

Abstract
Toxic metal pollution affects the composition and metal tolerance of soil bacterial communities. However, there is virtually no knowledge concerning the responses of members of specific bacterial taxa (e.g., phyla or classes) to metal toxicity, and contradictory results have been obtained regarding the impact of metals on operational taxonomic unit (OTU) richness. We used tag-coded pyrosequencing of the 16S rRNA gene to elucidate the impacts of copper (Cu) on bacterial community composition and diversity within a well-described Cu gradient (20 to 3,537 μg g−1) stemming from industrial contamination with CuSO4 more than 85 years ago. DNA sequence information was linked to analysis of pollution-induced community tolerance (PICT) to Cu, as determined by the [3H]leucine incorporation technique, and to chemical characterization of the soil. PICT was significantly correlated to bioavailable Cu, as determined by the results seen with a Cu-specific bioluminescent biosensor strain, demonstrating a specific community response to Cu. The relative abundances of members of several phyla or candidate phyla, including the Proteobacteria, Bacteroidetes, Verrumicrobia, Chloroflexi, WS3, and Planctomycetes, decreased with increasing bioavailable Cu, while members of the dominant phylum, the Actinobacteria, showed no response and members of the Acidobacteria showed a marked increase in abundance. Interestingly, changes in the relative abundances of classes frequently deviated from the responses of the phyla to which they belong. Despite the apparent Cu impacts on Cu resistance and community structure, bioavailable Cu levels did not show any correlation to bacterial OTU richness (97% similarity level). Our report highlights several bacterial taxa responding to Cu and thereby provides new guidelines for future studies aiming to explore the bacterial domain for members of metal-responding taxa.
INTRODUCTION
Soil is an extremely complex environment where bacteria, which commonly adhere to surfaces of minerals and organic matter, have to cope with continuously changing chemical and physical conditions. As a consequence of the many niches and the spatial isolation of bacterial populations created by this complexity, soil probably harbors the highest species-level bacterial diversity on earth (10), with one gram of soil containing at least several thousand species (44) and perhaps up to millions of species (18).
Knowledge of the composition of the soil bacterial community has improved considerably during recent years, initially due to the study of 16S rRNA gene clone libraries (23), but the more recent emergence of pyrosequencing technology has greatly expanded our ability to investigate soil microbial communities (38). Several studies report that members of the phyla Acidobacteria, Actinobacteria, Proteobacteria, Bacteriodetes, and Firmicutes dominate soil bacterial communities but that their relative contributions differ (17, 32). Recent studies have identified pH as an important driver for changes in community structure at both the continental and local scales (11, 25, 33), and the availability of carbon sources also seems to shape bacterial communities, in particular, the balance between copiotrophs and oligotrophs (17). However, the environmental factors that determine the abundance and distribution of soil bacteria are still far from being fully understood.
Toxic metals represent soil chemical factors with a high potential to affect soil bacterial communities. Indeed, soil bacteria are generally considered highly sensitive to metal pollution (19), although some bacteria have developed a range of metal resistance mechanisms that may be carried on mobile genetic elements (30). Copper (Cu) is an essential metal which is toxic at high concentrations. It has frequently been used as a source of perturbation in studies addressing the resistance and resilience of bacterial communities with respect to chronic stress (7, 20), but Cu is also an important environmental pollutant. In soil, Cu pollution may arise from mining and smelting activities and from industrial activities such as wood impregnation (45) but also from current agricultural practices such as the use of Cu-containing pesticides (24) and amendments with manure containing Cu (4).
The use of rRNA gene-based fingerprinting techniques has revealed that Cu can influence the structure of bacterial communities in soil (7, 12, 26, 43). In those studies, however, Cu caused only small differences in community fingerprints. In contrast to those results, Gans and coworkers provided evidence for a more than 1,000-fold reduction in bacterial diversity even in moderately metal-polluted soil based on the recalculation of DNA reassociation data from a previous study (18, 35).
The objective of the current study was to explore the long-term effects of Cu on soil bacterial composition and diversity across a Cu pollution gradient. We used tag-coded pyrosequencing of 16S rRNA genes to obtain a detailed survey of the soil bacterial communities in 16 soil samples along the Cu gradient. Thereby we were able to test the hypothesis that Cu exposure leads to a reduction of bacterial diversity in soil and explore specific taxonomic groups for their potential to serve as Cu impact indicators. DNA sequence information was further related to analysis of the corresponding pollution-induced community tolerance (PICT) to Cu, as the PICT response is one of the most sensitive specific indicators of a community response to this pollutant (7). Finally, we determined whether total Cu, water-extractable Cu, or bioavailable Cu, as determined by a whole-cell bacterial biosensor, constituted the optimal descriptor of the observed Cu effects.
MATERIALS AND METHODS
Soil sampling.
In June 2010, 32 soil samples were collected (0 to 15 cm depth) at the well-described 60- by 120-m Cu-polluted field site in Hygum, Denmark. The contamination stems from the usage of CuSO4 at a wood impregnation plant that was active between 1911 and 1924. The area was cultivated until 1993 and has since been maintained as a fallow field in order to conserve the unique soil Cu gradient for research purposes (40).
Samples were taken every 4 m in a straight line along the transversal 0-m coordinate. An additional sample was taken at the highly polluted transversal 32-m and longitudinal 48-m coordinates. Reductions in plant cover, biomass, and species richness at the most contaminated part of the Hygum Cu gradient have been described previously (40). To minimize indirect effects of plant cover on microbial communities, our sampling protocol emphasized sampling of bulk soil rather than rhizosphere soil by removal of roots and adhering soil. The sampled soils were air dried for 24 h at room temperature to facilitate subsequent sieving (2-mm mesh size) and mixing in plastic bags before use.
Soil characterization.
Total Cu, water-extractable Cu, pH, organic C, and total N were determined for soil samples as described previously (5). In brief, water-extractable Cu was determined from 1-g soil samples that were extracted with 5 ml of demineralized water (Milli-Q; Millipore) for 2 h at 200 rpm on a rotary shaker. Samples were centrifuged at 10,000 × g for 10 min before analysis of Cu in the soil extracts (supernatants) by graphite furnace atomic absorption spectrometry. Further data on soil characteristics can be found elsewhere (3, 40). Bioavailable Cu was determined by the use of Cu-specific bioluminescent whole-cell biosensor Pseudomonas fluorescens strain DF57-Cu15 (42) as described previously (5). Hence, bioavailable Cu was operationally defined as the water-extractable copper species (see above for extraction protocol) able to induce reporter gene expression (bioluminescence). Levels of bioavailable Cu in soil extracts, and in dilutions thereof, were determined in duplicate with reference to external standard dilutions of CuSO4 (0.005 to 1.25 μM). On the basis of data for bioavailable Cu, 16 samples evenly distributed over the Cu gradient were chosen for further analysis.
Assay for detection of pollution-induced community tolerance.
PICT to Cu was determined by applying a [3H]leucine incorporation-microcentrifugation method to bacteria extracted from soil (1) with modifications as described in reference 8. The water-extractable bacteria are assumed to be representative of the total bacterial community with regard to Cu tolerance (3, 7, 13, 14). Importantly, bacterial community tolerancewas measured in pH-buffered solutions to avoid pH-dependent Cu speciation artifacts during PICT detection (K. K. Brandt, unpublished results) and to ensure that the investigated soil bacterial suspensions were not acidified following Cu addition.
In brief, soil samples were preincubated for 24 h at 22°C before bacteria were extracted with MES buffer (4-morpholineethanesulfonic acid, 20 mM, pH 5.9; soil/buffer ratio of 1:10) for 15 min on a rotary shaker (250 rpm, 22°C). Following centrifugation (1,000 × g, 10 min, 22°C), soil bacterial suspensions (supernatants) were exposed to CuSO4 at a final concentration of 0 or 10 μM for 30 min at 22°C before an incubation for 2 h with [3H]leucine (Amersham) (2.59 TBq mmol−1, 35 MBq ml−1) and nonlabeled l-leucine to give a final leucine concentration of 200 nM and radioactivity of 6 kBq per assay. Incubations were stopped by adding ice-cold 50% trichloroacetic acid (TCA). Finally, [3H]leucine incorporated into TCA-precipitated protein was separated from nonincorporated [3H]leucine through a series of washing and centrifugation steps (1) and quantified by scintillation counting. Soil bacterial suspensions amended with TCA prior to addition of leucine served as killed control samples. The tolerance index for each soil sample was calculated as the mean value of leucine incorporation in triplicate samples performed with 10 μM Cu divided by the mean value of leucine incorporation in the corresponding triplicate samples without Cu.
16S rRNA gene pyrosequencing of bacterial communities.
DNA from 0.50-g soil subsamples was extracted using a FastDNA Spin Kit for Soil (MP Biomedicals) according to the manufacturer's instructions. Pyrosequencing of the 16S rRNA gene was performed as described previously using amplified DNA from three pooled PCRs (27). The primers used for pyrosequencing were 341F (5′CCTACGGGRBGCASCAG-3′) and 806R (5′GGACTACNNGGGTATCTAAT-3′) flanking the V3 and V4 regions of the 16S rRNA gene and have previously been reported to target both bacteria and archaea but with a higher average matching efficiency for bacteria (50). Sequence analysis of the V4 (and V3) region of the 16S rRNA gene has previously been reported to provide an appropriate estimate of operational taxonomic unit (OTU) richness compared to several other regions of the16S rRNA gene (47, 48). The amplified fragments with adapters and tags were quantified using a Qubit fluorometer (Invitrogen) and mixed in approximately equal concentrations (4 × 106 copies μl−1) to ensure near-equal representations of all samples. A two-region 454 sequencing run was performed on a GS FLX Titanium PicoTiterPlate using a GS FLX pyrosequencing system according to the manufacturer's instructions (Roche).
Data analysis.
Obtained DNA sequences were quality checked, including elimination of chimeras, sorted, and trimmed using the Qiime pipeline (http://qiime.org/) at settings of length ≥ 250 bp and quality score ≥ 25 as sequence quality criteria and trimming the amplification primers and sequencing adapters. The RDP Classifier at the RDP's Pyrosequencing Pipeline Pipeline (http://rdp.cme.msu.edu/) was subsequently used to assign 16S rRNA gene sequences to phylogenetically consistent higher-order bacterial taxonomy with a confidence threshold of 80%. A random subset of 6,538 sequences from each tag was processed with the following RDP services: Pyrosequencing Aligner (29), Complete Linkage Clustering, and Rarefaction (97%). The number of operational taxonomic units (OTUs) in the subset formed the basis for calculation of total species richness as predicted by the Chao 1 index (9), the diversity as estimated by the Shannon diversity index (H′), and the Simpson's evenness index (E), by the RDP service Shannon Index and Chao1 estimator. Principal component analyses (PCA) of the normalized pyrosequencing data at a taxonomic level of class were processed with the “prcomp” function in R version 2.9.1 (R Development Core Team, 2009). Linear correlations were calculated using the “lm” function, while nonlinear correlations were analyzed with the nonparametric Spearman rank correlation test using “cor.test” with the option method = “spearman” in R version 2.9.1 (R Development Core Team, 2009).
Nucleotide sequence accession numbers.
The 16S rRNA gene sequences derived from pyrosequencing have been deposited at NCBI under accession number SRA051283.2 (Hygum Cu gradient). Partial 16S rRNA gene sequences of each of the 16 different soil samples have been deposited at NCBI under accession numbers SRX131849 to SRX131864.
RESULTS
Soil characterization.
Initially, 16 soil samples from the Hygum Cu gradient were analyzed for total Cu, water-extractable Cu (i.e., Cu in aqueous soil extracts), and bioavailable Cu (biosensor) in aqueous soil extracts (Table 1). This was done to determine the Cu exposure levels and, ultimately, to be able to identify which of these descriptors optimally reflects Cu impacts on bacterial communities. Total Cu ranged from 20 to 1,611 μg g−1 of soil in samples taken along the Hygum site transversal 0-m coordinate (samples 1 to 14 and sample 16), where sample 1 represents nonpolluted soil. The sample corresponding to the highly polluted transversal 32-m and longitudinal 48-m coordinates (sample 15) contained as much as 3,537 μg g−1. Extractable Cu ranged between 0.062 and 3.82 μg g−1 of soil, while bioavailable Cu ranged between 0.006 and 1.90 μg g−1 of soil. All Cu descriptors were strongly correlated to each other (see Fig. S1 in the supplemental material; linear correlation; all P ≤ 4.87 × 10−5), even though Cu bioavailability (expressed as the concentration of bioavailable Cu divided by the concentration of water-extractable Cu) increased from 9.7% in the control soil to 63.5% in the sample with the largest amount of bioavailable Cu.
Table 1.
Determined Cu exposure descriptors and chemical characteristics of 16 Hygum soil Cu gradient samplesa
| Sample | pH (H2O) | C, organic % | N, total % | Cu (μg g−1) |
||
|---|---|---|---|---|---|---|
| Total | Water extractable | Bioavailable | ||||
| 1 | 6.6 | 4.0 | 0.30 | 20 | 0.062 | 0.006 |
| 2 | 6.4 | 5.9 | 0.41 | 363 | 0.47 | 0.052 |
| 3 | 6.7 | 5.6 | 0.42 | 511 | 0.63 | 0.12 |
| 4 | 6.4 | 6.1 | 0.46 | 487 | 0.82 | 0.14 |
| 5 | 6.2 | 5.5 | 0.44 | 439 | 0.58 | 0.18 |
| 6 | 6.2 | 4.9 | 0.39 | 479 | 0.76 | 0.19 |
| 7 | 6.1 | 4.3 | 0.32 | 346 | 0.53 | 0.21 |
| 8 | 5.9 | 5.0 | 0.37 | 557 | 0.90 | 0.28 |
| 9 | 6.0 | 6.0 | 0.45 | 853 | 1.45 | 0.44 |
| 10 | 6.1 | 4.6 | 0.35 | 1,069 | 1.74 | 0.54 |
| 11 | 6.1 | 3.6 | 0.28 | 870 | 1.16 | 0.57 |
| 12 | 5.7 | 6.0 | 0.43 | 1,160 | 2.13 | 1.10 |
| 13 | 5.8 | 4.2 | 0.32 | 1,196 | 2.38 | 1.42 |
| 14 | 5.8 | 5.0 | 0.39 | 1,434 | 2.79 | 1.46 |
| 15 | 5.8 | 4.5 | 0.31 | 3,537 | 3.82 | 1.81 |
| 16 | 5.5 | 4.6 | 0.40 | 1,611 | 2.99 | 1.90 |
Soils were ranked (numbered) according their contents of bioavailable Cu as quantified by measurements with a Cu-specific whole-cell Pseudomonas fluorescens biosensor strain.
Table 1 also presents data for soil pH, organic C, and total N content of the Cu gradient soil samples. The soil pH ranged between 6.7 and 5.5 and was significantly correlated to the Cu content of the soil regardless of the Cu descriptor chosen (see Fig. S1 in the supplemental material; linear correlation; all P ≤ 0.0119). In contrast, neither the organic C nor the total N content of the soils was correlated to any of the Cu descriptors.
PICT to Cu.
The tolerance of the bacterial communities to Cu generally increased with increasing Cu exposure (Fig. 1). An analysis of the relation between the PICT response (Cu tolerance index) and total Cu, water-extractable Cu, and bioavailable Cu showed that PICT had strong linear correlations to the logarithms of the concentrations of bioavailable Cu (P = 3.23 × 10−8) and water-extractable Cu (P = 4.44 × 10−8), while the correlation to the logarithm of total Cu concentration was weaker (P = 1.16 × 10−5). The measurement of bioavailable Cu relies on the response of one specific bacterial biosensor strain. Interestingly, our data show that bioavailable Cu was a valid descriptor of Cu impacts on the soil bacterial community as reflected by PICT, and we consequently used this descriptor for comparisons of Cu exposure levels and changes in bacterial community compositions in the remaining part of the current study.
Fig 1.

Correlation plots showing Cu tolerance index for the bacterial community as measured by the leucine incorporation method (mean values; n = 3) versus total Cu (left panel), water-extractable Cu (middle panel), and bioavailable Cu (right panel) measured using the Pseudomonas fluorescens DF57-Cu15 biosensor. Data for Cu are mean values (n = 3). Curves represent best fits using a logarithmic model.
Sequence analysis and bacterial diversity.
We obtained in the range of 6,538 to 34,754 16S rRNA gene sequences from the 16 soil samples across the Cu gradient. The average length of sequences used for phylogenetic analysis was 428 nucleotides (data not shown). Between 68% and 85% of the sequences were able to be classified at the 80% level in the RDP Classifier. Due to the higher matching efficiency of the primers for bacteria, archaeal sequences accounted for less than 0.01% of the total sequences and the remaining analyses consequently focus on the bacterial community only. The numbers of different bacterial OTUs at the 97% similarity level ranged between ca. 3,000 and 4,500 OTUs per sample (calculated at 6,538 randomly selected sequences per sample) but did not show a significant linear correlation to bioavailable Cu (Fig. 2; P = 0.12). Neither the diversity indices H′ and E nor the Chao 1 estimate of OTU richness was correlated to bioavailable Cu (data not shown).
Fig 2.
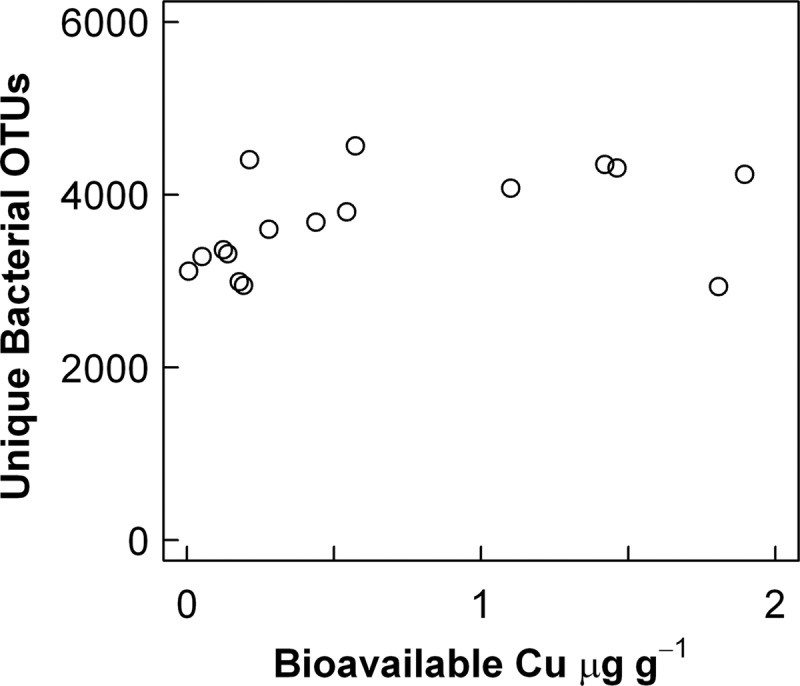
Bacterial diversity expressed as the number of unique operational taxonomic units (OTUs) defined at the ≥97% sequence similarity level for a sampling effort using 6,538 randomly selected 16S rRNA gene sequences versus bioavailable Cu in the 16 Hygum Cu gradient soil samples.
The rarefaction curves, showing the relation between the number of sequences and the number of unique OTUs at the 97% similarity level, did not reach an asymptote at either 6,538 sequences (see Fig. S2 in the supplemental material) or 34,754 sequences (data not shown) in accordance with the presence of highly diverse soil bacterial communities in all soils. High diversities were also reflected by the fact that the individual samples shared only in the range of 2.1% to 9.3% of the OTUs (see Fig. S3 in the supplemental material). Hence, there were no indications that the Cu contamination negatively affected the OTU richness of the bacterial communities.
Community composition.
Fig. 3 provides an overview of the bacterial community composition in the nonpolluted soil (sample 1). The Actinobacteria and Proteobacteria were the dominant phyla, representing about 40% and 25% of the bacterial sequences, respectively. The Acidobacteria accounted for ca. 12% of the sequences, while the Bacteriodetes, Firmicutes, Gemmatimonadetes, and Verrucomicrobia each represented 1% to 3% of the sequences. All other phyla accounted for less that 1% of the total bacterial sequences, and 21% of the sequences could not be assigned to known bacterial taxa.
Fig 3.

Relative abundances of dominant bacterial taxa in nonpolluted soil (sample 1) from the Hygum Cu gradient.
Cu contamination had a distinct impact on the bacterial community composition. A PCA of the sequence data at the taxonomic class level revealed that principal component 1 (PC1) and PC2 accounted for 34% and 11% of the variation in community composition, respectively. PC1 had a strong linear correlation (P < 0.001) to bioavailable Cu as shown in Fig. 4. Neither PC2 nor PC3 correlated with bioavailable Cu (data not shown).
Fig 4.

Bioavailable Cu as the explanatory variable plotted against the first principal component (PC1) accounting for 34% of the variation in a principal component analysis using 16S rRNA gene sequence data at the taxonomic level of class as the input for ordination. The linear relationship between bioavailable Cu and PC1 was statistically significant (***, P ≤ 0.001).
Figure 5 shows the relative abundances of the 10 most abundant phyla across the Cu gradient. Relative abundances of members of the most abundant phylum in the control soil, the Actinobacteria, and of the Firmicutes remained unaffected. Acidobacteria and the Gemmatimonadetes increased in relative abundance with increasing bioavailable Cu, while the relative abundances of all other phyla had negative correlations to bioavailable Cu (see Fig. 5 for significance levels).
Fig 5.

Relative abundances of the most abundant phyla and candidate phyla plotted against bioavailable Cu in the 16 Hygum Cu gradient soil samples. Correlation between the abundance of an individual taxa and bioavailable Cu was determined by the Spearman rank correlation test, and statistically significant correlations are indicated with NS (not significant), * (P ≤ 0.05), or ** (P ≤ 0.01).
Interestingly, changes in the relative abundances of classes frequently deviated from responses of the phyla to which they belong (Fig. 6). In members of the dominant phylum Actinobacteria, which overall did not respond to increasing bioavailable Cu, the two classes Acidimicrobidae and Rubrobacteridae were negatively correlated (Fig. 6A and B). Within the Proteobacteria, relative abundances of the Beta-, Gamma-, and Deltaproteobacteria were negatively correlated to bioavailable Cu (Fig. 6C, D, and E) and only the Alphaproteobacteria remained unaffected (see Fig. S4 in the supplemental material). The Acidobacteria were represented by several classes and subgroups, which responded differently to bioavailable Cu, as the relative abundances of subgroups 1, 2, and 3 were positively correlated whereas those of subgroups 6, 17, and 22 were negatively correlated to bioavailable Cu (Fig. 6F to K). The remaining subgroups 4, 7, and 16 remained unaffected (see Fig. S4 in the supplemental material). For the less abundant phyla Bacteroidetes, Verrucomicrobia, and Firmicutes, the responses of members of their respective subphyla or classes (Flavobacteria, Sphingobacteria—Verrucomicrobiae, subdivision 3—Bacilli, Clostridia) agreed with the responses at the phylum level (Fig. 6L to O; see also Fig. S4 in the supplemental material).
Fig 6.

Relative abundances of the most abundant Cu-affected classes plotted against bioavailable Cu in the 16 Hygum Cu gradient soil samples. A and B, phylum Actinobacteria. C to E, phylum Proteobacteria. F to K, phylum Acidobacteria. L and M, phylum Bacteriodetes. N and O, phylum Verrucomicrobia. Correlation between the abundance of an individual taxa and bioavailable Cu was determined by the Spearman rank correlation test, and statistically significant correlations are indicated with NS (not significant), * (P ≤ 0.05), ** (P ≤ 0.01), or *** (P ≤ 0.001).
Some bacterial groups, including the pseudomonads and the rhizobia, have been proposed as particularly sensitive indicators of metal or Cu pollution (6, 19). In the current study, the relative abundance of the genus Pseudomonas showed a significant, negative correlation to bioavailable Cu (Fig. 7) whereas the genus Rhizobium (data not shown) was too poorly represented to permit conclusions to be made. Further, the relative abundance of the genus Ilumatobacter within the class Actinobacteria showed a highly significant negative correlation with increasing Cu level (Fig. 7).
Fig 7.
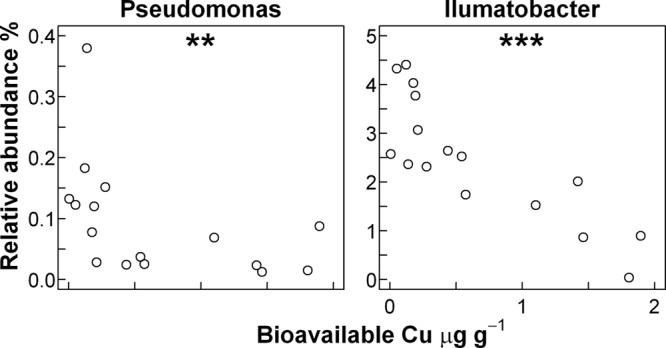
Relative abundances of selected genera plotted against bioavailable Cu in the 16 Hygum Cu gradient soil samples. Correlation between the abundance of an individual taxa and bioavailable Cu was determined by the Spearman rank correlation test, and statistically significant correlations are indicated with ** (P ≤ 0.01) or *** (P ≤ 0.001).
DISCUSSION
Cu effects on bacterial community tolerance.
The Hygum site, where soil texture has previously been reported to be highly uniform (37), was solely polluted with Cu due to the industrial use of CuSO4 as a wood preservative (3, 40). The site therefore provides a unique possibility to study the long-term effects of Cu contamination ranging from background Cu levels to severe Cu contamination without significant interference from other toxic elements or organic contaminants commonly used by the wood preservation industry (3, 45).
PICT development to Cu was evident even in the weakly contaminated part of the Cu gradient, with negligible differences in soil pH, indicating a specific response of the bacterial community to the elevated Cu levels. Our results are in line with previous studies showing that Cu has selected for tolerant populations of ammonia-oxidizing prokaryotes within the Hygum Cu gradient (16, 28) and with previous work showing that PICT development serves as a sensitive indicator for both short-term and long-term Cu impacts on soil microbial communities (7, 14).
We assessed multiple Cu exposure descriptors for their abilities to explain the overall Cu impact within the soil bacterial community as revealed by PICT. The strengths of the correlations between PICT and the logarithm of Cu concentration decreased in the order bioavailable Cu > water-extractable Cu ≫ total Cu. Hence, our results support previous studies showing that water-extractable Cu provides a better predictor of Cu impacts than total soil Cu does (34, 39). Importantly, our report demonstrates that bioavailable Cu, as determined using a P. fluorescens biosensor strain, is a robust predictor of PICT development by the entire bacterial community. This novel finding adds further credibility to the use of Cu-specific bacterial biosensors for environmental monitoring (5, 7). The levels of bioavailable Cu observed to induce a PICT response in the current study corresponded to the lowest observed effect concentrations for previous field experiments where PICT responses were observed at bioavailable Cu levels of 0.16 to 0.20 μg g−1 as determined using the P. fluorescens biosensor (see compilation of results in reference 21). However, for those previous experiments, the Cu exposure time was between 21 and 60 months whereas, in the current experiment, the bacterial community studied had been influenced by Cu for more than 85 years. Although Cu tolerance may develop within a few days following Cu amendment to soil (7, 13), complete community adaptation to toxic Cu levels may take years to develop (13, 16). Further, bioavailable Cu levels decrease with aging (7, 41). In case of the Hygum soils, we thus assume that the soil bacteria had previously been exposed to higher levels of bioavailable Cu than the current measured levels and that the bacterial communities had ample time to adapt to the prevailing Cu level.
Cu effects on diversity and composition of the soil bacterial community.
Our report marks the first attempt to study toxic metal impacts on soil bacterial communities based on high-throughput DNA sequencing of the 16S rRNA gene. Covering at least ca. 6,500 sequences per soil sample, it represents what was by far the deepest sequencing effort for studies of soil bacterial communities challenged by Cu toxicity. Previously, the most comprehensive study to date compared only 104 and 114 16S rRNA gene sequences from a Cu-impacted soil and a corresponding control soil, respectively (46). The bacterial communities present in the Hygum Cu gradient soils were dominated by members of phyla reported to be of major abundance in other soils worldwide, although the Bacteriodetes species were less abundant than seen in some other studies (49).
Our results did not demonstrate any significant correlation between bioavailable Cu and bacterial OTU richness; hence, our initial hypothesis that Cu reduces bacterial diversity in soil was not supported. A few previous studies employing rRNA gene-targeted community fingerprinting have reported specific Cu effects on bacterial diversity (22, 46). In the most comprehensive of those studies, Wakelin and coworkers used denaturing gradient gel electrophoresis (DGGE) to study effects of Cu concentrations comparable to those of the current study (46). Those authors reported an initial increase of bacterial diversity with increasing Cu followed by a steep decrease at total Cu levels above 560 μg g−1 of soil nearly 6 years after Cu amendment. However, those studies, and others before them, estimated community diversity based on rRNA gene-based community fingerprinting techniques that offer limited taxonomic resolution and suffer from serious drawbacks for quantification of species richness and evenness of complex bacterial communities (2). Further, the time aspect may be important and we speculate that Hygum soil bacterial diversity was initially reduced by Cu during the period in which Cu contamination occurred (1911 to 1924) and that Cu-adapted bacterial communities may have recovered their full diversity during the subsequent 85 years.
Although we could not detect a significant impact of Cu on OTU richness, our report demonstrates marked impacts of Cu on bacterial community composition. Members of a number of relatively high-abundance classes of the Actinobacteria and Proteobacteria were negatively influenced by increasing Cu levels, as were those of the less-abundant phyla and candidate phyla Bacteroidetes, Verrucomicrobia, Chloroflexi, WS3, and Planctomycetes. Several previous studies relying on cultivation-independent community fingerprinting methods such as automated ribosomal intergenic spacer analysis (ARISA), terminal restriction fragment length polymorphism (T-RFLP) analysis, and denaturing gradient gel electrophoresis (DGGE) have reported changes in community composition caused by short-term or long-term Cu pollution (7, 11, 12, 26, 43). However, only one previous study reported significant impacts of Cu on the abundance of specific taxonomic groups (46). At the taxonomic level of phyla, Wakelin and coworkers found a quite dramatic decrease of populations of Acidobacteria together with an increase in Firmicutes 6 years after a single Cu application. Although the total Cu concentration in their study was comparable to the highest Cu concentration within the soil Cu gradient of our study, we did not find comparable changes.
Only a few studies have addressed the changes in ecosystem functions caused by increasing Cu levels in the Hygum gradient. Hence, Sauvé (36) found inhibition of soil organic matter decomposition, while Mertens et al. (28) reported decreasing potential nitrification rates but unaltered functional resistance and resilience with respect to a range of environmental stressors. Hence, it is difficult to link changes in bacterial community composition to changes in ecosystem processes at the current time, but more research on this topic is obviously required. Among the Gammaproteobacteria, species of the genus Pseudomonas perform several beneficial functions in soil, including the suppression of plant pathogens as outlined in reference 6. Consistent with previous studies employing cultivation-dependent techniques (6, 15), we observed a reduced relative abundance of Pseudomonas rRNA gene sequences with increasing Cu level; therefore, this ecosystem service might be at risk in Cu-contaminated soils. The current report also highlights the more abundant actinobacterial genus Ilumatobacter, which might serve as an even more sensitive indicator of Cu impacts, but the significance of this genus for community function is unknown.
The development of PICT to Cu indicates that differential levels of Cu exposure have caused direct selection of bacterial communities across the Hygum gradient. However, it is possible that some of the observed Cu impacts on community composition have occurred due to indirect Cu effects. Cu salt amendment reduces soil pH, which is known to be a strong driver of changes in soil bacterial community composition (11, 25, 33). However, soil pH ranged only between 5.5 and 6.7 within the Hygum Cu gradient, while there was a more than 300-fold difference in levels of bioavailable Cu across the gradient, leading to a Cu-specific PICT response even in the weakly contaminated part of the Cu gradient with negligible differences in soil pH. Rousk and coworkers studied an extreme pH gradient (4.0 to 8.3) in a pyrosequencing study comparable to the current one (33). In the pH range between ca 5.5 and 6.7, those authors primarily observed differences in the abundance of Acidobacteria subgroups. Also, data from a recent study of long-term Cu and pH impacts on soil microbiota indicate the significance of pH (range 4.0 to 6.1) for the abundance of Acidobacteria but not other dominant soil bacterial groups. Hence, it is possible that the changes in relative abundance, in particular of Acidobacteria and some of its subgroups, are partly caused by indirect Cu effects on pH.
Plant cover, biomass, and species richness have also been shown to be affected by Cu in the studied soil Cu gradient (40). Although we omitted rhizosphere soil from our analysis, we cannot entirely rule out the possibility that some of the observed Cu impacts on bacterial community composition were mediated by Cu-induced changes of plant-microbe interactions rather than direct Cu toxicity. In line with several other researchers studying the Hygum Cu gradient site (3, 16, 28, 31, 36, 40) and the discussion above, we find it safe to conclude that differential Cu exposure was the dominant selective force affecting soil biota.
In conclusion, our data demonstrate that long-term Cu exposure may select for Cu-tolerant soil bacterial communities with changed community composition without affecting OTU richness at the relatively crude taxonomic level associated with studies of 16S rRNA gene fragments. Two plausible mechanisms may underlie our results: competitive exclusion of metal-sensitive strains by Cu-resistant strains and horizontal transfer of Cu resistance determinants carried on mobile genetic elements from resistant to sensitive strains (30). We predict that the first mechanism leads to reduced strain-level OTU richness in Cu-impacted soil, whereas the second mechanism does not. We therefore propose that future studies of Cu impacts on bacterial communities should focus on the strain and species levels. More detailed studies along these lines are also needed in order to understand the dominant mechanisms explaining Cu-induced coselection for antibiotic resistance in soil bacterial communities (3).
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Center for Environmental and Agricultural Microbiology (CREAM) funded by the Villum Kann Rasmussen Foundation.
We thank John Jensen from Department of Terrestrial Ecology, Aarhus University, Denmark, for granting access to the Hygum field site.
Footnotes
Published ahead of print 17 August 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Bååth E, Pettersson M, Söderberg KH. 2001. Adaptation of a rapid and economical microcentrifugation method to measure thymidine and leucine incorporation by soil bacteria. Soil Biol. Biochem. 33:1571–1574 [Google Scholar]
- 2. Bent SJ, Forney LJ. 2008. The tragedy of the uncommon: understanding limitations in the analysis of microbial diversity. ISME J. 2:689–695 [DOI] [PubMed] [Google Scholar]
- 3. Berg J, et al. 2010. Cu exposure under field conditions coselects for antibiotic resistance as determined by a novel cultivation-independent bacterial community tolerance assay. Environ. Sci. Technol. 44:8724–8728 [DOI] [PubMed] [Google Scholar]
- 4. Bolan NS, Adriano DC, Mahimairaja S. 2004. Distribution and bioavailability of trace elements in livestock and poultry manure by-products. Crit. Rev. Environ. Sci. Technol. 34:291–338 [Google Scholar]
- 5. Brandt KK, Holm PE, Nybroe O. 2008. Evidence for bioavailable Cu-dissolved organic matter complexes and transiently increased Cu bioavailability in manure-amended soils as determined by bioluminescent bacterial biosensors. Environ. Sci. Technol. 42:3102–3108 [DOI] [PubMed] [Google Scholar]
- 6. Brandt KK, Petersen A, Holm PE, Nybroe O. 2006. Decreased abundance and diversity of culturable Pseudomonas spp. populations with increasing copper exposure in the sugar beet rhizosphere. FEMS Microbiol. Ecol. 56:281–291 [DOI] [PubMed] [Google Scholar]
- 7. Brandt KK, Frandsen RJN, Holm PE, Nybroe O. 2010. Development of pollution-induced community tolerance is linked to structural and functional resilience of a soil bacterial community following a five-year field exposure to copper. Soil Biol. Biochem. 42:748–757 [Google Scholar]
- 8. Brandt KK, Sjøholm OR, Krogh KA, Halling-Sørensen B, Nybroe O. 2009. Increased pollution-induced bacterial community tolerance to sulfadiazine in soil hotspots amended with artificial root exudates. Environ. Sci. Technol. 43:2963–2968 [DOI] [PubMed] [Google Scholar]
- 9. Colwell RK, Coddington JA. 1994. Estimating terrestrial biodiversity through extrapolation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 345:101–118 [DOI] [PubMed] [Google Scholar]
- 10. Daniel R. 2005. The metagenomics of soil. Nat. Rev. Microbiol. 3:470–478 [DOI] [PubMed] [Google Scholar]
- 11. de Boer TE, et al. 2012. The influence of long-term copper contaminated agricultural soil at different pH levels on microbial communities and springtail transcriptional regulation. Environ. Sci. Technol. 46:60–68 [DOI] [PubMed] [Google Scholar]
- 12. Dell'Amico E, Mazzocchi M, Cavalca L, Allievi L, Andreoni V. 2008. Assessment of bacterial community structure in a long-term copper-polluted ex-vineyard soil. Microbiol. Res. 163:671–683 [DOI] [PubMed] [Google Scholar]
- 13. Diaz-Ravina M, Bååth E. 1996. Development of metal tolerance in soil microbial communities exposed to experimentally increased metal levels. Appl. Environ. Microbiol. 62:2970–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Díaz-Raviña M, Bååth E, Frostegård Å. 1994. Multiple heavy metal tolerance of soil bacterial communities and its measurement by a thymidine incorporation technique. Appl. Environ. Microbiol. 60:2238–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ellis RJ, et al. 2002. Similarity of microbial and meiofaunal community analysis for mapping ecological effects of heavy-metal contamination in soil. FEMS Microbiol. Ecol. 40:113–122 [DOI] [PubMed] [Google Scholar]
- 16. Fait G, Broos K, Zrna S, Lombi E, Hamon R. 2006. Tolerance of nitrifying bacteria to copper and nickel. Environ. Toxicol. Chem. 25:2000–2005 [DOI] [PubMed] [Google Scholar]
- 17. Fierer N, Bradford MA, Jackson RB. 2007. Toward an ecological classification of soil bacteria. Ecology 88:1354–1364 [DOI] [PubMed] [Google Scholar]
- 18. Gans J, Wolinsky M, Dunbar J. 2005. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309:1387–1390 [DOI] [PubMed] [Google Scholar]
- 19. Giller KE, Witter E, McGrath SP. 1998. Toxicity of heavy metals to microorganisms and microbial processes in agricultural soils: a review. Soil Biol. Biochem. 30:1389–1414 [Google Scholar]
- 20. Girvan MS, Campbell CD, Killham K, Prosser JI, Glover LA. 2005. Bacterial diversity promotes community stability and functional resilience after perturbation. Environ. Microbiol. 7:301–313 [DOI] [PubMed] [Google Scholar]
- 21. Hagerberg D, et al. 2011. Low concentration of copper inhibits colonization of soil by the arbuscular mycorrhizal fungus Glomus intraradices and changes the microbial community structure. Microb. Ecol. 61:844–852 [DOI] [PubMed] [Google Scholar]
- 22. Ippolito JA, Ducey T, Tarkalson D. 2010. Copper impacts on corn, soil extractability, and the soil bacterial community. Soil Sci. 175:586–592 [Google Scholar]
- 23. Janssen PH. 2006. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72:1719–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Komárek ME, Cadkova E, Chrastny V, Bordas F, Bollinger JC. 2010. Contamination of vineyard soils with fungicides: a review of environmental and toxicological aspects. Environ. Int. 36:138–151 [DOI] [PubMed] [Google Scholar]
- 25. Lauber CL, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75:5111–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lejon DPH, et al. 2008. Copper dynamics and impact on microbial communities in soils of variable organic status. Environ. Sci. Technol. 42:2819–2825 [DOI] [PubMed] [Google Scholar]
- 27. Masoud W, et al. 2011. Characterization of bacterial populations in Danish raw milk cheeses made with different starter cultures by denaturating gradient gel electrophoresis and pyrosequencing. Int. Dairy J. 21:142–148 [Google Scholar]
- 28. Mertens J, Wakelin SA, Broos K, McLaughlin MJ, Smolders E. 2010. Extent of copper tolerance and consequences for functional stability of the ammonia-oxidizing community in long-term copper-contamimated soils. Environ. Toxicol. Chem. 29:27–37 [DOI] [PubMed] [Google Scholar]
- 29. Nawrocki EP, Eddy SR. 2007. Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Comput. Biol. 3:e56 doi:10.1371/journal.pcbi.0030056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nies DH. 1999. Microbial heavy-metal resistance. Appl. Microbiol. Biotechnol. 51:730–750 [DOI] [PubMed] [Google Scholar]
- 31. Oorts K, Ghesquiere U, Swinnen K, Smolders E. 2006. Soil properties affecting the toxicity of CuCl2 and NiCl2 for soil microbial processes in freshly spiked soils. Environ. Toxicol. Chem. 25:836–844 [DOI] [PubMed] [Google Scholar]
- 32. Roesch LFW, et al. 2007. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 1:283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rousk J, et al. 2010. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4:1340–1351 [DOI] [PubMed] [Google Scholar]
- 34. Saeki K, Kunito T, Oyaizu H, Matsumoto S. 2002. Relationships between bacterial tolerance levels and forms of copper and zinc in soils. J. Environ. Qual. 31:1570–1575 [DOI] [PubMed] [Google Scholar]
- 35. Sandaa RA, et al. 1999. Analysis of bacterial communities in heavy metal-contaminated soils at different levels of resolution. FEMS Microbiol. Ecol. 30:237–251 [DOI] [PubMed] [Google Scholar]
- 36. Sauvé S. 2006. Copper inhibition of soil organic matter decomposition in a seventy-year field exposure. Environ. Toxicol. Chem. 25:854–857 [DOI] [PubMed] [Google Scholar]
- 37. Sauvé S, McBride MB, Norwell WA, Hendershot WH. 1997. Copper solubility and speciation of in situ contaminated soils: effects of copper level, pH and organic matter. Water Air Soil Pollut. 100:133–149 [Google Scholar]
- 38. Singh BK, Campbell CD, Sorensen SJ, Zhou J. 2009. Soil genomics is the way forward. Nat. Rev. Microbiol. 7:756–757 [DOI] [PubMed] [Google Scholar]
- 39. Smolders E, et al. 2009. Toxicity of trace metals in soil as affected by soil type and aging after contamination: using calibrated bioavailability models to set ecological standards. Environ. Toxicol. Chem. 28:1633–1642 [DOI] [PubMed] [Google Scholar]
- 40. Strandberg B, Axelsen JA, Pedersen MB, Jensen J, Attrill MJ. 2006. Effect of a copper gradient on plant community structure. Environ. Toxicol. Chem. 25:743–753 [DOI] [PubMed] [Google Scholar]
- 41. Tom-Petersen A, Hansen HCB, Nybroe O. 2004. Time and moisture effects on total and bioavailable copper in soil solutions. J. Environ. Qual. 33:505–512 [DOI] [PubMed] [Google Scholar]
- 42. Tom-Petersen A, Hosbond C, Nybroe O. 2001. Identification of copper-induced genes in Pseudomonas fluorescens and use of a reporter strain to monitor bioavailable copper in soil. FEMS Microbiol. Ecol. 38:59–67 [Google Scholar]
- 43. Tom-Petersen A, Leser T, Marsh TL, Nybroe O. 2003. Effects of copper amendment on the bacterial community in agricultural soil analysed by the T-RFLP technique. FEMS Microbiol. Ecol. 46:53–62 [DOI] [PubMed] [Google Scholar]
- 44. Torsvik V, Øvreås L, Thingstad TF. 2002. Prokaryotic diversity—magnitude, dynamics, and controlling factors. Science 296:1064–1066 [DOI] [PubMed] [Google Scholar]
- 45. Turpeinen R, Kairesalo T, Häggblom MM. 2004. Microbial community structure and activity in arsenic-, chromium- and copper-contaminated soils. FEMS Microbiol. Ecol. 47:39–50 [DOI] [PubMed] [Google Scholar]
- 46. Wakelin SA, Chu G, Lardner R, Liang Y, McLaughlin M. 2010. A single application of Cu to field soil has long-term effects on bacterial community structure, diversity, and soil processes. Pedobiologia 53:149–158 [Google Scholar]
- 47. Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Youssef N, et al. 2009. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl. Environ. Microbiol. 75:5227–5236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Youssef NH, Elshahed MS. 2009. Diversity rankings among bacterial lineages in soil. ISME J. 3:305–313 [DOI] [PubMed] [Google Scholar]
- 50. Yu Y, Lee C, Kim J, Hwang S. 2005. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 89:670–679 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.