

Abstract
PTH is a potent anabolic agent but the cellular mechanisms by which it increases bone mass are not fully understood. Dkk1 is an endogenous inhibitor of Wnt signaling and suppresses bone formation in vivo. We sought to determine if Dkk1 and anabolic PTH treatment interact in regulating bone mass. PTH treatment of primary murine osteoblasts for 24 hours reduced Dkk1 expression by 90% as quantified by real-time PCR, while PTH treatment in vivo reduced Dkk1 expression by 30% when give as a single daily subcutaneous dose. To directly determine whether Dkk1 modulates PTH’s anabolic response in vivo, we engineered transgenic (TG) mice expressing murine Dkk1 under the control of the 2.3-kb rat collagen alpha-1 promoter. TG mice had significantly reduced bone mass, which was accompanied by reduced histomorphometric parameters of bone formation (reduced OV/TV, ObS/OS and NOb/TAR). Treatment of TG and wild-type (WT) littermates with 95 ng/g body-weight of human (1–34) PTH daily for 34 days resulted in comparable increases in bone mass at all skeletal sites. Histomorphometric analyses indicated that PTH treatment increased numbers of both osteoblasts and osteoclasts in WT mice but only increased numbers of osteoblasts in TG mice. We conclude that overexpression of Dkk1 does not attenuate the anabolic response to PTH in vivo.
Keywords: PTH, Dkk1, osteoblasts, osteoclasts, Wnt
Introduction
The cell surface receptors, low-density lipoprotein receptor-related proteins 5/6 (LRP5 and LRP6) are key regulators of bone mass [1]. Available data indicate that members of the Wnt family bind both LRP5/6 and a seven-transmembrane spanning receptor, Frizzled, to form a functional ligand-receptor complex that then activates the canonical Wnt/β-catenin intracellular signaling pathway [1]. The signaling functions of LRP5 and LRP6 are tightly regulated by a large number of extracellular proteins including members of the Dickkopf (Dkk) family of proteins. Dkks are encoded by a four-member gene family in vertebrates, three of which, (Dkk1, 2, and 4), are reported to interact with LRP5 and/or LRP6 [2]. Gain-of-function mutations in LRP5 increase bone mass in part by conferring resistance to the inhibitory actions of Dkk1 [3]. Increased expression of Dkk1 by malignant plasma cells has been reported as the basis for reduced bone formation in multiple myeloma [4, 5]. A neutralizing antibody to Dkk1 prevents suppression of osteoblast function in a mouse model of myeloma [6]. Dkk1 TG mice have been reported to develop osteopenia [7]. Haploinsufficiency of the Dkk1 gene leads to an increase in bone mass in mice [8, 9]. Together, these findings suggest that Dkk1 suppresses bone formation in vivo.
PTH is a potent anabolic agent when administered as a single daily dose, but the cellular mechanisms by which it increases bone mass are incompletely understood. Kulkarni et al. reported that PTH treatment of osteoblast-like cells increased expression of LRP6 and Frizzled 1 and suppressed Dkk1 expression [10]. These effects were associated with an increase in cellular levels of β-catenin suggesting that the actions of PTH in bone could be mediated at least in part modulating the activity of the Wnt signaling pathway [10]. Consistent with that hypothesis, inhibition of protein kinase A suppressed PTH-induced activation of a Wnt-activated TCF/LEF reporter gene while forskolin mimicked the effects of PTH. Wan et al. have recently reported that PTH induces phosphorylation of LRP6, which results in recruitment of axin to LRP6 and stabilization of β-catenin [11]. Tobimatsu et al. have also reported that PTH increased levels of β-catenin in the mouse osteoblastic MC3T3-E1 cell line [12].
Although these findings suggest cross-talk between the PTH and the Wnt-β-catenin signaling cascades, there is less evidence for a direct interaction between these two pathways at the level of cell surface receptors in vivo. Thus, a 4-week course of intermittent PTH treatment increased skeletal mass to the same extent in LRP5−/− and LRP5+/+ mice, indicating that the anabolic effects of PTH do not require LRP5 signaling [13]. Consistent with this finding, Iwaniec et al. reported that treatment with PTH for 6 weeks resulted in similar increases in osteoblast surface and osteoclast surface in both LRP5−/− and LRP5+/+ mice, although curiously PTH did not augment cancellous bone volume in either genotype [14]. However, femur cortical thickness was 11% higher in PTH-treated mice in comparison with vehicle-treated mice, regardless of genotype [14]. These investigators concluded that LRP5 is not essential for the anabolic effects of PTH in bone [14]. Interestingly, Wan et al have reported that single doses of PTH administered daily for 28 days to mice, increased cellular levels of β-catenin and phosphorylated LRP5/6 in osteoblasts resident on trabecular bone [11, 15]. In contrast, continuous PTH treatment for the same period of time did not change levels of β-catenin or phosphorylated LRP5/6 [15]. Where and to what extent the PTH signaling cascades and those activated by Wnts intersect in bone cells is still being clarified.
To further explore role of Dkk1 in bone as well as its effects on the anabolic response to PTH treatment, we engineered TG mice in which Dkk1 was expressed under the control of the 2.3 kb rat collagen type I α promoter. We characterized the skeletal phenotype of these animals and their response to an anabolic PTH regimen.
Materials and Methods
Generation and identification of Dkk1 TG mice
The Dkk1 transgene was created using the same strategy previously reported for generating the transgene used to target expression of mCSF1 to osteoblasts [16]. The 1.1 kb cDNA for mouse Dkk1 was amplified by RT-PCR using mRNA isolated from primary murine osteoblasts [17]. The forward primer is: GAAGTTGAGGTTCCG CAGTC; and the reverse primer is: CAGGGGAGTTCCATCAAGAA. The Dkk1 cDNA was cloned 3′ of the 2.3 kb rat collagen type I α promoter. A 2.2-kb segment of the human growth hormone gene containing exons 1–5 and the intervening introns were added downstream of the cDNA to provide termination/polyadenylation signals and to increase expression efficiency.
The assembled transgene was microinjected into fertilized C57BL/6 X SJLF2 oocytes and the resultant TG mice identified by PCR amplification of a 171-bp sequence within exon 5 of the HGH portion of the transgene [18]. The integrity of genomic DNA was assessed by co-amplification of a 259-bp segment of the endogenous murine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene [18]. Two TG founders were identified. TG lines were generated by mating founder animals to CD-1 WT mice and the TG animals in subsequent generations were identified by PCR. All animals used for this study were 12–14 weeks old unless other indicated. The use of mice in this study was approved by the Yale Animal Care and Use Committee.
Measuring Dkk1 transgene expression
Transgene expression was assessed by quantitative RT-PCR (qRT-PCR) using the DNA Engine Opticon 2 System from M.J. Research (Waltham, MA) and the Brilliant® QRT-PCR Master Mix Kit from Stratagene (Lo Jolla, CA). To prepare total cellular RNA, bone and other tissues were rapidly dissected and snap frozen in liquid nitrogen, pulverized with a mortar and pestle in the presence of powdered dry ice to keep the tissue frozen, and then solubilized in Trizol reagent (GIBCO-BRL). The thermal cycling conditions comprised an initial 50°C for 30 min and a denaturation step at 95°C for 3 min, 40 cycles at 95°C for 30 s, 60°C for 1 min, and 72°C for 1 min. The forward primer for Dkk1 is: CCATCAAGCCAGCA ATTCTT; and the reverse primer for HGH is: GATTGGCCATACTGGGCTTA. This primer pair amplifies only the transgene. The transgene was detected in bone but not in lung, heart, skin, spleen, liver or kidney. The founder line expressing the highest level of Dkk1 was chosen for further study.
Dkk1 expression in bone was determined by qRT-PCR using the forward primer TCCCAGAAGAACCACACTGACTTC; and the reverse primer TCTTGGACCAGAAGT CTCTTGCAC. This primer pair amplifies both endogenous Dkk1 and the Dkk1 transgene and was therefore used to detect the total level of Dkk1 transcript expression in TG mice and WT littermates.
Bone density measurements
In vivo bone density measurements were performed by dual-energy x-ray absorptiometry (DXA) using a PIXImus densitometer (Lunar Corporation, Madison, WI). Anesthetized mice (ketamine, 30 mg/kg body wt and xylazine, 3 mg/kg body wt given IP) were placed in the prone position and scans performed with a 1.270-mm-diameter collimator, 0.762-mm line spacing, 0.380-mm point resolution, and an acquisition time of 5 minutes. The spine window is a rectangle spanning a length of the spine from T1 to the beginning of the sacrum. The femur window encompasses the entire right femur of each mouse. The coefficient of variation for total body BMD is approximately 1.5%.
Bone histomorphometry
Histomorphometry was performed as previously reported [19–21]. At the time of sacrifice the tibiae were removed, stripped of soft tissue, and fixed in 70% ethanol. Tibiae were then dehydrated through graded ethanol, cleared in toluene, infiltrated with increasing concentrations of methylmethacrylate, and embedded in methylmethacrylate according to previously described methods [19, 20]. Analyses were performed on 5 μM thick sections stained with toluidine blue, pH 3.7 using a Nikon microscope interfaced with the Osteomeasure system software and hardware (Osteometrics, Atlanta, GA). Measurements were obtained in an area of cancellous bone that measured approximately 2.5 mm2, containing only secondary spongiosa, and located 0.5–2.5 mm distal to the epiphyseal growth cartilage. Longitudinal sections (5 μm thick) taken in the frontal plane through the cancellous bone of the proximal tibia were prepared with a Leica RM2165 microtome, mounted on chrom-alum coated glass slides, and stained with toluidine blue, pH 3.7. All indices were defined according to the American Society of Bone and Mineral Research histomorphometry nomenclature [22].
Effects of PTH treatment on Dkk1 expression in vitro and in vivo
Primary murine osteoblasts were prepared from calvariae of 2 to 4-day-old mice by collagenase-dispase digestion as described previously [17]. Cells were grown in Alpha-Minimal Essential Medium (MEM) supplemented with 10% FBS, penicillin, streptomycin, L-glutamine, and 20 mM HEPES pH 7.4. At 75% confluence, cells were treated with 10−8 molar human (1–34) PTH. The cells were harvested at the indicated times after PTH treatment, RNA were isolated and used for qRT-PCR.
For anabolic PTH treatment in vivo, human (1–34) PTH was reconstituted in 10 mM acetic acid containing 2% heat-inactivated mouse serum and given at a dose 95 ng/g body weight daily for 34 days as we have previously reported [21]. Vehicle-treated animals received 10 mM acetic acid containing 2% heat-inactivated mouse serum. Animals were weighed every 7 days and the dose of PTH adjusted for changes in weight. On days 1 and 34 of treatment, total body and regional bone mineral density (BMD) were determined by DXA. The left tibiae were then harvested and processed for histomorphometric analyses.
Results
High level expression of Dkk1 in bone from TG mice
The level of total Dkk1 transcript expression (endogenous plus transgene) was examined by qRT-PCR as described in the Methods. The pattern and level of Dkk1 transcript expression in heart, liver, spleen, lung and kidney was not significantly different in the transgenic line reported in this manuscript when compared to WT littermates. In contrast, there was a 9-fold increase in Dkk1 transcript expression (combined transgene and endogenous Dkk1) in the bone of the transgenic line described in this report.
Over-expression of Dkk1 reduces BMD and suppresses bone formation
Dkk1 TG mice showed 6% (p<0.01); 6% (p<0.01); and 4% (p<0.05) reductions in femoral, spinal and total BMD respectively compared to their WT littermates (Fig 1). Histomorphometric analyses revealed a significant reduction in trabecular bone volume, consistent with the densitometric findings. As shown in Table 1, there was also suppression in measures of osteoblast function with reductions in osteoblast surface per bone surface and in the number of osteoblasts per total bone area in the TG animals compared to controls. In contrast, overexpression of Dkk1 did not affect osteoclast surface per bone surface or the number of osteoclasts per total bone area.
Figure 1.

Dkk1 TG mice have lower bone density compared to WT mice. 43 Dkk1 TG mice and 44 age-matched WT littermates had spinal, femur and total body BMD measured by DXA (PIXImus). *= p<0.05, **= p<0.01 compared to WT.
Table 1.
Bone histomorphometry in wild type and Dkk1 transgenic mice
| Wght (gms) | N | Sex (M/F) | BV/TV (%) | OV/TV (%) | ObS/BS (%) | ObS/OS | NOb/TAR (#) | OcS/BS (%) | TbSp | TbN | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Wt | 36±6 | 6 | 3/3 | 16.7±1.4 | 0.85±0.15 | 21.6±4.2 | 52.9±3.6 | 380±61 | 5.33±1.05 | 133±10 | 6.4±0.4 |
| Tg | 37±4 | 6 | 3/3 | 7.6±1.1 | 0.42±0.06 | 16.4±2.7 | 41.7±4.1 | 180±28 | 7.92±0.06 | 256±37 | 3.9±0.5 |
| pvalue | NS | < 0.001 | 0.02 | NS | 0.05 | 0.01 | 0.06 | 0.01 | 0.001 |
PTH suppress Dkk1 expression in osteoblasts in vitro and in vivo
Since normal bone remodeling is regulated by PTH, we wondered if there is an interaction between PTH and Dkk1 in vitro and in vivo. When murine primary osteoblasts were cultured in the presence of 10−8 molar PTH for 24 hours, there was a time-dependent significant decline in the level of Dkk1 expression as quantified by qRT-PCR. As shown in Fig. 2, there was a greater than 90% suppression of Dkk1 expression after 24 hours of PTH treatment. We next examined the effects of an in vivo anabolic PTH treatment regimen on Dkk1 expression in WT animals. After 34 days of single daily PTH administration, there was a moderate but significant, 33% reduction in Dkk1 transcript expression in bone from PTH-treated as compared to vehicle-treated WT animals (Fig. 3). In the TG animals, there was no change in the level of total Dkk1 with PTH treatment indicating that expression of the Dkk1 transgene (which largely dictates Dkk1 levels in the TG animals) was not affected by administration of PTH (Fig. 3).
Figure 2.
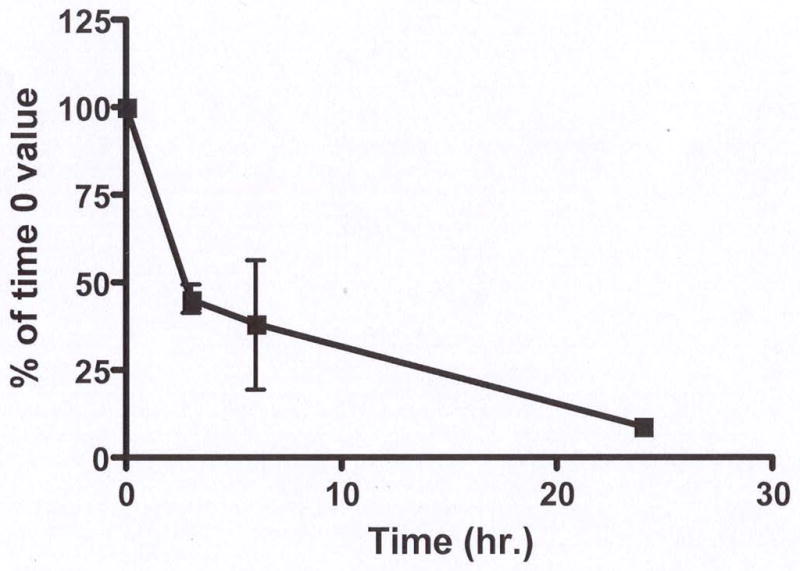
PTH suppresses Dkk1 mRNA expression in vitro in osteoblasts. Total RNA was isolated from murine primary osteoblasts treated with 10−8 M PTH for the indicted times. Levels of Dkk1 expression were quantified by qRT-PCR and expressed as a percent of the time 0 value. The results represent the Mean ± SEM of four separate experiments.
Figure 3.
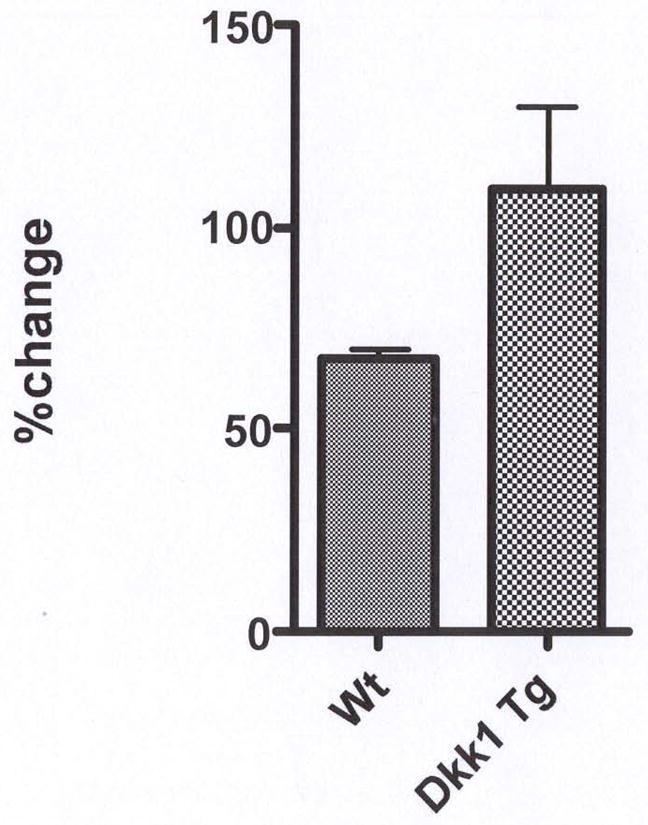
PTH suppresses Dkk1 mRNA expression in vivo in WT mice. Eight WT and eight TG mice, were treated with either single daily doses of human (1–34) PTH (95 ng/g) or vehicle for 34 days (4 animals/group), femurs harvested and RNA extracted as described previously (16). Levels of Dkk1 expression were quantified by qRT-PCR and expressed as a percent of the value in vehicle-treated animals.
The anabolic response to PTH is not impaired in Dkk1 TG mice
As shown in Fig. 4A, daily subcutaneous PTH administration led to a significant increase in spinal, femur and total body BMD in TG and WT mice. Interestingly, TG overexpression of Dkk1 did not impair the anabolic response to PTH. At the end of 34 days of PTH treatment, spinal BMD had increased by 12.9% over baseline in the WT animals and by an equivalent 14.9 % in the PTH-treated TG mice. Similarly, femur BMD increased by 10.9 % and total body BMD by 8.4 % over baseline in the WT animals and increased by 18.9 % and 11.9 % respectively in the TG animals. Further, the net increase in spinal BMD (mean PTH-treated value minus the mean vehicle-treated value) was 8.5 % in the WT animals and 9.7 % in the TG mice. The mean percent increases in femur and total body BMD were 4.1 % and 10.7 % in WT animals and 5.9 % vs. 6.2 % in TG. As shown in Fig 4B, although there is a lower trabecular bone mass in the transgenic animals at baseline, both TG and WT animals showed robust increases in bone mass with PTH treatment.
Figure 4.


Anabolic effect of PTH in WT and TG mice. A. Percent change from baseline in spine, femur and total body BMD determined by DXA (PIXImus) in 40 WT (20 PTH-treated and 20 vehicle-treated) and 39 TG (18 PTH-treated and 21 vehicle-treated) mice. The data are the mean values after 34 days of treatment with either 95 ng/g of human (1–34) PTH or vehicle. B 4X view of the distal tibiae of WT (upper two panels) and TG (lower two panels) mice treated with vehicle or PTH for 34 days. Arrowheads point to trabeculae, which are increased in number and/or thickness with PTH treatment. Arrows point to cortical bone which is increased in thickness with PTH treatment.
Mean body weights were comparable at the end of the treatment period in all four groups, WT-vehicle: 34.6 ± 1.6 g; WT-PTH: 32.0 ± 1.4 g; TG-vehicle: 35.8 ± 1.6 g; TG-PTH: 35.6 ± 1.6 g.
Consistent with the bone density results, histomorphometric analyses showed that PTH increased trabecular bone volume (BV/TV) in Dkk1 Tg mice and their WT littermates by comparable extents (38.6% vs. 35.1% respectively). ObS/BS and NOb/BPm increased significantly in response to PTH treatment in both groups indicating an ability of PTH to overcome or by-pass the suppression in bone formation induced by the transgene (Fig 5A). In contrast to the similarity in changes induced by PTH in histomorphometric parameters of bone formation in the two groups, there was a clear difference in parameters of bone resorption. While PTH induced increases in OcS/BS, Noc/TAR and the Noc/BP in WT mice, none of these parameters increased significantly in the TG animals (Fig 5B).
Figure 5.

Effect of daily PTH administration on cellular parameters of osteoblast and osteoclast activity in WT and TG mice. Histomorphometric analyses were performed in 16 PTH-treated and 17 vehicle-treated TG animals as well as 16 PTH-treated and 14 vehicle-treated WT mice. The data for WT mice are summarized in panel A and for TG mice in panel B. Results are expressed as Mean ± SEM
*= p<0.05
Discussion
We found that selective overexpression of Dkk1 in osteoblasts leads to low bone mass. This observation is consistent with the report of Li et al., who observed a low bone density in transgenic mice in which Dkk1 was expressed under the control of the rat collagen 1A1 promoter [7]. Our data indicate this effect appears to be due to suppression of bone formation with a trend toward increased resorptive activity. As shown in Table 1, ObS/BS and NOb/TAR were suppressed in TG animals as compared to WT controls, changes also observed by Li and colleagues [7]. In our mice the suppression in bone formation was accompanied by a trend towards an increase in resorptive activity as reflected by an increased OcS/BS. However, despite the baseline suppression in bone formation, the anabolic response to PTH was not attenuated in Dkk1 TG animals.
The relationship between the PTH and the Wnt signaling cascades has been an area of intense recent investigative interest and a number of observations have suggested an interaction between these two pathways [10–12]. While these data strongly suggest an interaction between the PTH and canonical Wnt-signaling pathways, the transmembrane Wnt receptor LRP5 does not seem to be required for PTH’s anabolic effect. Two groups, have reported that mice with genetic absence of LRP5 have a normal anabolic response to single daily subcutaneous administration of parathyroid hormone in vivo [13, 14]. Thus, the level at which the PTH and Wnt signaling cascades intersect remains somewhat uncertain.
Dkk1 is an important inhibitor of Wnt signaling and acts to interdict the interaction of LRP5/6 and Frizzled with Wnt ligand. Another transmembrane protein Kremin is thought to be necessary for Dkk1 to effect this inhibition although this model has recently been called into question [23]. The importance of Dkk1 as a regulator of bone mass in vivo has been unequivocally established by studies in mice with haploinsufficiency of the Dkk1 gene. These mice show a marked increase in bone mass [8, 9]. Underscoring the relevance of these findings in mice to humans, is the observation that Dkk1 appears to play a central role mediating the inhibitory effect of myeloma cells on bone formation [4]. To determine whether Dkk1 and PTH interact in any important way in vivo we initially explored the effects of PTH on Dkk1 expression in osteoblasts. Consistent with the findings of Onyia et al. [24], we showed that PTH profoundly suppressed Dkk1 expression in osteoblasts continuously exposed to the hormone (Fig 2). We also found that intermittent administration of PTH inhibited Dkk1 expression albeit by a more modest 33 % (Fig 3). These data indicate that one potentially important mechanism by which PTH might induce bone anabolism is by suppressing Dkk1 expression in vivo. We reasoned that if this was the case, transgenic overexpression of Dkk1 in bone should abrogate the anabolic effects of PTH since down regulation of Dkk1 would not occur. Our Dkk1 transgene achieved high-level expression in bone and was indeed unresponsive to PTH (Fig 3) allowing us to test this hypothesis. Our data demonstrate that there is no attenuation in the anabolic response to PTH in Dkk1 TG animals. In two independent experiments; both the increase from baseline and the net change in BMD (mean BMD value after PTH – mean BMD value after vehicle) at all three skeletal sites were comparable in the WT and TG animals (Fig 4). These data clearly indicate that suppression of canonical Wnt signaling at the level of the LRP5/6 receptors does not attenuate the anabolic response to PTH. This suggests that if there is a non-redundant interaction between the PTH and Wnt signaling cascades it must be occur at a post receptor site.
Kramer et al. have recently reported that transgenic overexpression of sclerostin attenuated the anabolic response to PTH [25]. Since sclerostin, like DKK1, is also an inhibitor of LRP5\6 this would appear to implicate a role for LRP5\6 in PTH-dependent bone anabolism. It may be that sclerostin is a more potent inhibitor. It is also possible that sclerostin could affect PTH’s anabolic effect via mechanisms other than inhibition of LRP5\6. In this context it is worth nothing that the anabolic response to PTH is not attenuated in LRP5 knock out mice despite the fact that these animals are markedly osteopenic [13]. Finally, there are methodological differences in the two studies. Our transgenic animals were 2 months old and we treated them with PTH for one month while Kramer at al studied 6-month old transgenic animals and treated them for 2 months. Further studies using carefully controlled protocols will be needed to reconcile these findings.
A recent report indicating that the site of LRP5 action in regulating bone may not be in osteoblasts but rather in the enterochromaffin cells of the duodenum, make our findings all the more interesting [26]. The work of Yadav et al would suggest that since the principal site of action of LRP5 is the enterochromaffin cell, the Dkk1 overexpressed by our TG animals is acting systemically to suppress Wnt signaling in those target cells. This would lead to an increase in serotonin secretion, which in turn suppresses osteoblast function. If this is the case, then anabolic PTH would be circumventing the suppressive effect of serotonin in osteoblasts in our Dkk1 TG animals. However, it is still possible that Dkk1 is acting directly to suppress osteoblasts in the local environment of bone. Our findings do not distinguish between these two possibilities.
The mechanism by which PTH achieves an anabolic response in WT animals has been extensively studied and our results are consistent with previous findings from our lab as well as those from a number of other groups. Namely, the anabolic response is characterized by an increase in both parameters of bone formation and resorption. It is presumed that the formative response precedes the increase in resorptive activity of osteoclasts and it is this “uncoupling” of bone turnover which leads to the initial increase in bone mass during treatment. It is speculated that the eventual increase in osteoclastic activity attenuates the anabolic response to PTH with continued treatment. Surprisingly, in the Dkk1 TG animals, anabolic PTH treatment did not result in an increase in osteoclastic activity. Baseline osteoclastic activity was higher in the TG animals. However, it seems unlikely that this somewhat higher baseline resorptive activity is the explanation why osteoclast activity did not increase further with PTH treatment. In contrast to the histomorphometric findings in osteoclasts, indices of bone formation, ObS/Bs and NOb/Bpm, did increase significantly with anabolic PTH treatment in the TG animals.
This change, in conjunction with the lack of an effect on osteoclasts, suggests that the combined effects were the cellular bases for the increase in bone mass in the Dkk1 TG animals. Indeed, one might have anticipated a more robust increase in bone mass given that there was no change in osteoclast activity. The reason this was not the case may be because the PTH-induced increment in bone formation was somewhat less in the Dkk1 TG animals although still statistically significant.
The fact that osteoclastic activity did not increase in the TG animals raises the interesting possibility that Dkk1 could be used therapeutically to augment the anabolic response to PTH albeit this seems somewhat counterintuitive. Nonetheless, as noted, the increase in resorptive activity that attends the anabolic response to PTH has been suggested as one of the factors limiting the therapeutic in response to this drug. If Dkk1 in some fashion suppresses the resorptive response to PTH it may be that sequential use of these two agents could result in a more sustained anabolic response to PTH.
Acknowledgments
This work was supported by grants from the National Institutes of Health DK45228, and DE12459 (to KLI) and in part by the Yale Core Center for Musculoskeletal Disorders (P30; AR46032).
References
- 1.Williams BO, Insogna KL. Where Wnts Went: The Exploding Field of Lrp5 and Lrp6 Signaling in Bone. J Bone Miner Res. 2009;24:171. doi: 10.1359/jbmr.081235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25:7469–7481. doi: 10.1038/sj.onc.1210054. [DOI] [PubMed] [Google Scholar]
- 3.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 4.Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD., Jr The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 5.Qiang Y-W, Barlogie B, Rudikoff S, Shaughnessy JD., Jr Dkk1-induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone. 2008;42:669–680. doi: 10.1016/j.bone.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Heath DJ, Chantry AD, Buckle CH, Coulton L, Shaughnessy JD, Evans HR, Snowden JA, Stover DR, Vanderkerken K, Croucher PI. Inhibiting Dickkopf-1 (Dkk1) Removes Suppression of Bone Formation and Prevents the Development of Osteolytic Bone Disease in Multiple Myeloma. J Bone Miner Res. 2009;24:425–436. doi: 10.1359/jbmr.081104. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, Grisanti M, Morony S, Adamu S, Geng Z, Qiu W, Kostenuik P, Lacey DL, Simonet WS, Bolon B, Qian X, Shalhoub V, Ominsky MS, Zhu Ke H, Li X, Richards WG. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone. 2006;39:754–766. doi: 10.1016/j.bone.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 8.Morvan F, Boulukos K, Clément-Lacroix P, Roman Roman S, Suc-Royer I, Vayssière Ba, Ammann P, Martin P, Pinho S, Pognonec P, Mollat P, Niehrs C, Baron R, Rawadi G. Deletion of a Single Allele of the Dkk1 Gene Leads to an Increase in Bone Formation and Bone Mass. J Bone Miner Res. 2006;21:934. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- 9.MacDonald BT, Joiner DM, Oyserman SM, Sharma P, Goldstein SA, He X, Hauschka PV. Bone mass is inversely proportional to Dkk1 levels in mice. Bone. 2007;41:331–339. doi: 10.1016/j.bone.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulkarni NH, Halladay DL, Miles RR, Gilbert LM, Frolik CA, Galvin RJ, Martin TJ, Gillespie MT, Onyia JE. Effects of parathyroid hormone on Wnt signaling pathway in bone. J Cell Biochem. 2005;95:1178–1190. doi: 10.1002/jcb.20506. [DOI] [PubMed] [Google Scholar]
- 11.Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes & development. 2008;22:2968–2979. doi: 10.1101/gad.1702708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tobimatsu T, Kaji H, Sowa H, Naito J, Canaff L, Hendy GN, Sugimoto T, Chihara K. Parathyroid hormone increases beta-catenin levels through Smad3 in mouse osteoblastic cells. Endocrinology. 2006;147:2583–2590. doi: 10.1210/en.2005-1627. [DOI] [PubMed] [Google Scholar]
- 13.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, Warman ML, Turner CH. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281:23698–23711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- 14.Iwaniec UT, Wronski TJ, Liu J, Rivera MF, Arzaga RR, Hansen G, Brommage R. PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. 2007;22:394–402. doi: 10.1359/jbmr.061118. [DOI] [PubMed] [Google Scholar]
- 15.Wan M, Yang C, Yuan H, Wu X, Lu C, Chang C, Cao X. Parathyroid hormone activates beta-catenin signaling through LRP5/6. J Bone Miner Res. 2007;22:S83. [Google Scholar]
- 16.Yao GQ, Wu JJ, Sun BH, Troiano N, Mitnick MA, Insogna K. The cell surface form of colony-stimulating factor-1 is biologically active in bone in vivo. Endocrinology. 2003;144:3677–3682. doi: 10.1210/en.2002-221071. [DOI] [PubMed] [Google Scholar]
- 17.Yao GQ, Sun BH, Weir EC, Insogna KL. A role for cell-surface CSF-1 in osteoblast-mediated osteoclastogenesis. Calcif Tissue Int. 2002;70:339–346. doi: 10.1007/s00223-001-1079-x. [DOI] [PubMed] [Google Scholar]
- 18.Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci U S A. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baron R, Vignery A, Neff L, Silvergate A, Santa Maria A. Bone Histomorphometry. In: Recker R, editor. Techniques and Interpretation. CRC Press; Boca Raton, FL: 1983. pp. 31–32. [Google Scholar]
- 20.Insogna KL, Stewart AF, Vignery AM, Weir EC, Namnum PA, Baron RE, Kirkwood JM, Deftos LM, Broadus AE. Biochemical and histomorphometric characterization of a rat model for humoral hypercalcemia of malignancy. Endocrinology. 1984;114:888–896. doi: 10.1210/endo-114-3-888. [DOI] [PubMed] [Google Scholar]
- 21.Knopp E, Troiano N, Bouxsein M, Sun BH, Lostritto K, Gundberg C, Dziura J, Insogna K. The effect of aging on the skeletal response to intermittent treatment with parathyroid hormone. Endocrinology. 2005;146:1983–1990. doi: 10.1210/en.2004-0770. [DOI] [PubMed] [Google Scholar]
- 22.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 23.Semenov MV, Zhang X, He X. DKK1 Antagonizes Wnt Signaling without Promotion of LRP6 Internalization and Degradation. J Biol Chem. 2008;283:21427–21432. doi: 10.1074/jbc.M800014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onyia JE, Helvering LM, Gelbert L, Wei T, Huang S, Chen P, Dow ER, Maran A, Zhang M, Lotinun S, Lin X, Halladay DL, Miles RR, Kulkarni NH, Ambrose EM, Ma YL, Frolik CA, Sato M, Bryant HU, Turner RT. Molecular profile of catabolic versus anabolic treatment regimens of parathyroid hormone (PTH) in rat bone: an analysis by DNA microarray. J Cell Biochem. 2005;95:403–418. doi: 10.1002/jcb.20438. [DOI] [PubMed] [Google Scholar]
- 25.Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid Hormone (PTH) Induced Bone Gain is Blunted in SOST Overexpressing and Deficient Mice. J Bone Miner Res. 2009 doi: 10.1359/jbmr.090730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, Mann JJ, Hen R, Ducy P, Karsenty G. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum.(see comment. Cell. 2008;135:825–837. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]