

Abstract
The activating receptor NKG2D plays an important role in the development of type-1 diabetes. Exploiting a natural phenomenon observed in tumors, plasmid DNA encoding for a soluble ligand to NKG2D (sRAE-1γ) was isolated and engineered into a plasmid expression system. A polymeric gene delivery system was developed to deliver the soluble RAE-1 plasmid to the pancreatic islets. The bioreducible cationic polymer poly(cystamine bisacrylamide–diamino hexane) (p(CBA-DAH)) was modified with poly(ethylene glycol) (PEG) and the targeting peptide CHVLWSTRC, known to target the EphA2 and EphA4 receptors. We observed a higher uptake of the targeting polymer Eph-PEG-p(CBA-DAH) in the pancreas of NOD mice compared to non-targeting controls. To evaluate the efficacy of preventing diabetes, the Eph-PEG-p(CBA-DAH)/RAE-1 complex (polyplex) was intravenously injected into 6-week-old female NOD mice. Within 17 weeks blood glucose levels were stabilized in animals injected with polyplex, while those treated without therapeutic plasmid developed progressive hyperglycemia. Additionally, the degree of insulitis and the infiltration of CD8+ T-cells in the polyplex treated group were improved over the targeting polymer only treated group. The current study suggest that the therapy of the Eph-PEG-p(CBA-DAH) delivering therapeutic sRAE-1 gene may be used to protect β-cells from autoimmune destruction and prevent type-1 diabetes.
Keywords: gene therapy, drug delivery, retinoic acid early inducible gene-1 (RAE-1), type 1 diabetes
INTRODUCTION
In the progression of the disease Type-1 diabetes, it is known that insulin-producing β-cells are destroyed by autoreactive cellular immunity [1]. Effective protection of β-cells from autoimmune destruction and specific targeting of pancreatic islets still remain difficult challenges [2]. The CD4+ and CD8+ T-cells are considered key players in the development of type-1 diabetes, and autoreactive CD8+ T-cells are required for both initiation and progression of type-1 diabetes [3, 4]. Additionally, the interaction of co-stimulatory receptors between T-cells and ligands on beta-cells are thought to be critical for the autoimmune response. The activating receptor natural killer group 2, member D (NKG2D) is thought to be directly involved in the progression of type-1 diabetes [5, 6]. NKG2D is a heterodimeric co-stimulatory receptor expressed by natural killer (NK) cells, CD8+ T-cells, and γδ T-cells [7, 8]. Expression of NKG2D can be observed in activated or memory CD8+ T-cells but not in naïve CD8+ nor CD4+ T-cells [9]. The ligands of NKG2D are stress-inducible proteins expressed in response to intra- or extra-cellular stimulation, such as cellular transformation and infection [10]. Retinoic acid early inducible gene-1 (RAE-1) has been identified as high affinity ligand of the NKG2D receptor [11].
The expression of RAE-1 on pancreatic islet beta-cells indicates a close relationship between this receptor and the pathogenesis of type-1 diabetes in the non-obese diabetic (NOD) mouse model [12]. It has been shown that systemic blockage of the NKG2D receptor with monoclonal antibodies impairs the expansion and function of autoreactive CD8+ T-cells, and consequently, prevents the progression of type-1 diabetes in the NOD mouse [13].
In a process known as immune evasion, a number of tumors protect themselves from the attack of NK cells and CD8+ T-cells by the action of tumor-derived soluble MIC ligands, a human analogue of mouse RAE-1 [14]. The soluble ligands interact with NKG2D receptors on NK cells and cancer-specific effector T-cells, which induces the endocytosis and degradation of the NKG2D receptor and concomitant down-regulation. However, systemic blockage of the NKG2D receptor is not clinically reasonable, due to the critical role of NKG2D in the immune system. Systemic administration of sRAE-1 could cause undesirable results, such as systemic non-function of NK cells and CD8+ T-cells [15–17]. Therefore, it is essential to limit active sRAE-1 expression to the pancreatic tissues, thereby minimizing side effects associated with systemic therapeutics.
We previously reported the applicatory potential of bioreducible cationic polymers, especially poly(cystamine bisacrylamide–diamino hexane) (p(CBA-DAH)), as promising candidates for efficient gene delivery [18, 19]. To specifically target the pancreas, the p(CBA-DAH) is modified with poly(ethylene glycol) (PEG) linked with targeting peptide CHVLWSTRC, known to target EphA2 and EphA4 receptors. The PEG serves to improve stability in the blood stream, while the peptide will target EphA2 and EphA4, which is overexpressed in the pancreatic islet microvasculature [20]. The specific targeting ability of Eph-PEG-p(CBA-DAH) to islet microvasculature endothelial cells was verified [21].
In this study, we evaluated the delivery of a plasmid coding for soluble RAE-1γ to the pancreatic islet microvasculature using a targeted bioreducible polymeric carrier in the NOD mouse. Specifically, we investigated the targeting ability of Eph-PEG-p(CBA-DAH), as well as the ability to reduce interactions between the beta-cells and infiltrating NKG2D positive lymphocytes, and effectively protect beta-cells from autoimmune destruction and prevent type-1 diabetes.
EXPERIMENTAL METHODS
Animals and diabetes incidence
The non-obese diabetic (NOD) mouse is a model of human type 1 diabetes in which autoreactive T cells mediate the destruction of pancreatic islet beta cells. Female 6-week-old NOD mice were purchased from the Jackson Laboratories (Bar Harbor, ME) and were maintained under specific pathogen free conditions. For gene therapy, polymer/plasmid DNA complex at the weight ratio of 10 was administered into the tail vein of a mouse at a dose of 40 μg of DNA per capita. After the first injection (day 0), each animal received the second injection (day 2), after which blood sampling was begun and the experimental data was collected. To evaluate diabetes incidence, blood samples were obtained once a week from the tail vein. Blood glucose levels were analyzed by glucometry using Accu-Check instant glucometer (Roche Diagnostics Co., Indianapolis, IN). Two consecutive glucose levels higher than 13.9 mmol/L (~250 mg/dL) considered hyperglycemia. NOD mice were sacrificed at scheduled times by cervical dislocation following anesthetization with methoxyflurane (Schering-Plough Animal Health, Union, NJ) inhalation. Statistical analysis will be performed using one-way ANOVA with Tukey’s post-hoc.
Preparation of Eph-PEG-poly(CBA-DAH)/DNA complex
The targeting polymer Eph-PEG-poly(CBA-DAH) and therapeutic RAE-1 plasmid were prepared and characterized as previously described [21]. Briefly, to synthesize the targeting polymer Eph-PEG-p(CBA-DAH), NHS-PEG-Mal (PEG Mw 2000) was added to the p(CBA-DAH) solution drop wise at a 1.5:1 molar ratio and reacted in the dark stirring overnight. The polymer was purified with dialysis (MWCO 3500). Eph peptide (NH2-GGGCHVLWSTRC) was added with stirring to Traut’s reagent in a 1:2 molar ratio for 1 h to thiolate the amine residue. Finally, the thiolated peptide Eph-SH was added to the distal end of the PEG chain previously conjugated to poly(CBA-DAH). Eph-PEG-poly(CBA-DAH) was dissolved in HEPES buffer solution (20 mM HEPES, pH 7.4, 5% glucose). Diluted Eph-PEG-poly(CBA-DAH) solution was slowly dropped into prepared DNA plasmid solution and incubated for 30 min to form complex. The complex formation of Eph-PEG-poly(CBA-DAH)/DNA was routinely monitored by 1.0% agarose gel electrophoresis.
Biodistribution of targeting polymer in organs
The biodistribution of delivered gene was characterized by luciferase assay using gWiz-Luc plasmid (Aldevron, Fargo, ND). At day 2 post injection, mice were sacrificed and major organs (liver, spleen, lungs, heart, kidneys and pancreas) were collected and homogenized. Luciferase protein expression was determined by luciferase assay. The luciferase protein was measured on a luminometer (Dynex Technologies, Inc) using a luciferase assay kit (Promega, Madison, WI).
The evaluation of autoimmune insulitis
Computer morphometry was performed as described previously [22, 23], on hematoxylin and eosin (H&E)-stained 4 micrometer thick section of formalin-fixed paraffin embedded pancreas tissue. In brief, the slides were scanned at 0.25 um/pixel resolution using an Aperio ScanScope XT system (Aperio Technologies, Vista, CA). The islet cell and peripheral lymphocytic infiltrate areas were manually segmented and quantified by positive pixel count algorithm using the Aperio ImageScope software (version 10; Aperio Technologies). Inflammatory infiltrate was quantified and expressed as a fraction of total tissue area occupied by islet cells and inflammatory cells. More than 30 islets from each pancreas were examined using a double-blind method for each animal group. To characterize the progression of insulitis, each islet was assigned a grade by using following insulitis grading system: grade 0, normal islets; grade 1, peripheral mononuclear cell infiltration (less than 25 % of the islets showing mononuclear infiltration; grade 2, 25–50 % of the islets showing mononuclear infiltration; grade 3, over 50 % of the islets showing mononuclear infiltration; grade 4; small retracted islets with few mononuclear cells.
Immunohistochemistry
The paraffin-embedded pancreas sections were prepared as described above. Pretreatment for epitope retrieval and immunostaining was carried out on an automated immunostainer (Ventana ES, Ventana Medical Systems, Tucson, AZ). The pretreatment and staining conditions were as follows: The enzyme-induced epitope retrieval was carried out by treating hydrated sections with Protease 2 (20 mg/mL, Ventana Medical Systems, Tucson, AZ) for 4 min. The sections were incubated with primary antibody for mouse CD8 monoclonal antibody (rat monoclonal IgG, Thermo Scientific, Rockford, IL) at 1:100 dilution for 30 min at room temperature and then incubated with Rabbit anti-rat immunoglobulin (1:100 dilution, Dako, Carpinteria, CA) for 30 min at room temperature. The colorimetric detection of microvessels is performed by using an IView DAB detection kit (Ventana Medical Systems), following the manufacturer’s recommendation. The tissue sections are counterstained with hematoxylin (Ventana Medical Systems) and dehydrated in graded ethanol concentrations (50 %, 70 %, 95 %, and 100 %).
RESULTS
Construction and characterization of Eph-PEG-p(CBA-DAH)
Our group has previously reported the construction and in-vitro characterization of an effective polymeric gene delivery system to deliver a plasmid coding for soluble RAE-1γ to the pancreatic islet microvasculature [21]. The Eph-PEG-poly(CBA-DAH) polymer shows selective uptake by pancreatic islet endothelial cells (MS1s) with enhanced cellular uptake efficiency. Additionally, expression and secretion of sRAE-1γ in the target cells were verified. Here, we evaluated targeting properties of Eph-PEG-poly(CBA-DAH) and therapeutic efficacy of delivered sRAE-1 gene by targeting polymer in NOD mice. The Eph-PEG-poly(CBA-DAH)/plasmid complex was prepared at a 10:1 weight ratio for systemic administration to NOD mice. The formation of complex was confirmed by gel retardation assay and dynamic light scattering (data not shown).
Biodistribution of Eph-PEG-p(CBA-DAH) in organs
Biodistribution of delivered DNA by the targeted polymer was confirmed by luciferase assay. As shown in Fig. 1, the uptake of gWiz-Luc DNA by the targeting polymer Eph-PEG-p(CBA-DAH) was no different between the major organs (liver, spleen, lungs, heart, kidneys) when compared with that of p(CBA-DAH). In contrast, the expression of luciferase protein in the pancreas of Eph-PEG-p(CBA-DAH) injected animals were significantly higher than the p(CBA-DAH) as backbone polymer (P<0.01). These results indicate that the ability of Eph-PEG-p(CBA-DAH) to deliver therapeutic genes specifically to the targeted tissue of the pancreatic islet microvasculature.
Figure 1.

Luciferase expression in major organs of NOD mice injected with g-Wiz Luc delivered with backbone polymer p(CBA-DAH) (CD, n=7), and targeting polymer Eph-PEG-p(CBA-DAH) (EPH, n=5) after 2 days. Polyplexes were formed at a polymer/pDNA weight ratio of 10. *p<0.05 vs. targeting polymer treated animals.
Decrease of diabetes incidence
The ability of the delivery of sRAE-1 plasmid to decrease diabetes incidence in NOD mice was studied. The change of body weight and survival rate was also monitored. To evaluate diabetes incidence, blood glucose levels were measured every week after treatment up to 25 weeks of age. The blood glucose levels of mice injected with the plasmid only and targeting polymer only were significantly increased compared with polyplex treated mice from 8 weeks after injection (P<0.01). The blood glucose levels of the polyplex treated mice showed a delay in the onset of diabetes (Fig 2A). The normalization of blood glucose level after 14 weeks is due to the death of diabetic animals (Fig. 3B). It should be noted that only 60–80% of NOD mice spontaneously develop diabetes [31–33]. In the current study, about 20% of the tested mice did not develop diabetes, which is also attributed to large deviation in certain data points (Fig. 2A). NOD mice treated with the polyplex also showed a delay in diabetes incidence compared to control and EPH groups (Fig. 2B).
Figure 2.

Blood glucose level (A) and cumulative incidence of diabetes (B) in NOD mice after treatment. Control (plasmid treated, n=8), EPH (targeting polymer treated, n=7) and RAE-1 (polyplex treated, n=10) animals were monitored weekly for blood glucose levels until 19 weeks after injection (25 weeks-old), respectively. *, p<0.05 vs targeting polymer treated animals.
Figure 3.

The body weight (A) and survival rate (B) in NOD mice after treatment. All animals were monitored with blood glucose levels. *, p<0.05 vs targeting polymer treated animals.
The body weight and survival rate from each group were also monitored. All mice displayed normal weight gain with 100% survival until 8 weeks. The body weight of mice who received injections of plasmid only and targeting polymer only was significantly decreased from 13 weeks (Fig 3A). The survival rate of these groups was similar to the body weight. Around 80% of animals in the plasmid only and targeting polymer only groups died by 15 weeks after treatments, compared to 100% survival of polyplex treated mice after 19 weeks (Fig 3B). These results indicate the specifically delivered RAE-1 plasmid by Eph-PEG-p(CBA-DAH) effectively protected beta-cells from autoimmune destruction and prevented type-1 diabetes.
Prevention of autoimmune insulitis
The insulitis level of each injection group was evaluated 9 weeks after the injection. The pancreas was hematoxylin-eosin stained and the insulitis level was evaluated using by the Computer morphometry (fig. 4). The degree of insulitis of the polyplex injection group (RAE-1) was improved compared with the targeting polymer only injection group. The degree of insulitis observed between the control and EPH group was not different.
Figure 4.
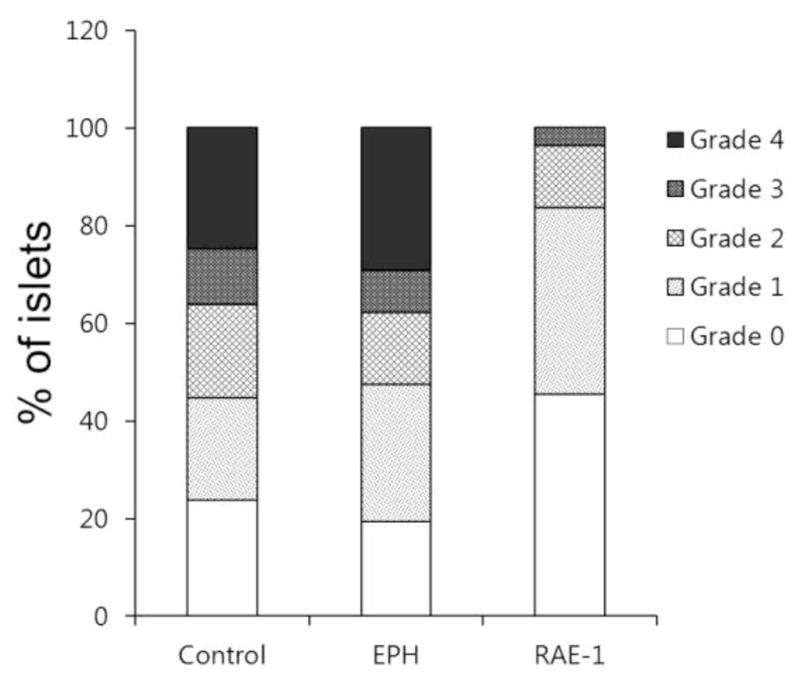
Insulitis in NOD mice. Formalin fixed pancreata were harvested from mice of Control (plasmid treated, n=3), EPH (targeting polymer treated only, n=4) and RAE-1 (polyplex treated, n=3) group at 9 weeks after treatment. More than 30 islets from each pancreas was examined using a double-blind method for each animal group. Scoring categories are as follows: 0, normal islets; 1, peripheral mononuclear cell infiltration (less than 25 % of the islets showing mononuclear infiltration); 2, 25–50 % of the islets showing mononuclear infiltration; 3, over 50 % of the islets showing mononuclear infiltration; 4, small retracted islets with few mononuclear cells.
It has been shown that the blockade of the NKG2D receptor with a monoclonal antibody prevents the onset of type-1 diabetes in the NOD mouse, indicating the blockade impairs the function of CD8+ T cells [13]. As can be seen in Fig. 5, normal mice (BALB/c) showed no observed mononuclear cell infiltration in normal (BALB/c) mice (A, E), whereas 18-week-old female NOD mice treated with plasmid alone (B, F) and targeting polymer alone (C, G) showed mild to severe mononuclear cell and CD8+ T-cell infiltration in and around the islets. In contrast, polyplex treated animals (D,H) showed limited peri-islet mononuclear cell and CD8+ T-cell infiltration in significantly fewer numbers of islets.
Figure 5.

Photomicrograph of representative H&E (top) and CD8+ (bottom) micrographs demonstrate infiltration of mononuclear cell changes in plasmid, targeting polymer and polyplex treated NOD mice compared with normal (BALB/c) mice. Normal mice (BALB/c) showed no observed mononuclear cell infiltration (A, E), whereas in plasmid (B, F) and targeting polymer treated (C, G) 18-week-old female non-obese diabetic mice showed mild to severe mononuclear cell and CD8+ T-cell infiltration in and around the islets. In contrast, polyplex treated animals (D, H) showed limited peri-islet mononuclear cell and CD8+ T-cell infiltration in significantly fewer numbers of islets. Scale bar indicates 100 um.
DISCUSSION
Evolution of efficient plasmid DNA strategies and delivery are key points for gene therapy. A large number of plasmid DNA treatments for diabetes have been investigated. The plasmid DNA encoding for a soluble ligand to NKG2D (sRAE-1γ) is being magnified as one of effectual candidate for type-1 diabetes. However, this therapeutic plasmid hasn’t taken center stage, even though the systemic blockage of the NKG2D receptor with monoclonal antibodies can prevent the occurrence of type-1diabetes in the NOD mouse [13]. This therapeutic plasmid needs to tissue specific delivery for better efficacy and safety. A large number of polymers which effectively and safely gene delivery for diabetes treatment have been investigated [24]. However, due to non-specific targeting in systemic injections make many of these polymers poor candidate for widespread use.
One of the important points in this study is that the polymer Eph-PEG-p(CBA-DAH) has the ability of in-vivo specific targeting to pancreas with therapeutic plasmid. In our previously studies, the problems related to pancreas specific delivery of plasmid were solved by conjugation of targeting peptide NH2-GGGCHVLWSTRC (Eph) to efficient and safe PEGylated gene carrier, PEG-p(CBA-DAH). This peptide is known as a targeting moiety to the receptors EphA2 and A4, which are overexpressed in the pancreatic islet microvasculature [25–27]. The targeting polymer Eph-PEG-p(CBA-DAH) has the verified ability to condense, unpackage and to aid endosomal escape of the therapeutic plasmid sRAE-1γ, soluble ligand of NKG2D receptor [24].
The targeting ability of Eph-PEG-p(CBA-DAH) with reporter gene to the pancreatic islet microvasculature was verified in various tissues of NOD mice after i.v. administration through tail vein. Typically, once a bioreducible polymer is opsonized and removed from the bloodstream, it is sequestered and concentrated heavily in the liver and spleen. To increase the stability of the gene carrier, the addition of PEGylation can push biodistribution toward the spleen [28–30]. Results from this study show the biodistribution profile of targeting polymer Eph-PEG-p(CBA-DAH) with a high degree of pancreas accumulation compared to the other major organs, as can be seen in Fig. 1. This result was observed only in animals injected twice with polyplex, not once.
Another important point in the study is that locally delivered soluble RAE-1 to the pancreas by Eph-PEG-p(CBA-DAH) will result in the binding and internalization of the NKG2D receptor of infiltrating lymphocytes and will halt the progression of type-1 diabetes. The blood glucose level of therapeutic plasmid and targeting carrier only injected NOD mice were showed continuously increasing incidence of diabetes similar to previous reports. Female NOD mice have an incidence of diabetes around 80% and begin the autoimmune response at roughly 3–5 weeks of age [31–33]. Polyplex treated mice showed a delay in the progression of autoimmune diabetes (Fig. 2).
Typically, NOD mice spontaneously increase weight irrespective of disease [34]. In this study, control animals treated with either plasmid alone or polymer alone showed a slight decrease in body weight from 11 weeks after injection, and the animals consequently died within a month (Fig. 3). It is thought that sudden body weight loss and death of the control animals were due to diabetic symptoms like as polyuria, polydipsia, hyperglycemia, glycosuria and hypercholesteremia [35].
The insulitis in the polyplex treated group were dramatically decreased compared to the control groups (Fig. 4). It is expected that each animal in the plasmid treated and targeting polymer treated control group will develop diabetes by 25 weeks, but the delivery of pCMV-RAE-1 to the islet microvasculature delays the onset of diabetes, reduces insulitis, and reduces infiltrating CD8+ T-cells as compared to control. Insulitis in NOD mice begins at the age of 3 to 5 weeks, with its extent gradually increasing until the age of 16 to 20 weeks [36]. Development of insulitis, characterized by accumulation of lymphocytes in and around the islets of Langerhans, is a prerequisite for autoimmune destruction of the beta cells. The types of mononuclear cells infiltrating the islets during insulitis can vary widely. CD8+ T cells, CD4+ T cells, B cells, NK cells, dendritic cells, and macrophages can all be found infiltrating pancreatic islets in the initial stages of disease [37–40]. Moreover, it is known that both initiation and progression of type-1 diabetes are dependent on CD8+ T-cells [3, 41]. The insulitis study in the animal model system suggested that pancreas specific targeted therapeutic plasmid blockade impairs the function of CD8+ T cells. As can be seen in Fig. 5, the CD8+ T-cell infiltration in and around the islets shows striking contrast between both the control group and the polyplex treated group.
With the development of a targeting polymeric gene carrier designed to target the pancreatic islet microvasculature and the development of a therapeutic plasmid designed to interfere with the immune-mediated attack of the islet beta cells, it is expected that this combined therapy can be used to prevent the incidence of type 1 diabetes in the NOD mouse.
CONCLUSION
In the present study, we investigated the delivery of a plasmid coding for soluble RAE-1γ to the pancreatic islet microvasculature using a targeted bioreducible polymeric carrier, Eph-PEG-p(CBA-DAH). This targeting polymer was able to specific deliver plasmid DNA to pancreas by systemic administration through tail vein. The delivered therapeutic RAE-1 plasmid by targeting polymer showed delay in the incidence of diabetes without a sharp decline of body weight and survival rate. Autoimmune insulitis was reduced by polyplex treatment compared with plasmid and targeting polymer injection. In histological results, plasmid and targeting polymer injected animals showed severe mononuclear cell infiltration on the islets, whereas polyplex treated animals showed limited peri-islet mononuclear cell infiltration in fewer numbers of islets. These results suggest that the specifically delivered therapeutic RAE-1 plasmid by Eph-PEG-p(CBA-DAH) effectively protected beta-cells from autoimmune destruction and prevented type-1 diabetes.
Acknowledgments
This work was financially supported by grants from the National Institutes of Health (NIH) (DK085075).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Atkinson MA, Maclaren NK. The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med. 1994;331(21):1428–1436. doi: 10.1056/NEJM199411243312107. [DOI] [PubMed] [Google Scholar]
- 2.Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7(6):727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- 3.Amrani A, Verdaguer J, Serra P, Tafuro S, Tan R, Santamaria P. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature. 2000;406(6797):739–742. doi: 10.1038/35021081. [DOI] [PubMed] [Google Scholar]
- 4.Nagata M, Santamaria P, Kawamura T, Utsugi T, Yoon JW. Evidence for the role of CD8+ cytotoxic T cells in the destruction of pancreatic beta-cells in nonobese diabetic mice. J Immunol. 1994;152(4):2042–2050. [PubMed] [Google Scholar]
- 5.Maasho K, Opoku-Anane J, Marusina AI, Coligan JE, Borrego F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J Immunol. 2005;174:4480–4484. doi: 10.4049/jimmunol.174.8.4480. [DOI] [PubMed] [Google Scholar]
- 6.Ogasawara K, Lanier LL. NKG2D in NK and T cell-mediated immunity. J Clin Immunol. 2005;25:534–540. doi: 10.1007/s10875-005-8786-4. [DOI] [PubMed] [Google Scholar]
- 7.Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. 2000;1(2):119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 8.Cerwenka A, Bakker AB, McClanahan T, Wagner J, Wu J, Phillips JH, Lanier LL. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12:721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 9.Diefenbach A, Tomasello E, Lucas M, Jamieson AM, Hsia JK, Vivier E, et al. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol. 2002;3(12):1142–1149. doi: 10.1038/ni858. [DOI] [PubMed] [Google Scholar]
- 10.Cerwenka A, Lanier LL. Ligands for natural killer cell receptors: redundancy or specificity. Immunol Rev. 2001;181:158–169. doi: 10.1034/j.1600-065x.2001.1810113.x. [DOI] [PubMed] [Google Scholar]
- 11.Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, Spies T. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics. 2001;53:279–287. doi: 10.1007/s002510100325. [DOI] [PubMed] [Google Scholar]
- 12.Ogasawara K, Hamerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, Pertel T, Carnaud C, Bluestone JA, Lanier LL. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 13.Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, Lanier LL. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20:757–767. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419(6908):734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 15.Mistry AR, O’Callaghan CA. Regulation of ligands for the activating receptor NKG2D. Immunology. 2007;121:439–447. doi: 10.1111/j.1365-2567.2007.02652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burgess SJ, Maasho K, Masilamani M, Narayanan S, Borrego F, Coligan JE. The NKG2D receptor: immunobiology and clinical implications. Immunol Res. 2008;40:18–34. doi: 10.1007/s12026-007-0060-9. [DOI] [PubMed] [Google Scholar]
- 17.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 18.Jeong JH, Kim SH, Christensen LV, Feijen J, Kim SW. Reducible poly(amido ethylenimine)-based gene delivery system for improved nucleus trafficking of plasmid DNA. Bioconjug Chem. 2010;21(2):296–301. doi: 10.1021/bc9003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SH, Ou M, Bull DA, Kim SW. Reductive degradation behavior of bioreducible poly(disulfide amine) for enhancing SiRNA efficiency. Macromol Biosci. 2010;10(8):898–905. doi: 10.1002/mabi.200900482. [DOI] [PubMed] [Google Scholar]
- 20.Yao VJ, Ozawa MG, Trepel M, Arap W, McDonald DM, Pasqualini R. Targeting pancreatic islets with phage display assisted by laser pressure catapult microdissection. Am J Pathol. 2005;166(2):625–636. doi: 10.1016/S0002-9440(10)62283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blevins KS, Jeong JH, Ou M, Brumbach JH, Kim SW. EphA2 targeting peptide tethered bioreducible poly(cystamine bisacrylamide-diamino hexane) for the delivery of therapeutic pCMV-RAE-1γ to pancreatic islets. J Control Release. 2012;158(1):115–122. doi: 10.1016/j.jconrel.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teman CJ, Wilson AR, Perkins SL, Hickman K, Prchal JT, Salama ME. Quantification of fibrosis and osteosclerosis in myeloproliferative neoplasms: a computer-assisted image study. Leuk Res. 2010;34:871–876. doi: 10.1016/j.leukres.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drakos SG, Kfoury AG, Hammond EH, Reid BB, Revelo MP, Rasmusson BY, Whitehead KJ, Salama ME, Selzman CH, Stehlik J, Clayson SE, Bristow MR, Renlund DG, Li DY. Impact of mechanical unloading on microvasculature and associated central remodeling features of the failing human heart. J Am Coll Cardiol. 2010;56:382–391. doi: 10.1016/j.jacc.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SW. Polymeric gene delivery for diabetic treatment. Diabetes Metab J. 2011;35(4):317–326. doi: 10.4093/dmj.2011.35.4.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mudali SV, Fu B, Lakkur SS, Luo M, Embuscado EE, Iacobuzio-Donahue CA. Patterns of EphA2 protein expression in primary and metastatic pancreatic carcinoma and correlation with genetic status. Clin Exp Metastasis. 2006;23(7–8):357–65. doi: 10.1007/s10585-006-9045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aspord C, Rome S, Thivolet C. Early events in islets and pancreatic lymph nodes in autoimmune diabetes. J Autoimmun. 2004;23:27–35. doi: 10.1016/j.jaut.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 27.Yao VJ, Ozawa MG, Trepel M, Arap W, McDonald DM, Pasqualini R. Targeting pancreatic islets with phage display assisted by laser pressure catapult microdissection. Am J Pathol. 2005;166:625–636. doi: 10.1016/S0002-9440(10)62283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brumbach JH, Lee YW, Kim SW, Yockman JW. Functional properties and biodistribution of poly(triethylenetetramine/cystamine bisacrylamide) and poly(triethylenetetramine/cystamine bisacrylamide)- poly(ethylene glycol) mixtures formed with nucleic acid. J Control Release. 2012;159(1):111–119. doi: 10.1016/j.jconrel.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishikawa M, Takakura Y, Hashida M. Pharmacokinetic evaluation of polymeric carriers. Adv Drug Deliv. 1996;21:135–155. Rev. [Google Scholar]
- 30.Owens DE, III, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Leiter EH, Prochazka M, Coleman DL. The non-obese diabetic (NOD) mouse. Am J Pathol. 1987;128(2):380–383. [PMC free article] [PubMed] [Google Scholar]
- 32.Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7(6):727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez C, Ménissier De Murcia J, Janiak P, Bidouard JP, Beauvais C, Karray S, Garchon HJ, Lévi-Strauss M. Unexpected sensitivity of nonobese diabetic mice with a disrupted poly(ADP-Ribose) polymerase-1 gene to streptozotocin-induced and spontaneous diabetes. Diabetes. 2002;51(5):1470–1476. doi: 10.2337/diabetes.51.5.1470. [DOI] [PubMed] [Google Scholar]
- 34.The Jackson Laboratory. Physiological Data Summary NOD/ShiLtJ (001976) 2007 ( http://jaxmice.jax.org/support/phenotyping/NODShiLtJdata001976.pdf)
- 35.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29(1):1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 36.Signore A, Procaccini E, Toscano AM, Ferretti E, Williams AJ, Beales PE, Cugini P, Pozzilli P. Histological study of pancreatic beta-cell loss in relation to the insulitis process in the non-obese diabetic mouse. Histochemistry. 1994;101(4):263–269. doi: 10.1007/BF00315913. [DOI] [PubMed] [Google Scholar]
- 37.Ludewig B, Odermatt B, Ochsenbein AF, Zinkernagel RM, Hengartner H. Role of dendritic cells in the induction and maintenance of autoimmune diseases. Immunol Rev. 1999;169:45–54. doi: 10.1111/j.1600-065x.1999.tb01305.x. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi K, Honeyman MC, Harrison LC. Impaired yield, phenotype, and function of monocyte-derived dendritic cells in humans at risk for insulin-dependent diabetes. Journal of Immunology. 1998;161(5):2629–2635. [PubMed] [Google Scholar]
- 39.Mallone R, Martinuzzi E, Blancou P, Novelli G, Afonso G, Dolz M, et al. CD8+ T-cell responses identify beta-cell autoimmunity in human type 1 diabetes. Diabetes. 2007;56(3):613–621. doi: 10.2337/db06-1419. [DOI] [PubMed] [Google Scholar]
- 40.Martinuzzi E, Lemonnier FA, Boitard C, Mallone R. Measurement of CD8 T cell responses in human type 1 diabetes. Annals of the New York Academy of Sciences. 2008;1150:61–67. doi: 10.1196/annals.1447.015. [DOI] [PubMed] [Google Scholar]
- 41.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clinical and Experimental Immunology. 2009;155(2):173–181. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]