;169(6):2254–65. doi: 10.2353/ajpath.2006.060196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom BR, Ransom CB. Astrocytes: multitalented stars of the central nervous system. Methods Mol Biol. 2012;814:3–7. doi: 10.1007/978-1-61779-452-0_1. [DOI] [PubMed] [Google Scholar]
- Reddy MA, Natarajan R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc Res. 2011;90(3):421–9. doi: 10.1093/cvr/cvr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero MP, Khosrotehrani K. Skin wound healing modulation by macrophages. Int J Clin Exp Pathol. 2010;3(7):643–53. [PMC free article] [PubMed] [Google Scholar]
- Rowley WR, Bezold C. Creating Public Awareness: State 2025 Diabetes Forecasts. Popul Health Manag. 2012 Jan 27; doi: 10.1089/pop.2011.0053. Epub. [DOI] [PubMed] [Google Scholar]
- Roy S, Cagliero E, Lorenzi M. Fibronectin overexpression in retinal microvessels of patients with diabetes. Invest Ophthalmol Vis Sci. 1996;37(2):258–66. [PubMed] [Google Scholar]
- Roy S, Driggs J, Elgharably H, Biswas S, Findley M, Khanna S, et al. Platelet-rich fibrin matrix improves wound angiogenesis via inducing endothelial cell proliferation. Wound Repair Regen. 2011;19(6):753–66. doi: 10.1111/j.1524-475X.2011.00740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbia-Brandt L, Mentha G, Desmoulière A, Alto Costa AM, Giostra E, Molas G, et al. Hepatic stellate cells reversibly express alpha-smooth muscle actin during acute hepatic ischemia. Transplant Proc. 1997;29(5):2390–5. doi: 10.1016/s0041-1345(97)00415-6. [DOI] [PubMed] [Google Scholar]
- Rustad KC, Wong VW, Sorkin M, Glotzbach JP, Major MR, Rajadas J, et al. Enhancement of mesenchymal stem cell angiogenic capacity and stemness by a biomimetic hydrogel scaffold. Biomaterials. 2012 Jan;33(1):80–90. doi: 10.1016/j.biomaterials.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Geniez M, Maharaj AS, Walshe TE, Tucker BA, Sekiyama E, Kurihara T, et al. Endogenous VEGF is required for visual function: evidence for a survival role on müller cells and photoreceptors. PLoS One. 2008;3(11):e3554. doi: 10.1371/journal.pone.0003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Abe R, Fujita Y, Ando S, Inokuma D, Shimizu H. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J Immunol. 2008;180(4):2581–7. doi: 10.4049/jimmunol.180.4.2581. [DOI] [PubMed] [Google Scholar]
- Saunders WB, Bohnsack BL, Faske JB, Anthis NJ, Bayless KJ, Hirschi KK, et al. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J Cell Biol. 2006;175(1):179–91. doi: 10.1083/jcb.200603176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlingemann RO, Rietveld FJ, Kwaspen F, van de Kerkhof PC, de Waal RM, Ruiter DJ. Differential expression of markers for endothelial cells, pericytes, and basal lamina in the microvasculature of tumors and granulation tissue. Am J Pathol. 1991;138(6):1335–47. [PMC free article] [PubMed] [Google Scholar]
- Schlingemann RO, Oosterwijk E, Wesseling P, Rietveld FJ, Ruiter DJ. Aminopeptidase a is a constituent of activated pericytes in angiogenesis. J Pathol. 1996;179(4):436–42. doi: 10.1002/(SICI)1096-9896(199608)179:4<436::AID-PATH611>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Schultz GS, Davidson JM, Kirsner RS, Bornstein P, Herman IM. Dynamic reciprocity in the wound microenvironment. Wound Repair Regen. 2011;19(2):134–48. doi: 10.1111/j.1524-475X.2011.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142(3):873–81. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva GV, Litovsky S, Assad JA, Sousa AL, Martin BJ, Vela D, et al. Mesenchymal stem cells differentiate into an endothelial phenotype, enhance vascular density, and improve heart function in a canine chronic ischemia model. Circulation. 2005;111(2):150–6. doi: 10.1161/01.CIR.0000151812.86142.45. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5(2):146–56. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Simpson DM, Ross R. The neutrophilic leukocyte in wound repair a study with antineutrophil serum. J Clin Invest. 1972;51(8):2009–23. doi: 10.1172/JCI107007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims DE. The pericyte--a review. Tissue Cell. 1986;18(2):153–74. doi: 10.1016/0040-8166(86)90026-1. [DOI] [PubMed] [Google Scholar]
- Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, Gabbiani G. A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol. 1986;103(6 Pt 2):2787–96. doi: 10.1083/jcb.103.6.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32(12):638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowiej A, Biswas P, Graesser D, Madri JA. Lack of platelet endothelial cell adhesion molecule-1 attenuates foreign body inflammation because of decreased angiogenesis. Am J Pathol. 2003;162(3):953–62. doi: 10.1016/S0002-9440(10)63890-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell JM, Caplan AI. Fibroblast heterogeneity: more than skin deep. J Cell Sci. 2004;117(Pt 5):667–75. doi: 10.1242/jcs.01005. [DOI] [PubMed] [Google Scholar]
- Sponheim J, Pollheimer J, Olsen T, Balogh J, Hammarström C, Loos T, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol. 2010;177(6):2804–15. doi: 10.2353/ajpath.2010.100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57(6):563–81. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Subramaniam M, Saffaripour S, Van De Water L, Frenette PS, Mayadas TN, Hynes RO, et al. Role of endothelial selectins in wound repair. Am J Pathol. 1997;150(5):1701–9. [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5(4):434–8. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- Thomas WE. Brain macrophages: on the role of pericytes and perivascular cells. Brain Res Brain Res Rev. 1999;31(1):42–57. doi: 10.1016/s0165-0173(99)00024-7. [DOI] [PubMed] [Google Scholar]
- Tigges U, Hyer EG, Scharf J, Stallcup WB. FGF2-dependent neovascularization of subcutaneous Matrigel plugs is initiated by bone marrow-derived pericytes and macrophages. Development. 2008;135(3):523–32. doi: 10.1242/dev.002071. [DOI] [PubMed] [Google Scholar]
- Tilton RG, Hoffmann PL, Kilo C, Williamson JR. Pericyte degeneration and basement membrane thickening in skeletal muscle capillaries of human diabetics. Diabetes. 1981;30(4):326–34. doi: 10.2337/diab.30.4.326. [DOI] [PubMed] [Google Scholar]
- Tilton RG, Faller AM, Burkhardt JK, Hoffmann PL, Kilo C, Williamson JR. Pericyte degeneration and acellular capillaries are increased in the feet of human diabetic patients. Diabetologia. 1985;28(12):895–900. doi: 10.1007/BF00703132. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol Symp Proc. 2000;5(1):40–6. doi: 10.1046/j.1087-0024.2000.00014.x. [DOI] [PubMed] [Google Scholar]
- Tout S, Chan-Ling T, Holländer H, Stone J. The role of Müller cells in the formation of the blood-retinal barrier. Neuroscience. 1993;55(1):291–301. doi: 10.1016/0306-4522(93)90473-s. [DOI] [PubMed] [Google Scholar]
- Traktuev DO, Merfeld-Clauss S, Li J, Kolonin M, Arap W, Pasqualini R, et al. A population of multipotent CD34-positive adipose stromal cells share pericyte and mesenchymal surface markers, reside in a periendothelial location, and stabilize endothelial networks. Circ Res. 2008;102(1):77–85. doi: 10.1161/CIRCRESAHA.107.159475. [DOI] [PubMed] [Google Scholar]
- Tu Z, Li Y, Smith DS, Sheibani N, Huang S, Kern T, et al. Retinal pericytes inhibit activated T cell proliferation. Invest Ophthalmol Vis Sci. 2011;52(12):9005–10. doi: 10.1167/iovs.11-8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetz D, Ponzoni M, Schiraldi M, Ferreri AJ, Bertoni F, Doglioni C, et al. Perivascular expression of CXCL9 and CXCL12 in primary central nervous system lymphoma: T-cell infiltration and positioning of malignant B cells. Int J Cancer. 2010;127(10):2300–12. doi: 10.1002/ijc.25236. [DOI] [PubMed] [Google Scholar]
- Verbeek MM, Westphal JR, Ruiter DJ, de Waal RM. T lymphocyte adhesion to human brain pericytes is mediated via very late antigen-4/vascular cell adhesion molecule-1 interactions. J Immunol. 1995;154(11):5876–84. [PubMed] [Google Scholar]
- Vogeli KM, Jin SW, Martin GR, Stainier DY. A common progenitor for haematopoietic and endothelial lineages in the zebrafish gastrula. Nature. 2006;443(7109):337–9. doi: 10.1038/nature05045. [DOI] [PubMed] [Google Scholar]
- Voisin MB, Pröbstl D, Nourshargh S. Venular basement membranes ubiquitously express matrix protein low-expression regions: characterization in multiple tissues and remodeling during inflammation. Am J Pathol. 2010;176(1):482–95. doi: 10.2353/ajpath.2010.090510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, et al. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. 2006;203(6):1519–32. doi: 10.1084/jem.20051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller A, Isenmann S, Vestweber D. Cloning of the mouse endothelial selectins. Expression of both E- and P-selectin is inducible by tumor necrosis factor alpha. J Biol Chem. 1992;267(21):15176–83. [PubMed] [Google Scholar]
- Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179(6):1311–23. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfin A, Voisin MB, Nourshargh S. Recent developments and complexities in neutrophil transmigration. Curr Opin Hematol. 2010;17(1):9–17. doi: 10.1097/MOH.0b013e3283333930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HS, Shin J, Bhang SH, Shin JY, Park J, Im GI, et al. Enhanced skin wound healing by a sustained release of growth factors contained in platelet-rich plasma. Exp Mol Med. 2011;43(11):622–9. doi: 10.3858/emm.2011.43.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Bai L, Yan ZQ, Wang YH, Jiang ZL. Shear stress and vascular smooth muscle cells promote endothelial differentiation of endothelial progenitor cells via activation of Akt. Clin Biomech (Bristol, Avon) 2008;23 (Suppl 1):S118–24. doi: 10.1016/j.clinbiomech.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Yoshimura K, Shigeura T, Matsumoto D, Sato T, Takaki Y, Aiba-Kojima E, et al. Characterization of freshly isolated and cultured cells derived from the fatty and fluid portions of liposuction aspirates. J Cell Physiol. 2006;208(1):64–76. doi: 10.1002/jcp.20636. [DOI] [PubMed] [Google Scholar]