

Sir,
Oxidative stress occurs when oxidative stress-related molecules, generated exceed intacellular antioxidant defences.[1] The liver has a critical role in the metabolism, digestion, detoxification, and elimination of substances from the body. Liver disease generally present clinically as hepatocellular, cholestatic (obstructive), or mixed.[2] The liver diseases produce reactive oxygen species that are involved in the transcription and activation of a large series of cytokines and growth factors which, in turn, can contribute to further production of Reactive Oxgen Species ROS and Reactive Nitrogen Species, RNS, continuing the vicious cycle.[3] The extent of cellular damage is manifested by either an increase in the oxidation products or a decrease in the antioxidant levels or both. Hence by measuring the oxidative stress parameters, the extent of liver damage can be assessed. Alcoholic and viral hepatitis, both these forms of hepatitis lead to hepatic damage of varying intensity. Although a lot of information is available regarding the biochemical changes and oxidative stress parameters in alcohol-induced hepatitis and hepatitis B virus infection separately, but a comparative study, taking both the forms of hepatitis into consideration, is lacking. This study aims to reveal differences in the extent of oxidative stress caused by chemical and biological agents causing hepatitis.
The study was conducted after obtaining usual permission from ethical committee and consent from subjects and controls were taken before commencing the study. The control group (Group 1) consisted of 100 normal, healthy individuals between the age group of 20–75 years from Kolkata. The second group consists of 100 clinically proven alcoholic hepatitis patients, aged between 20 and 75 years and the third group consisted of 100 clinically and serologically proven cases of hepatitis B patients admitted in hospital. Smokers, chronic drug users, pregnant, painters, and diabetes patients were excluded from the study. A total of 100 patients with clinically proven alcoholic hepatitis in the age group 20-75 years formed (Group 2). A total of 100 patients with clinically and serologically proven Hepatitis B infection (Group 3) were included in the study. About 5 ml of venous sample was taken from groups 1, 2 and 3 under aseptic precautions. Of the 5 ml of blood sample, 3 ml was collected in ethylenediaminetetraacetic acid (EDTA) containing vacutainer for analysis of oxidative stress parameters and 2 ml in plain vacutainer for analyzing liver enzymes. Blood sample from the EDTA containing vacutainer was centrifuged at 3000 rpm for 10 min and supernatant plasma was used for ascorbic acid estimation using 2,4-Dinitrophenyl hydrazine (DNPH). The buffy coat was discarded. The packed cells were suspended in equal volume of cold phosphate buffer saline and re-centrifuged. The supernatant was discarded. The washing of packed cells was repeated twice and the packed cells were used for analysis of glutathione (GSH) and malondialdehyde (MDA). The blood sample from plain vacutainers was centrifuged at 3000 rpm for 10 min; the serum was separated and was further used for analyzing liver enzymes. The oxidative stress parameters that were assayed included – erythrocyte GSH which was estimated by 5,5′-Di Thiobis 2-Nitrobenzoic Acid (DTNB) method.[4] MDA in erythrocyte was evaluated by measuring the thiobarbituric acid reacting substances (TBARs).[5] Plasma ascorbic acid was estimated by DNPH method.[6] Aspartate amino transferase (AST), alanine amino transferase (ALT), gamma glutamyl transferase (GGT), and alkaline phosphatase (ALP) were measured by using standard methods adapted in the clinical laboratory. The values of all parameters were statistically analyzed using Statistical Package for the Social Sciences developed by IBM Corporation, SPSS 15.0 software. A comparison of the mean values of oxidative stress parameters and liver enzymes were done separately between groups 1and 2, groups 1 and 3, and groups 2 and 3 using independent t test. Pearson correlation was done to compare the oxidative stress parameters with liver enzymes.
The mean values of GSH and MDA of groups 2 and 3 patients were found to be significant (P < 0.005) and different from that of group 1, but the difference was not significant. For ascorbic acid, the mean value of group 2 patients was found to be significant (P < 0.005) and different from groups 1 and 3, but the difference between Groups 1 and 3 was not significant. The values are depicted in Table 1. The differences of means of the liver enzymes between the groups (controls, group 2, and group 3) were statistically significant (P < 0.05). On comparison, it was found that the mean values of AST and ALP were not significantly different from those of group 3. The values are depicted in Table 2. Pearsons correlation showed that erythrocyte GSH and plasma ascorbic acid correlated negatively with all the liver enzymes and it was highly significant (P < 0.005) in both alcoholic hepatitis and hepatitis B cases as depicted in Table 3. Erythrocyte MDA also correlated positively with liver enzymes and was also significant (P < 0.005) in both the groups 2 and 3 as depicted in Table 3.
Table 1.
Mean values and S.D of Erythrocyte (GSH), (MDA) and Plasma Ascorbic Acid in groups 1, 2 and 3. Comparison of Oxidative stress parameters in group1 (Control), group 2 (Alcoholic Hepatitis) and group 3 (Hepatitis B)

Table 2.
Mean values and SD of AST, ALT, ALP and GGT in Groups 1, 2 and 3
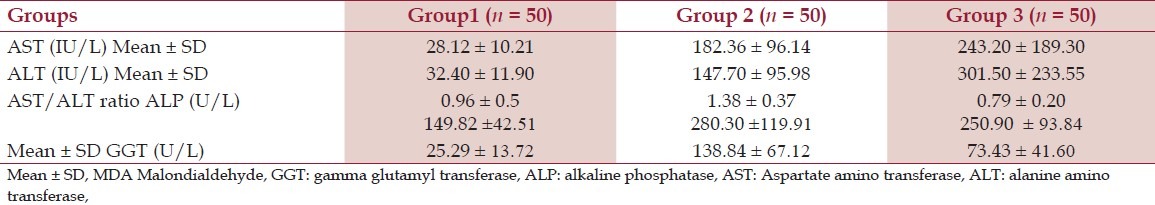
Table 3.
Correlation between the oxidative stress parameters and liver enzymes
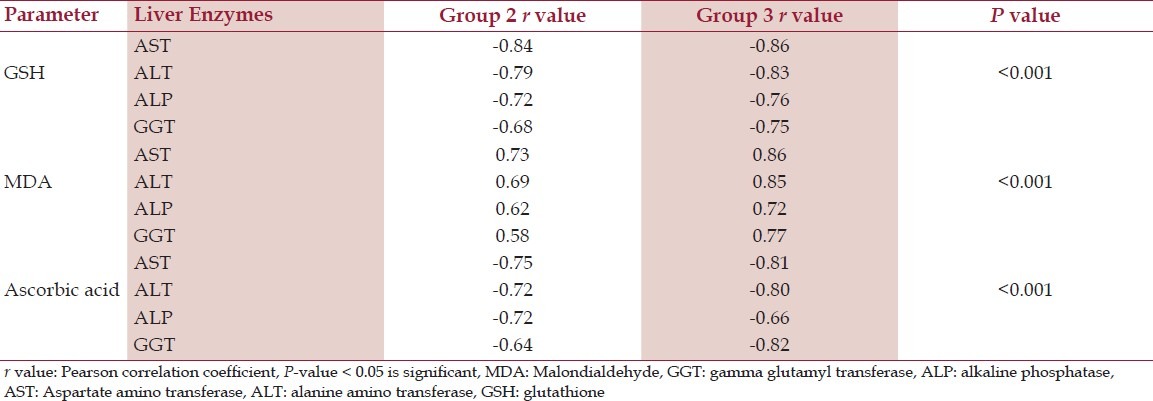
Increased levels of reactive oxygen species and toxic degradation products have been reported in patients with liver disease. The present study revealed that the patients suffering from liver disease either due to biological agent (hepatitis B virus infection) or chemical agent (excessive alcohol intake) showed significant depletion (P < 0.005) of GSH level when compared with controls, which is in agreement with other studies.[7] Several factors contribute to the fall in GSH level. Most important is oxidative stress, which consumes GSH. Depletion of GSH renders the cell more susceptible to oxidative stress.[8] Decreased GSH reductase activity may be predominant cause of GSH depletion within red blood cell (RBC) leading to serious consequences like increased lipid peroxidation and hemolysis. The other reasons for GSH depletion being, it acts as a co-factor for glutathione transferase (GST) during detoxification of xenobiotics including alcohol and also suppression of glutathione synthesis by ethanol.[9,10] However, when compared between groups 2 and 3, the mean values of GSH showed no significance (P > 0.05). The present study revealed a significant rise in the MDA levels (P < 0.005) in both groups 2 and 3 patients compared with controls. Raised MDA level reflects the oxidative injury to the liver. Free radical formation that subtracts hydrogen atoms from lipoproteins causing lipid peroxidation, which leads to elevated MDA levels. However, the levels of MDA did not vary significantly (P > 0.05) when compared in groups 2 and 3 cases. It has been found that the ascorbic acid levels were significantly lowered (P < 0.005) in alcoholic patients compared with controls, but the levels were insignificantly reduced (P > 0.05) in case of hepatitis B virus infection. These results corroborated well with previous studies.[7,10] However, a comparison of the ascorbic acid levels in groups 2 and 3, showed high significance (P < 0.005). The possible explanation for this is that apart from the free radicals mediated depletion of ascorbic acid in hepatitis; alcohol directly reduces ascorbic acid levels in blood by impairing its absorption, disabling transport into the blood, and altering vitamin C metabolism and utilization.[11–13] The measure of liver enzymes revealed a significant rise in the levels of AST (P < 0.005), ALT (P < 0.005), ALP (P < 0.005) and GGT (P < 0.005) in both hepatitis B virus infection and alcoholic hepatitis, compared with controls, which also confirms the literature evidence.[14–16] The AST/ALT ratio was found to be < 1 in controls as well in hepatitis B, but > 1 in alcoholic hepatitis patients. This is due to release of mitochondrial AST by alcohol itself or through its toxicity by its metabolites and/or oxidative stress.[17] Whereas in case of hepatitis B, ALT is typically higher than AST because of slower clearance.[3] Thus the present study revealed a certain degree of oxidative stress and free radicals mediated damage in both alcoholic hepatitis and hepatitis B virus infection. Previous study by Di Sario et al. revealed that liver fibrosis may be considered as a dynamic and integrated cellular response to chronic liver injury. The activation of hepatic stellate cells and the consequent deposition of large amounts of extracellular matrix play a major role in the fibrogenic process, but it has been shown that other cellular components of the liver are also involved. Although the pathogenesis of the liver damage usually depends on the underlying disease, oxidative damage of biologically relevant molecules might represent a common link between different forms of chronic liver injury and hepatic fibrosis. In fact, oxidative stress-related molecules may act as mediators able to modulate all the events involved in the progression of liver fibrosis. In addition, chronic liver diseases are often associated with decreased antioxidant defenses.[18] Earlier authors like Cederbaum and Mormone et al. postulated in their study that chronic HBV infection and long-term consumption of alcohol induce cell damage through increased generation of ROS. Indeed, oxidative stress, which favors mitochondrial permeability transition, is able to promote hepatocyte necrosis and/or apoptosis. In some clinically relevant conditions, generation of ROS within hepatocytes may represent an altered metabolic state as in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis, or significant ethanol metabolism as it occurs in alcoholic steatohepatitis. ROS are generated mainly via the mitochondrial electron transport chain via activation of cytochrome P450-mostly cytochrome P450 2E1-, NADPH oxidase, xanthine oxidase, or via mitochondrial damage. The ROS generated can directly affect the hepatic stellate cells and myofibroblasts behavior. ROS up-regulate the expression of critical fibrosis associated gene such as COL1A1, COL1A2, MCP1, and TIMP1 via activation of signal transduction pathways and transcription factors, including Jun Kinase JNK, activator protein-1, and NFκB. ROS generation in hepatic stellate cells and myofibroblasts occur in response to several unknown pro-fibrogenic mediators, including angiotensinII, Platelet derived growth factor, PDGF, TGFβ, and leptin. Overall a decrease in the antioxidant defense such as GSH, catalase or Superoxide dismutase, SOD, in conjunction with enhanced lipid peroxidation leads to a pro-fibrogenic response by enhancing collagen I protein expression.[19,20] Thus hepatic ethanol overload is followed both by an increase in reactive oxygen species and by a decline of antioxidants. Obviously the latter can further deteriorate once lipid peroxidation has been stimulated. Previous studies have shown that in hepatitis B virus infection, HBx, the X gene product of hepatitis B virus genome, interacts with an outer mitochondrial voltage-dependent anion channel (VDAC3) and this association leads to a decrease in the mitochondrial membrane potential and causes elevation of reactive oxygen species. In chronic hepatitis B and C there is a significant chronic liver injury with subsequent progression to advanced liver fibrosis and in many cases cirrhosis, a condition characterized by distortion of the normal architecture, septae and nodule formation, altered blood flow, portal hypertension, hepatocellular carcinoma, and ultimately liver failure. While hepatitis B virus can be integrated into the host genome leading to changes in genomic function or chromosomal instability, Hepatitis C cannot integrate into the host genome. Various hepatitis C virus (HCV) proteins, including the HCV core protein, the envelope, and nonstructural proteins present oncogenic properties. This study further revealed, in hereditary hemochromatosis, the excessive absorption and accumulation of iron in tissues and liver as ferritin is related to mutations in the HFE (High-iron gene).[21] When compared between oxidative stress parameters and the liver enzymes in both groups of patients, erythrocyte GSH and plasma ascorbic acid correlates negatively and plasma MDA correlates positively with all the liver enzymes in our study. The extent of damage caused due to oxidative stress was similar in both chemically and biologically induced hepatitis patients.
This study has several limitations. First, it is a cross-sectional study comprising a small group; therefore the directionality of associations cannot be clearly established. Second, better characterization and stratification of patients were not possible clinically. Third, this study was carried out purely on biochemical parameters and neither histological study nor parameters that could reveal inflammatory status of the patients were studied. Hence largescale clinical trials to establish a relation between oxidative stress parameters and liver diseases is worth undertaking. This study suggests that supplementation of antioxidants, along with other judicious medical intervention may improve the condition of the patients suffering from such diseases.
Acknowledgments
We would like to thank the following technical staff for their active participation in our study, Mr Arup Ratan Chowdhury and Mr. Subir Mondal. Special thanks to the management of Medical College, Kolkata for their active participation and patronage.
References
- 1.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 2.Krishnamurthy N, Ghosh C, Sumathi ME. Biological vs chemically induced hepatitis-a comparative study of oxidative stress parameters. Biomed Res. 2012;23:289–94. [Google Scholar]
- 3.Loguercio C, Federico A. Oxidative stress in viral and alcoholic hepatitis. Free Radic Biol Med. 2003;34:1–10. doi: 10.1016/s0891-5849(02)01167-x. [DOI] [PubMed] [Google Scholar]
- 4.Pastore A, Piemonte F, Locatelli M, Lo Russo A, Gaeta LM, Tozzi G, et al. Determination of blood total, reduced and oxidized glutathione in pediatric subjects. Clin Chem. 2001;47:1467–9. [PubMed] [Google Scholar]
- 5.Premanand R, Kumar S, Mohan A. Study of thiobarbituric reactive substances and total reduced glutathione as indices of oxidative stress in chronic smokers with and without chronic obstructive pulmonary disease. Indian J Chest Dis Allied Sci. 2007;49:9–12. [PubMed] [Google Scholar]
- 6.Burtis CA, Ashwood ER, Bruns DE. Teitz text book of Clinical Chemistry and Molecular Diagnostics. 4th ed. Vol. 30. Suanders (An imprint of Elseiver); 2006. pp. 1105–7. [Google Scholar]
- 7.Das SK, Vasudevan DM. Monitoring oxidative stress in patients with non-alcoholic and alcoholic liver diseases. Indian J Clin Biochem. 2005;20:24–8. doi: 10.1007/BF02867396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ortega AL, Mena S, Estrela JM. Glutathione in cancer cell death. Cancers. 2011;3:1285–310. doi: 10.3390/cancers3011285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Checa JC, Hirano T, Tsukamoto H, Kaplowitz N. Mitochondrial glutathione deflection in alcoholic liver disease. Alcohol. 1993;10:469–75. doi: 10.1016/0741-8329(93)90067-x. [DOI] [PubMed] [Google Scholar]
- 10.Afiong A, Etim H, Maisie H, Etukudo Ascorbic acid levels in hepatitis and non-hepatitis subjects in university of Calabar Teaching Hospital (UCTH), Calabar. Pak J Nutr. 2006;5:490–1. [Google Scholar]
- 11.Feinman L. Absorption and utilization of nutrients in alcoholism. Alcohol Health Res World. 1989;13:207–10. [Google Scholar]
- 12.Halsted CH. Nutrition and alcoholic liver disease. Semin Liver Dis. 2004;24:289–304. doi: 10.1055/s-2004-832941. [DOI] [PubMed] [Google Scholar]
- 13.Shenbagam M, Nalini N. Dose response effect of rutin a dietary antioxidant on alcohol-induced prooxidant and antioxidant imbalance–a histopathologic study. Fundam Clin Pharmacol. 2011;25:493–502. doi: 10.1111/j.1472-8206.2010.00861.x. [DOI] [PubMed] [Google Scholar]
- 14.Pratt DS, Kaplan MM. Evaluation of liver function. In: Longo, Fauci, Kasper, Hauser, Jameson, Loscalzo, editors. Harrison's principles of internal medicine. 18th ed. United States of America: Mc Graw Hill; 2011. pp. 2527–31. [Google Scholar]
- 15.Dienstag JL. Acute viral hepatitis. In: Longo, Fauci, Kasper, Hauser, Jameson, Loscalzo, editors. Harrison's principles of internal medicine. 18th ed. United States of America: Mc Graw Hill; 2011. pp. 2537–57. [Google Scholar]
- 16.Mailliard ME, Sorrell MF. Alcoholic liver disease. In: Longo, Fauci, Kasper, Hauser, Jameson, Loscalzo, editors. Harrison's principles of internal medicine. 18th ed. United States of America: Mc Graw Hill; 2011. pp. 2589–91. [Google Scholar]
- 17.Gupta S, Pandey R, Katyal R. Lipid peroxide levels and antioxidant status in alcoholic liver disease. Indian J Clin Biochem. 2005;20:67. doi: 10.1007/BF02893045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Sario A, Candelaresi C. Omenetti vitamin E in chronic liver diseases and liver fibrosis. Vitam Horm. 2007;76:551–73. doi: 10.1016/S0083-6729(07)76021-1. [DOI] [PubMed] [Google Scholar]
- 19.Cederbaum AI, Yang L, Wang X, Wu D. CYP2E1 sensitizes the liver to LPS- and TNF a-induced toxicity via elevated oxidative and nitrosative stress and activation of ASK-1 and JNK mitogen-activated kinases. Int J Hepatol. 2012;2012:582790. doi: 10.1155/2012/582790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mormone E, George J, Nieto N. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem Biol Interact. 2011;193:225–31. doi: 10.1016/j.cbi.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu L, Chen L, Li L, Sun HY, Yang G, Chang Y, et al. Hepatitis B virus X protein enhances cisplatin-induced hepatotoxicity via a mechanism involving degradation of Mcl-1. J Virol. 2011;85:3214–28. doi: 10.1128/JVI.01841-10. [DOI] [PMC free article] [PubMed] [Google Scholar]