

Abstract
Corynebacterium glutamicum amylomaltase (CgAM) catalyzes the formation of large-ring cyclodextrins (LR-CDs) with a degree of polymerization of 19 and higher. The cloned CgAM gene was ligated into the pET-17b vector and used to transform Escherichia coli BL21(DE3). Site-directed mutagenesis of Tyr-172 in CgAM to alanine (Y172A) was performed to determine its role in the control of LR-CD production. Both the recombinant wild-type (WT) and Y172A enzymes were purified to apparent homogeneity and characterized. The Y172A enzyme exhibited lower disproportionation, cyclization, and hydrolysis activities than the WT. The kcat/Km of the disproportionation reaction of the Y172A enzyme was 2.8-fold lower than that of the WT enzyme. The LR-CD product profile from enzyme catalysis depended on the incubation time and the enzyme concentration. Interestingly, the Y172A enzyme showed a product pattern different from that of the WT CgAM at a long incubation time. The principal LR-CD products of the Y172A mutated enzyme were a cycloamylose mixture with a degree of polymerization of 28 or 29 (CD28 or CD29), while the principal LR-CD product of the WT enzyme was CD25 at 0.05 U of amylomaltase. These results suggest that Tyr-172 plays an important role in determining the LR-CD product profile of this novel CgAM.
INTRODUCTION
The 4-α-glucanotransferase (4αGTase) family is mainly involved in starch metabolism (38). Amylomaltase (AM; EC 2.4.1.25), a member of the 4αGTase family, catalyzes the hydrolysis of an α-1,4-glucosidic linkage and the transfer of a glucosyl group to another linear oligosaccharide that is often referred to as an intermolecular transglycosylation or disproportionation reaction. The enzyme also catalyzes intramolecular transglycosylation or cyclization to produce a cyclic glucan product (cycloamylose [CA] or large-ring cyclodextrin [LR-CD]) with a degree of polymerization (DP) of 16 and up (6, 38). LR-CDs are highly soluble in water and are assumed to form a single helical V amylose conformation and a toroidal shape with an anhydrophilic channel-like cavity (11). They can form inclusion complexes with inorganic (19) and organic (38) molecules and have several potential applications in pharmaceuticals, food science, and biotechnology (6, 41). In addition, LR-CDs have been reported to act as an artificial chaperone for protein refolding, and as such, they are added as an ingredient in commercial protein refolding kits (26).
The X-ray structures of several AMs have been reported (2, 3, 13, 31, 42). The AM from the thermophilic bacterium Thermus aquaticus (TaAM) was shown to have another substrate binding site called the second binding site, in addition to the enzyme active site. This second binding site was proposed to help form a CA by the hydrophobic interaction of Tyr-54 and Tyr-101 with the substrate (37). The mutation of these residues affected the hydrolysis activity of the TaAM enzyme, which played an important role in product formation (8, 9). A novel AM from the mesophilic bacterium Corynebacterium glutamicum ATCC 13032 (CgAM) has recently been reported, and interestingly, it has only a 28% amino acid sequence similarity to TaAM (36). In addition, the LR-CD production profile of CgAM was different from that of the well-characterized TaAM enzyme (40). By amino acid sequence alignment, the Tyr-172 residue of CgAM was proposed to correspond to the Tyr-54 residue in TaAM (36). The aim of the present study was to investigate the importance of Tyr-172 of CgAM in the LR-CD production profile.
MATERIALS AND METHODS
Bacterial strains, plasmids, and chemicals.
C. glutamicum ATCC 13032 was obtained from the Culture Collection Center of the Thailand Institute of Scientific and Technological Research (TISTR). The CgAM gene (2,121 bp) cloned in the pET19b vector (pET-CgAM) (36) was available in-house. Escherichia coli BL21(DE3) and the pET-17b expression vector were from Novagen (Germany). Restriction enzymes, DNA ligase, and DNA polymerase were the products of New England BioLabs Inc. HiTrap DEAE Fast Flow (FF) and phenyl FF chromatography columns were from GE Healthcare (United Kingdom). Maltotriose and the glucoamylase from a Rhizopus sp. were purchased from Sorachim (France). A cycloamylose mixture with a degree of polymerization of from 22 to 50 (CD22 to CD50) was from Ezaki Glico Co., Ltd. (Japan). Pea starch was kindly provided by Emsland-Stärke GmbH (Emlichheim, Germany), while soluble potato starch was a product of Scharlau (Spain). A Roti-Quant assay kit was obtained from Carl Roth GmbH (Germany). A commercial glucose oxidase test kit was from Human Gesellschaft für Biochemica und Diagnostica mbH (Germany). All other chemicals used were of analytical grade.
Construction of recombinant wild-type (WT) and Y172A CgAM.
pET-CgAM (a plasmid pET-19b containing the C. glutamicum amylomaltase gene) (36) was extracted from E. coli BL21(DE3) cells using a standard method (1). The CgAM gene was double digested with NdeI and XhoI and then resolved by agarose gel electrophoresis and eluted from the gels. This CgAM gene was ligated into the pET-17b vector, creating p17CgAM. The recombinant plasmid obtained was analyzed by 0.7% (wt/vol) agarose gel electrophoresis and compared to a 1-kb DNA ladder (New England BioLabs Inc.). Transformation of E. coli BL21(DE3) was then performed, and the transformants were selected by plating on LB plates containing 100 μg/ml ampicillin. The nucleotide sequences of the randomly selected positive clones were checked.
Mutated CgAM was constructed by PCR-mediated site-directed mutagenesis using the p17CgAM plasmid as the template and the mutagenic primers with a mutation at the desired position (primers Y172A_FWD [CTGCTGATAAGGCGCTTGATTCCC] and Y172A_REV [GGGAATCAAGCGCCTTATCAGCAG]) to create the Y172A substitution (the Y172A codon that was changed is underlined). PCR conditions were an initial denaturation at 95°C for 2 min, followed by 16 cycles of amplification, each at 95°C for 1 min, 60°C for 1 min, and 72°C for 12 min. The reaction mixture was then treated with DpnI. Transformation of the mutated Y172A into E. coli BL21(DE3) was then performed. Transformants with Y172A were selected on LB plates containing 100 μg/ml ampicillin, and the introduced mutation was confirmed by sequencing.
Expression and purification of amylomaltase.
E. coli cells harboring the WT or the Y172A CgAM gene were grown in LB medium containing 100 μg/ml ampicillin at 37°C for 14 to 18 h. The expression of both the wild-type and mutated enzymes was induced by 0.4 mM isopropylthio-β-d-galactoside (IPTG) when the A600 of the culture reached 0.4. After 2 h, cells were harvested and sonicated using a Bandelin Sonoplus ultrasonic homogenizer (Bandelin, Germany). Bacterial cell debris was removed by centrifugation at 12,000 × g and 4°C for 30 min. The supernatant obtained was used as the crude enzyme for further study.
The crude WT or Y172A CgAM was loaded onto a HiTrap DEAE FF column (GE Healthcare, United Kingdom), and unbound proteins were removed by washing with 50 mM phosphate buffer (pH 7.4) containing 0.01% (vol/vol) β-mercaptoethanol. Stepwise elution by the same buffer but supplemented with 0.2, 0.3, or 1 M NaCl at a flow rate of 1 ml/min was made. The AM activity-containing fractions were pooled and dialyzed against 50 mM phosphate buffer (pH 7.4). Then, ammonium sulfate was added to a final concentration of 1 M and the mixture was loaded onto a HiTrap phenyl FF column (GE Healthcare, United Kingdom). The column was washed with 50 mM phosphate buffer (pH 7.4) containing 1 M ammonium sulfate and 0.01% (vol/vol) β-mercaptoethanol. Stepwise elution was made with the same buffer with 1, 0.2, or 0 M ammonium sulfate. The AM activity-containing fractions were collected.
Assay of amylomaltase.
All assays were performed for both recombinant wild-type and mutated enzymes.
(i) Starch-degrading activity.
The starch-degrading activity of the enzyme was measured by the iodine method using 0.3% (wt/vol) soluble potato starch as the substrate. The incubation with enzyme in a total volume of 250 μl was performed at pH 6.0 and 30°C for 10 min. The reaction was stopped by adding 500 μl of 1 N HCl. Then, a 100-μl aliquot was withdrawn and mixed with 900 μl iodine solution (0.005% [wt/vol] I2 in 0.05% [wt/vol] KI), and the absorbance at 660 nm was monitored. One unit is defined as the amount of enzyme required to degrade 1 mg starch/ml in a 10-min reaction time under the described conditions (modified from those described in reference 10).
(ii) Starch transglycosylation activity.
Starch transglycosylation activity was measured by the iodine method using soluble potato starch and maltose as the substrates as previously described (36). One unit is defined as the amount of enzyme required to degrade 1 mg starch/ml per min under the described conditions (modified from those described in reference 30).
(iii) Disproportionation activity.
Disproportionation activity was measured by the glucose oxidase method (27). The 50-μl reaction mixture, containing 3% (wt/vol) maltotriose and enzyme in 50 mM phosphate buffer, pH 6.0, was incubated at 30°C for 10 min, and then 30 μl of 1 N HCl was added to stop the reaction. The glucose oxidase reagent was added to a final volume of 1.0 ml, the mixture was incubated at room temperature for 10 min, and the absorbance at 505 nm was measured. One unit is defined as the amount of enzyme required for the production of 1 μmol of glucose per min under the described conditions.
(iv) Hydrolytic activity.
Hydrolytic activity was measured by the bicinchoninic acid assay (34). The reaction mixture, containing 30 μl of 0.5 mg/ml mixture of the LR-CDs (CD22 to CD50), was incubated with the enzyme in 50 mM phosphate buffer, pH 6.0, at 30°C for 4 h, and then the reaction was stopped by adding 30 μl of 1 N HCl. Bicinchoninic acid reagent was added to a final volume of 1.0 ml, the mixture was incubated at 80°C for 25 min, and the reaction was inactivated on ice for 5 min prior to measuring the absorbance at 562 nm. One unit is defined as the amount of enzyme required for the production of 1 μmol of reduced glucose per min under the described conditions.
(v) Cyclization activity.
Cyclization activity was measured by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) analysis (21). The reaction mixture, containing 150 μl of 2% (wt/vol) pea starch, 50 μl of the enzyme, and 1.3 ml of 50 mM phosphate buffer, pH 6.0, was incubated at 30°C for 90 min and then boiled for 10 min to stop the reaction. Treatment by 8 U of glucoamylase was performed at 40°C for 30 min and then boiling for 10 min again to inactivate the enzyme. The reaction mixture was then cooled down and analyzed by HPAEC-PAD. One unit is defined as the amount of enzyme required for the production of 1 nC of CD34 per min under the described conditions.
(vi) Coupling activity.
Coupling activity is a reverse of cyclization activity. The reaction mixture contained a mixture of LR-CDs and cellobiose as the substrates. The amount of sugar released was determined by three different methods: 3,5-dinitrosalicylic acid assay (5), glucose oxidase method (27), and bicinchoninic acid assay (34). One unit is defined as the amount of enzyme required for the production of 1 μmol of reduced glucose per min under the described conditions.
Synthesis of LR-CDs.
A 1.0-ml reaction mixture containing 100 μl of 2% (wt/vol) pea starch and 0.15 U (starch-degrading activity) of recombinant amylomaltase in 50 mM phosphate buffer, pH 6.0, was incubated at 30°C for various times, and the reaction was stopped by boiling for 10 min. Then, the mixture was incubated with 8 U of glucoamylase at 40°C for 30 min and inactivated by boiling for 10 min. The reaction product was analyzed as described below.
Analysis of LR-CDs.
HPAEC-PAD was used to analyze the LR-CD products with a model ICS 3000 system (Dionex) using a Carbopac PA-100 column (4 by 250 mm; Dionex). A 25-μl sample was loaded onto the column and eluted with six continuous-step linear gradients of 200 mM sodium nitrate in 150 mM NaOH at a flow rate of 1 ml/min as previously described (21, 36). The size of the LR-CD products was compared with the sizes of the standard LR-CDs that had been confirmed by matrix-assisted laser desorption ionization–time of flight analysis.
Substrate specificity.
Substrate specificity for the disproportionation reaction was determined using malto-oligosaccharides (maltose [G2] to maltoheptaose [G7]) as the substrate. The reaction mixtures contained 50 mM substrate and 0.2 U of starch transglycosylation activity in 50 mM phosphate buffer, pH 6.0. Incubation was at 30°C for 10 min, and the reaction was stopped by boiling. Then, the amount of glucose was determined by the glucose oxidase method (27).
Analysis of kinetic parameters.
The kinetics of the disproportionation reaction were investigated. The purified recombinant amylomaltase in 50 mM phosphate buffer, pH 6.0, was incubated at 30°C with various concentrations of maltotriose (0 to 200 mM) for 5 min, and then the reaction was stopped by boiling for 10 min. The rate of product formation was analyzed by the glucose oxidase method (27). The kinetic parameters Km and Vmax were determined from a Lineweaver-Burk plot, and then the kcat and kcat/Km values were calculated.
Electrophoresis and protein determination.
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) (7.5% [wt/vol]) was carried out on a Bio-Rad Mini-Protein III gel apparatus (Bio-Rad Laboratories, Hercules, MA) using the Laemmli buffer system (25 mM Tris, 192 mM glycine, 0.1% [wt/vol] SDS, pH 8.3) (22). The molecular mass markers used were phosphorylase b (97.0 kDa), bovine serum albumin (66.0 kDa), ovalbumin (45.0 kDa), carbonic anhydrase (30.0 kDa), trypsin inhibitor (20.1 kDa), and α-lactalbumin (14.4 kDa) (GE Healthcare, United Kingdom). The protein concentrations were measured by the Roti-Quant assay (Carl Roth GmbH, Germany) using bovine serum albumin as the standard.
RESULTS
Cloning and expression of the WT and Y172A CgAM enzymes.
The CgAM gene was cloned into the pET-17b vector, and expression was induced using IPTG, as described in Materials and Methods. After induction, crude WT CgAM showed a specific activity of 1.18 mU/mg protein (Table 1). Y172A CgAM was generated by PCR-mediated site-directed mutagenesis, and the mutation was confirmed by nucleotide sequencing. Crude Y172A CgAM enzyme, expressed in E. coli BL21(DE3), had a specific activity of 0.62 mU/mg protein (Table 1).
Table 1.
Purification of WT and Y172A CgAM enzymes
| CgAM enzyme | Total protein (mg) | Total activitya (mU) | Sp acta (mU/mg) | Purification (fold) | Yield (%) |
|---|---|---|---|---|---|
| WT | |||||
| Crude extract | 305 | 360 | 1.18 | 1.0 | 100 |
| DEAE FF preparation | 13.7 | 105 | 7.66 | 6.5 | 29.1 |
| Phenyl FF preparation | 2.24 | 33.3 | 14.9 | 13 | 9.25 |
| Y172A | |||||
| Crude extract | 506 | 314 | 0.62 | 1.0 | 100 |
| DEAE FF preparation | 17.5 | 37.0 | 2.11 | 3.4 | 11.8 |
| Phenyl FF preparation | 0.99 | 13.2 | 13.3 | 21 | 4.20 |
Assayed by starch transglycosylation activity.
Purification and characterization of recombinant amylomaltase.
Both the WT and Y172A CgAM enzymes were purified by fast-performance liquid chromatography on a DEAE ion-exchange column and by phenyl hydrophobic column chromatography (Table 1). The final purified enzymes exhibited a single band on SDS-PAGE (Fig. 1) with an apparent molecular mass of 81 kDa. The optimum temperature, when screened using the starch transglycosylation assay, was 30°C for both enzymes. They are not thermostable, losing almost all activity after incubation at temperatures higher than 50°C for 10 min (35). Likewise the pH-activity profiles were the same for the WT and Y172A mutated enzymes, with the optimum pH being 6.0 from a tested pH range of 4.0 to 9.0. The enzymes are stable when incubated over a pH range of from 5.5 to 9.0 at 30°C for 1 h (35).
Fig 1.
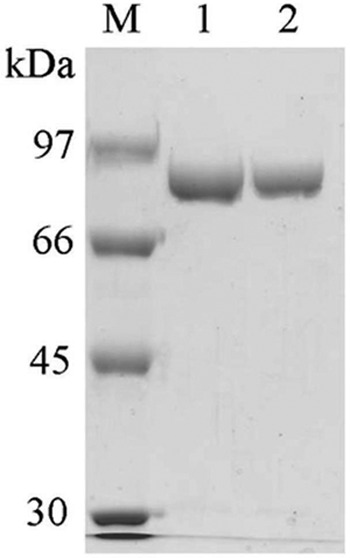
SDS-PAGE analysis of the purified recombinant WT and Y172A CgAM after phenyl FF column chromatography. Lane M, molecular mass marker; lane 1, 3 μg of WT CgAM; lane 2, 3 μg of Y172A CgAM.
All four activities of the WT and Y172A CgAM enzymes were measured and are summarized in Table 2. The Y172A mutated enzyme showed significantly decreased disproportionation (3.5-fold), hydrolysis (1.9-fold), and cyclization (1.8-fold) activities. For the coupling reaction, activity was not detected for either the WT or Y172A mutated enzyme by any of the several different assay methods tried in this study, perhaps due to trace amounts of AM coupling activity. Comparison of the glucose released from the hydrolytic activity of the two enzymes at various reaction times (Fig. 2) showed that the amount of glucose released by WT CgAM was significantly increased during the first 6 h and kept on increasing gradually until 24 h, while that released by Y172A CgAM showed no significant change throughout the period.
Table 2.
Activities of WT and Y172A CgAM enzymes
| Reaction | Sp acta (U/mg protein) |
|
|---|---|---|
| WT CgAM | Y172A CgAM | |
| Disproportionation | 21.8 ± 0.59 | 6.18 ± 0.05 |
| Hydrolysis | 8.05 ± 0.04 | 4.22 ± 0.02 |
| Cyclization | 0.502 ± 0.06 | 0.283 ± 0.03 |
| Coupling | NDb | ND |
Data are presented as the mean ± standard deviation and are derived from three independent repeats.
ND, not detectable.
Fig 2.
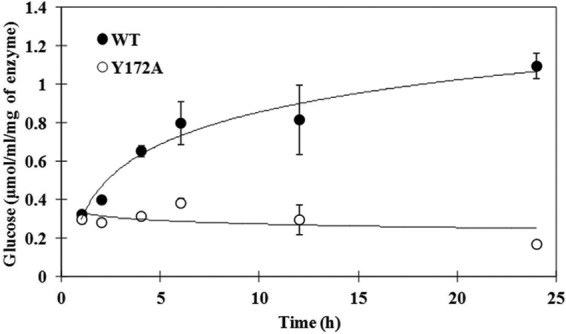
Time course of LR-CD hydrolysis reaction by WT and Y172A CgAMs at 30°C and pH 6.0 determined by glucose release. The reaction conditions are described in the “Assay of amylomaltase” section of Materials and Methods. Data are shown as the mean ± SD and are derived from 3 independent experiments.
Synthesis of LR-CDs.
To compare the LR-CD production profile of WT and Y172A CgAMs, both enzymes (at 0.05 U/ml) were incubated with pea starch substrate and the amounts of LR-CDs were measured by HPAEC-PAD (Fig. 3). At a short incubation time (1 h), the product profiles were relatively similar, with CD28 to CD30 having the highest proportion of a somewhat broad and symmetrical size distribution frequency curve (Fig. 3A and C). The two enzymes, however, differed in the amounts of products that they formed, with Y172A producing a noticeably smaller amount than the WT enzyme. In contrast, at a long incubation time (24 h), the amounts of products from both enzymes were similar, while the product size distribution profiles were different (Fig. 3B and D). The span of DPs of the products from WT CgAM at 24 h was broad, with a relatively high level of low-DP species (CD24 to CD28), while the principal products of the Y172A mutated enzyme had higher DPs (CD28 to CD29).
Fig 3.

Large-ring cyclodextrin products analyzed by HPAEC-PAD. Data for WT CgAM (A and B) and Y172A CgAM (C and D) after incubation at 1 h (A and C) and 24 h (B and D) with the indicated amounts of enzymes are shown as the mean ± SD and are derived from 3 independent experiments.
The enzyme concentration had a distinct effect on the LR-CD production profile for both enzymes, but the effect occurred in a differential manner (Fig. 3). At a short incubation time, the amounts of LR-CDs corresponded to the enzyme activity level for both enzymes, with CD28 to CD30 being the principal products (Fig. 3A and C). At 24 h of incubation, the amount of LR-CDs produced in the presence of the highest enzyme level (0.15 U/ml) by the WT enzyme was lower than that produced in the presence of lower enzyme levels (0.10 U and 0.05 U) (Fig. 3B). In addition, the principal LR-CD produced from a high enzyme activity level (0.15 U/ml) was smaller (CD23) than that produced from a lower enzyme activity level (CD24 and CD25 for 0.10 and 0.05 U/ml, respectively). For the Y172A CgAM enzyme, a different product pattern was observed, especially at a lower enzyme activity level, where larger CDs (CD28 and CD29) were obtained (Fig. 3D).
Substrate specificity.
The substrate specificities of the WT and Y172A CgAM enzymes in the disproportionation reactions of various malto-oligosaccharides (G2 to G7) were compared (Fig. 4). It was found that maltotriose (G3) is the best substrate for both enzymes. For recombinant wild-type enzyme, the descending order of preferred substrate was G3 > G4 > G5 > G6 > G7 ≈ G2, while the Y172A mutated enzyme showed a substrate order of G3 > G4 > G5 > G6 > G7 > G2. It was noticed that the wild-type enzyme could use maltose (G2) better than the Y172A mutated enzyme.
Fig 4.
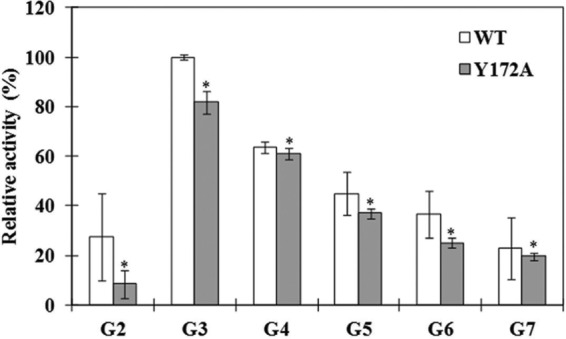
Substrate specificity of WT and Y172A CgAMs in disproportionation reaction using malto-oligosaccharide (maltose [G2] to maltoheptaose [G7]) as the substrate. Data are shown as the mean ± SD and are derived from 3 independent experiments. *, P < 0.05 (Student's t test) with respect to the disproportionation reaction of WT CgAM.
Kinetic parameters.
The kinetic parameters of the WT and Y172A CgAM enzymes were determined for the disproportionation reaction using maltotriose, the most efficient substrate. Under these conditions, the mutated enzyme showed lower Km (1.5-fold) and kcat (4.3-fold) values for maltotriose than the wild type (Table 3). In addition, a 2.8-fold lower catalytic efficiency of the disproportionation reaction was observed with the Y172A CgAM.
Table 3.
Kinetic parameters of WT and Y172A CgAM enzymes derived from the disproportionation reaction using maltotriose as the substratea
| CgAM enzyme | Km (mM) | kcat (min−1 [103]) | kcat/Km (mM−1min−1 [103]) |
|---|---|---|---|
| Wild type | 19.6 ± 3.2 | 9.37 ± 1.1 | 0.479 ± 0.1 |
| Y172A mutated | 12.9 ± 0.8 | 2.17 ± 0.2 | 0.168 ± 0.1 |
Data shown are the mean ± standard deviation and are derived from Lineweaver-Burk plots of the results of three independent sets of different enzyme-substrate concentrations with product analysis at different time points.
DISCUSSION
Investigation of LR-CD-producing enzyme is required due to known potential applications for large-ring cyclodextrins (6, 14). The gene encoding the amylomaltase of the mesophilic organism C. glutamicum has been previously characterized (12, 14). Recently, CgAM was successfully expressed in E. coli as the recombinant enzyme with His-tag residues at the N terminus and was purified to homogeneity (36). However, after the removal of the His-tag residues by enterokinase, the mature amylomaltase enzyme showed no activity. In this study, a new transformant (p17CgAM) was constructed using an expression vector without a His tag (pET-17b).
For structure-function studies, the Tyr-54 and Tyr-101 at the secondary binding site of TaAM were reported to be important residues for the formation of the LR-CD product due to hydrophobic interaction with the substrate (8, 37). Since CgAM is very different from the T. aquaticus enzyme in terms of molecular size and amino acid composition (36), it is reasonable to investigate CgAM at the same position found to be important for the activity of TaAM. Tyr-172 in CgAM, corresponding to Tyr-54 in TaAM, is the target for site-directed mutagenesis (36). Tyr-172 of CgAM was changed to alanine by PCR amplification.
The WT and Y172A CgAMs were purified to homogeneity. The optimum temperature for both enzymes is 30°C, which is close to values for 4αGTases from other mesophilic bacteria, such as E. coli IFO3806, Synechocystis sp. PCC 6803, and Pseudomonas stutzeri (18, 23, 33). Both enzymes exert stability over a wide range of pHs (pH 5.5 to 9.0), similar to TaAM (pH 4.0 to 10.0) (40). The temperature and pH activity profiles of WT and Y172A CgAM are not different from those of WT CgAM-His6 (36). These results suggest that the His tag and mutagenesis at Tyr-172 did not result in any notable changes to these characteristics. However, changes in activities were observed. Both the inter- and intramolecular transglycosylation reactions were affected by the Y172A substitution in the CgAM enzyme. Despite using the same catalytic mechanism, these reactions involve different donor and acceptor molecules (31, 37). The Y172A CgAM displays characteristics different from those reported for Y54A TaAM, where the disproportionation, hydrolysis, and coupling activities were decreased but the cyclization activity was increased (8, 9). For disproportionation and hydrolytic activities, TaAM with the replacement of Tyr-54 by Ala showed 4.4- and 2.6-fold lower activity, respectively, than the wild type. A similar trend was observed with Y172A CgAM, but activities in relation to those of its wild type were 3.5- and 1.7-fold lower, respectively. A different result in the cyclization reaction was clearly shown, whereby Y54A TaAM gave rise to 1.9-fold higher activity but the Y172A CgAM showed 1.8-fold lower activity than the corresponding wild type.
The WT and Y172A CgAMs display significant differences in hydrolytic activity at various time points using LR-CDs as the substrate. The WT enzyme shows an increase in activity during a 24-h reaction time, while no significant change in activity was observed with the mutated enzyme. Hence, the hydrolytic ability of the enzyme significantly decreases after mutation. The observation with TaAM is in sharp contrast to the observations made in this study, since the Y54F/K/P/W and Y101G mutated enzymes with lower hydrolytic activity also showed time-dependent increases in activity, though it was less than that of the wild type.
Since LR-CD production is a notable characteristic of amylomaltase, the effect of the Y172A substitution on the formation of LR-CDs was investigated. We found that the size and the amount of LR-CD products are influenced by the amount of enzyme and the incubation time. In addition, the product patterns of the WT and Y172A CgAMs at long incubation times are different, changing from a larger to a smaller ring size, as was similarly observed with CgAM-(His)6 (36), TaAM (40), and the potato D enzyme (39). We also observed that the rate of change in principal product formation from larger- to smaller-ring CDs is faster with the WT CgAM than the Y172A CgAM.
Substrate specificity studies suggest that maltotriose is the most preferable substrate for both WT and Y172A enzymes. Interestingly, the WT CgAM utilizes maltose better than the Y172A mutated enzyme. Similar substrate specificity has been reported for the amylomaltase from Thermus thermophilus HB8 (15). On the contrary, the descending order of preferred substrate is different from that of the Pyrobaculum aerophilum IM2 amylomaltase, which was G3 > G4 > G7 > G5 > G6 (16). When the kinetic parameters of the WT and Y172A CgAMs were analyzed, the lower Km of the Y172A enzyme suggests that the enzyme-substrate binding affinity is increased in the mutant, while the reaction rate kcat decreases. Indeed, the kcat/Km value of Y172A CgAM is about 2.8-fold lower than that of the WT enzyme. The Y172A substitution thus contributes to the difference in catalytic efficiency of the enzyme. In TaAM, Tyr-54 is located in the B2 subdomain between the second and third barrel strands of the enzyme (31). It has been suggested that this position might trigger the enzyme structure to the active conformation (9). In CGTase, an enzyme that resembles AM in the catalytic reaction but that differs in structure, the aromatic amino acid residue involved in the reaction specificity was identified to be outside the catalytic-site region (17). A second substrate binding site has also been reported in the α-amylase family, such as in bacteria (20, 25), archaea (24), and plants (32). The decrease in kcat and the modest decrease in the observed Km toward malto-oligosaccharides were observed in the Y380A barley R α-amylase I, whereby position 380 is the binding site outside the catalytic region (4, 28). It was proposed that oligosaccharide interaction at a site distant from the catalytic region of this enzyme is required to yield full activity toward amylose (4, 29). The formation of productive enzyme-substrate complexes with longer α-glucan substrates not only is mediated by the active-site cleft but also is enhanced by starch binding sites SBS1 and SBS2 in α-amylase (28). In addition, the secondary binding site might also contribute to allosteric activation (28). Thus, from the overall previous information and from our results on the Y172A mutation of CgAM presented in this study, we propose that Tyr-172 is a part of the secondary binding site of this amylomaltase and plays an important role in its catalytic activities. To gain more information on the structure and mechanism of CgAM, determination of the crystal structure of the enzyme is under way.
ACKNOWLEDGMENTS
W.S. was supported by a Royal Golden Jubilee Ph.D. fellowship from the Thailand Research Fund. She presently receives a postdoctoral fellowship under the Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University. Financial support from the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission (FW650B), and from a Research Institution Partnership Grant of the Alexander von Humboldt Foundation is acknowledged. We also acknowledge the support from the Thai Government Stimulus Package 2 (TKK 2555) under Project PERFECTA.
Special thanks go to Robert Butcher of the Publication Counseling Unit of the Faculty of Science for editing the manuscript.
Footnotes
Published ahead of print 3 August 2012
REFERENCES
- 1. Ausubel FM. 1995. Short protocols in molecular biology. John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 2. Barends TRM, et al. Structural influences on product specificity in amylomaltase from Aquifex aeolicus, in press [Google Scholar]
- 3. Barends TRM, et al. 2007. Three-way stabilization of the covalent intermediate in amylomaltase, an α-amylase-like transglycosylase. J. Biol. Chem. 282:17242–17249 [DOI] [PubMed] [Google Scholar]
- 4. Bozonnet S, et al. 2007. The ‘pair of sugar tongs’ site on the non-catalytic domain C of barley α-amylase participates in substrate binding and activity. FEBS J. 274:5055–5067 [DOI] [PubMed] [Google Scholar]
- 5. Chaplin MF, Kennedy JF. 1994. Carbohydrate analysis: a practical approach, 2nd ed Oxford University Press Inc., New York, NY [Google Scholar]
- 6. Endo T, Zheng M, Zimmermann W. 2002. Enzymatic synthesis and analysis of large-ring cyclodextrins. Aust. J. Chem. 55:39–48 [Google Scholar]
- 7. Reference deleted.
- 8. Fujii K, et al. 2005. Use of random and saturation mutagenesis to improve the properties of Thermus aquaticus amylomaltase for efficient production of cycloamyloses. Appl. Environ. Microbiol. 71:5823–5827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujii K, et al. 2007. Function of second glucan binding site including tyrosines 54 and 101 in Thermus aquaticus amylomaltase. J. Biosci. Bioeng. 103:167–173 [DOI] [PubMed] [Google Scholar]
- 10. Fuwa H. 1954. A new method for microdetermination of amylase activity by the use of amylose as the substrate. J. Biochem. 41:583–603 [Google Scholar]
- 11. Gessler K, et al. 1999. V-amylose at atomic resolution: X-ray structure of a cycloamylose with 26 glucose residues (cyclomaltohexaicosaose). Proc. Natl. Acad. Sci. U. S. A. 96:4246–4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ikeda M, Nakagawa S. 2003. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 62:99–109 [DOI] [PubMed] [Google Scholar]
- 13. Jung JH, et al. 2011. Structural and functional analysis of substrate recognition by the 250s loop in amylomaltase from Thermus brockianus. Proteins 79:633–644 [DOI] [PubMed] [Google Scholar]
- 14. Kalinowski J, et al. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5–25 [DOI] [PubMed] [Google Scholar]
- 15. Kaper T, et al. 2007. Identification of acceptor substrate binding subsites +2 and +3 in the amylomaltase from Thermus thermophilus HB8. Biochemistry 46:5261–5269 [DOI] [PubMed] [Google Scholar]
- 16. Kaper T, et al. 2005. Amylomaltase of Pyrobaculum aerophilum IM2 produces thermoreversible starch gels. Appl. Environ. Microbiol. 71:5098–5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kelly RM, et al. 2008. Elimination of competing hydrolysis and coupling side reactions of a cyclodextrin glucanotransferase by directed evolution. Biochem. J. 413:517–525 [DOI] [PubMed] [Google Scholar]
- 18. Kitahata S, Murakami H, Okada S. 1989. Purification and some properties of amylomaltase from Escherichia coli IFO 3806. Agric. Biol. Chem. 53:2653–2659 [Google Scholar]
- 19. Kitamura S, Nakatani K, Takaha T, Okada S. 1999. Complex formation of large-ring cyclodextrins with iodine in aqueous solution as revealed by isothermal titration calorimetry. Macromol. Rapid. Commun. 20:612–615 [Google Scholar]
- 20. Knegtel RMA, et al. 1995. Crystallographic studies of the interaction of cyclodextrin glycosyltransferase from Bacillus circulans strain-251 with natural substrates and products. J. Biol. Chem. 270:29256–29264 [DOI] [PubMed] [Google Scholar]
- 21. Koizumi K, Sanbe H, Kubota Y, Terada Y, Takaha T. 1999. Isolation and characterization of cyclic α-(1→4)-glucans having degrees of polymerization 9-31 and their quantitative analysis by high-performance anion-exchange chromatography with pulsed amperometric detection. J. Chromatogr. A 852:407–416 [DOI] [PubMed] [Google Scholar]
- 22. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 23. Lee BH, Oh DK, Yoo SH. 2009. Characterization of 4-α-glucanotransferase from Synechocystis sp. PCC 6803 and its application to various corn starches. N. Biotechnol. 26:29–36 [DOI] [PubMed] [Google Scholar]
- 24. Linden A, Mayans O, Meyer-Klaucke W, Antranikian G, Wilmanns M. 2003. Differential regulation of a hyperthermophilic R-amylase with a novel (Ca,Zn) two-metal center by zinc. J. Biol. Chem. 278:9875–9884 [DOI] [PubMed] [Google Scholar]
- 25. Lyhne-Iversen L, Hobley TJ, Kaasgaard SG, Harris P. 2006. Structure of Bacillus halmapalus R-amylase crystallized with and without the substrate analogue acarbose and maltose. Acta Crystallogr. F 62:849–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Machida S, et al. 2000. Cycloamylose as an efficient artificial chaperone for protein refolding. FEBS Lett. 486:131–135 [DOI] [PubMed] [Google Scholar]
- 27. Miwa I, Okuda J, Maeda K, Okuda G. 1972. Mutarotase effect on colorimetric determination of blood glucose with β-d-glucose oxidase. Clin. Chim. Acta 37:538–540 [DOI] [PubMed] [Google Scholar]
- 28. Nielsen MM, et al. 2009. Two secondary carbohydrate binding sites on the surface of barley R-amylase 1 have distinct functions and display synergy in hydrolysis of starch granules. Biochemistry 48:7686–7697 [DOI] [PubMed] [Google Scholar]
- 29. Oudjeriouat N, et al. 2003. On the mechanism of α-amylase: acarbose and cyclodextrin inhibition of barley amylase isozymes. Eur. J. Biochem. 270:3871–3879 [DOI] [PubMed] [Google Scholar]
- 30. Park JH, et al. 2007. The action mode of Thermus aquaticus YT-1 4-α-glucanotransferase and its chimeric enzymes introduced with starch-binding domain on amylose and amylopectin. Carbohydr. Polym. 67:164–173 [Google Scholar]
- 31. Przylas I, et al. 2000. X-ray structure of acarbose bound to amylomaltase from Thermus aquaticus: implications for the synthesis of large cyclic glucans. Eur. J. Biochem. 267:6903–6913 [DOI] [PubMed] [Google Scholar]
- 32. Robert X, et al. 2003. The structure of barley R-amylase isozyme 1 reveals a novel role of domain C in substrate recognition and binding: a pair of sugar tongs. Structure 11:973–984 [DOI] [PubMed] [Google Scholar]
- 33. Schmidt J, John M. 1979. Starch metabolism in Pseudomonas stutzeri. II. Purification and properties of a dextrin glucosyltransferase (D-enzyme) and amylomaltase. Biochim. Biophys. Acta 566:100–114 [DOI] [PubMed] [Google Scholar]
- 34. Sinner M, Puls J. 1978. Non-corrosive dye reagent for detection of reducing sugars in borate complex ion-exchange chromatography. J. Chromatogr. 156:197–204 [Google Scholar]
- 35. Srisimarat W. 2010. Characterization of a novel amylomaltase from Corynebacterium glutamicum ATCC 13032. Ph.D. thesis Chulalongkorn University, Bangkok, Thailand [Google Scholar]
- 36. Srisimarat W, et al. 2011. A novel amylomaltase from Corynebacterium glutamicum and analysis of the large-ring cyclodextrin products. J. Incl. Phenom. Macrocycl. Chem. 70:369–375 [Google Scholar]
- 37. Sträter N, et al. 2002. Structural basis of the synthesis of large cycloamyloses by amylomaltase. Biologia 57(Suppl 11):93–99 [Google Scholar]
- 38. Takaha T, Smith SM. 1999. The function of 4-α-glucanotransferase and their use for the production of cyclic glucans. Biotechnol. Genet. Eng. Rev. 16:257–280 [DOI] [PubMed] [Google Scholar]
- 39. Takaha T, Yanase M, Takata H, Okada S, Smith SM. 1996. Potato D-enzyme catalyzes the cyclization of amylose to produce cycloamylose, a novel cyclic glucan. J. Biol. Chem. 271:2902–2908 [DOI] [PubMed] [Google Scholar]
- 40. Terada Y, Fujii K, Takaha T, Okada S. 1999. Thermus aquaticus ATCC 33923 amylomaltase gene cloning and expression and enzyme characterization: production of cycloamylose. Appl. Environ. Microbiol. 65:910–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tomono K, et al. 2002. Interaction between cycloamylose and various drugs. J. Incl. Phenom. Macrocycl. Chem. 44:267–270 [Google Scholar]
- 42. Uitdehagg JCM, Enverink GJ, van der Veen BA, van der Maarel M, Dijkstra BW. Structure and mechanism of the amylomaltase from Thermus thermophilus HB8, in press [Google Scholar]