

Abstract
Microbial associations with corals are common and are most likely symbiotic, although their diversity and relationships with environmental factors and host species remain unclear. In this study, we adopted a 16S rRNA gene tag-pyrosequencing technique to investigate the bacterial communities associated with three stony Scleractinea and two soft Octocorallia corals from three locations in the Red Sea. Our results revealed highly diverse bacterial communities in the Red Sea corals, with more than 600 ribotypes detected and up to 1,000 species estimated from a single coral species. Altogether, 21 bacterial phyla were recovered from the corals, of which Gammaproteobacteria was the most dominant group, and Chloroflexi, Chlamydiae, and the candidate phylum WS3 were reported in corals for the first time. The associated bacterial communities varied greatly with location, where environmental conditions differed significantly. Corals from disturbed areas appeared to share more similar bacterial communities, but larger variations in community structures were observed between different coral species from pristine waters. Ordination methods identified salinity and depth as the most influential parameters affecting the abundance of Vibrio, Pseudoalteromonas, Serratia, Stenotrophomonas, Pseudomonas, and Achromobacter in the corals. On the other hand, bacteria such as Chloracidobacterium and Endozoicomonas were more sensitive to the coral species, suggesting that the host species type may be influential in the associated bacterial community, as well. The combined influences of the coral host and environmental factors on the associated microbial communities are discussed. This study represents the first comparative study using tag-pyrosequencing technology to investigate the bacterial communities in Red Sea corals.
INTRODUCTION
The Red Sea, notable for its perennial high temperature and salinity, is a unique and largely unexplored marine ecosystem. An extensive area of 2,000 km of coral reef is found along the coastline of the sea, from which approximately 200 coral species were reported (42). Previous studies revealed diverse and abundant microbial populations in corals from a wide range of environments (8, 33, 62, 63, 64, 69). However, no in-depth survey of microbial diversity has been carried out on Red Sea corals. It has been suggested that corals provide ecological niches and nutrients for the colonization of microbes (18, 77). In return, the associated microbes could fix and cycle nitrogen, carbon, sulfur, and other nutrients for their hosts (34, 41, 56, 61, 67, 72). They could also act as a source of secondary metabolites to defend their hosts against predation and microbial infection (12, 25).
In addition to investigating the function of this symbiotic relationship, ecologists have attempted to uncover the diversity and variation in these microbial communities. Early studies on coral-associated microbes relied on culture techniques (17, 18). In 2001, Rohwer et al. (64) were the first to employ culture-independent molecular techniques to study coral-associated bacterial communities, and they discovered 75 bacterial ribotypes in the Caribbean coral Montastraea franksi. Since 2004, about 1,000 distinct microbial ribotypes covering 16 bacterial divisions have been recovered from corals (61). In a more recent study, Mouchka et al. (50) analyzed 6,774 16S rRNA gene sequences that were retrieved from 32 coral microbial ecology studies and reported that healthy and bleached corals harbored similar dominant taxa, while certain genera were found to be more abundant in bleached/diseased corals.
The accumulation of information on coral-derived microbial sequences has sparked a controversial debate on the nature and specificity of coral-associated microbes. The sequencing of over 1,000 bacterial 16S rRNA genes from three massive coral species, M. franksi, Diploria strigosa, and Porites astreoides, from Panama and Bermuda revealed unique bacterial communities for each coral species but found that geographically separate conspecific corals shared similar communities, supporting the argument for a conserved coral-microbial consortium (64, 65). Bourne and Munn (8) further found that different individuals of the coral Pocillopora damicornis harbored a small uniform microbial population that was absent from the surrounding seawater and coral mucus. Recently, Hong et al. (30) reported high species specificity in the bacterial communities associated with corals from the Caribbean Sea. These studies suggest that coral-associated microbial communities are most likely coral species specific (62). In contrast, however, recent studies have found that the communities may shift in response to environmental conditions (7, 35, 45, 52, 80). Using a combination of molecular techniques, Webster and Bourne (80) studied the bacteria associated with the Antarctic soft coral Alcyonium antarcticum and found that they resembled those from the Antarctic seawater and sponges. In 2008, Neulinger and colleagues (52) suggested that colors and sampling sites of the cold-water coral Lophelia pertusa from Norway could affect the associated microbial communities. In a more recent study, Littman and coworkers (45) showed that location plays a more crucial role than coral species in the bacterial composition in three acroporid corals from the Great Barrier Reef. Variation of the associated bacterial communities with time as opposed to coral species was also demonstrated in corals from Western Australia (13). The nature and specificity of microbial communities in corals are not yet clearly understood, and a more comprehensive survey is required.
The inconsistency of the findings on the specificity of coral-associated microbial communities may be due to differences in the methods employed. All methods have certain biases and thus may have failed to capture a complete picture of the microbial communities. The development of high-throughput pyrosequencing technology has revolutionized the traditional cloning and capillary Sanger sequencing technique (6, 48, 66, 68). Using such a sequencing technique combined with barcoded PCR primers, Sogin and coworkers concluded that the genetic diversity, community composition, relative abundance, and distribution of microbes in the natural environment remain undersampled (73). In 2010, Sunagawa et al. (74) were the first to employ the 454 sequencing technique to explore the microbial diversity in the Caribbean corals and found that corals of the same genus or family harbored similar bacterial communities. However, at higher taxonomic levels, the correlation of similar bacterial communities with phylogenetically closely related corals was not evident. This result led to the questioning of the evolutionary stability of coral-microbial associations. In this study, we used the same sequencing technique to study the microbial communities associated with corals from the Red Sea. We compared bacterial communities associated with different species of corals from different locations to understand the specificity of such associations and the impact of environmental conditions on the communities.
MATERIALS AND METHODS
Sample collection, environmental-parameter measurement, and identification of corals.
Five different coral species were collected from three different locations along the Red Sea coast of Saudi Arabia by snorkeling or scuba diving in April 2009 (Fig. 1 and Table 1). Sites 1 and 2 were close to the mouth of Sharm Obhur (1.6 km from the mouth and 1.2 km inside the mouth, respectively). The sharm continuously receives sewage discharge from anthropogenic and domestic sources along its two sides (personal observation). Therefore, these two sites were considered “disturbed” sites. Site 3 was about 50 km northwest of the sharm mouth and 10 km from the coastline. It sat in an open-water coral reef (named Abu Madafi Reef) and represented an apparently “pristine” area. At each sampling location, 3 different individual colonies for a single coral species were detached from the substrata at the basal plate using a hammer and chisel and immediately transported back to the laboratory in sterile plastic bags with seawater. CITES (Convention on International Trade in Endangered Species) permits for collecting Pocillopora verrucosa and Stylophora pistillata were obtained from the Department of Permits, Commission for Wildlife Conservation and Development (permit numbers 09-SA-00289-CO and 09-SA-00291-CO; Riyadh, Kingdom of Saudi Arabia). Environmental parameters were measured (three replicated readings) on site by using a multiparameter water quality sonde (YSI 6600 Sonde), and 1 liter of seawater (for the three replicates) was collected at each site for subsequent nutrient analysis in the laboratory by using a TOC (total organic carbon) device (TOCV-CPH, Shimadzu, Japan) and a nutrient analyzer (Skalar 4000, Breda, The Netherlands) following the manufacturers' protocols. Heavy metal contents in sediments collected from each sampling location were measured. Briefly, 0.25-g freeze-dried samples were digested with 2 ml of 75% nitric acid and 6 ml 37% hydrochloric acid, using the microwave assistant method (Environmental Protection Agency [EPA] method 3051; 175°C; 10 min) (77). The digested samples were measured with a Perkin Elmer atomic absorption analyzer (AAS 800) for Cd, Zn, Cu, Pb, and As. A CETAC QuickTrace Mercury Analyzer measured total Hg. One-way analysis of variance (ANOVA) was performed to test if there was a significant difference in any of the measured parameters among sites. Nonparametric Kruskal-Wallis one-way ANOVA was performed with ranked data when the data failed to pass the Shapiro-Wilk normality and equal variance tests. Hard-coral specimens were identified based on colony morphological characteristics and the structure and size of the sclerites, whereas soft-coral specimens were identified by the 18S rRNA gene sequence (Table 1; see Fig. S1 in the supplemental material).
Fig 1.

Maps showing the sampling locations. Site 1 was outside but close to (1.6 km) the mouth of Sharm Obhur, while site 2 was located within the sharm, 1.2 km inside its mouth and 9 km from its head. Site 3 was about 50 km northwest of the sharm mouth and was an open-water coral reef named Abu Madafi Reef.
Table 1.
Coral species collected from different locations along the coast of the Red Sea
| Site | Location | Depth (m) | Coral species | Coral type | Sample identifier |
|---|---|---|---|---|---|
| 1 | 21°42.32′N, 39°04.23′E | 15 | Pocillopora verrucosa | Stony | S1-Pv |
| Astreopora myriophthalma | Stony | S1-Am | |||
| 2 | 21°42.64′N, 39°05.69′E | 8.0 | P. verrucosa | Stony | S2-Pv |
| Sarcophyton sp. 1 | Soft | S2-Sa-I | |||
| Sarcophyton sp. 2 | Soft | S2-Sa-II | |||
| 3 | 22°03.66′N, 38°46.07′E | 19 | P. verrucosa | Stony | S3-Pv |
| Sarcophyton sp. 1 | Soft | S3-Sa-I | |||
| A. myriophthalma | Stony | S3-Am | |||
| Stylophora pistillata | Stony | S3-Sp |
Extraction of microbial community DNA.
Upon arrival at the laboratory, the coral specimens were flushed thoroughly with 0.22-μm-filtered seawater to remove loosely attached bacteria and mucus from the surface. The coral specimens were homogenized with a sterile mortar, and then about 0.5 ml (measured by water displacement) of coral homogenates from each replicate was frozen in 0.8 ml of extraction buffer (100 mM Tris-HCl, 100 mM Na2-EDTA, 100 mM Na2HPO4, 1.5 M NaCl, 1% CTAB [cetyltrimethylammonium bromide], pH 8) for community DNA extraction. The total genomic DNA was extracted according to the modified SDS-based method described by Lee et al. (39) and purified with a Mo Bio soil DNA isolation kit (Mo Bio Laboratories, Inc., Carlsbad, CA). The quality and quantity of the DNA were checked with a NanoDrop spectrophotometer (ND-1000; NanoDrop). Purified DNA samples were kept at −20°C for future use.
PCR amplification of the 16S rRNA gene and pyrosequencing of barcoded amplicons.
As DNA fingerprinting terminal restriction fragment length polymorphism (TRFLP) analysis revealed similar microbial communities among the replicates (see Fig. S2 in the supplemental material), equal amounts of DNA from the three replicates were pooled for PCR to generate the 16S rRNA gene amplicons. Pooling DNA from different replicates for subsequent PCR can reduce intrasample variations (70). Different samples were PCR amplified using primers with unique barcodes. A set of primers was designed by adding a 6-nucleotide (nt) barcode unique to each sample (see Table S1 in the supplemental material) to the universal forward primer U341F (5′-CCTACGGGRSGCAGCAG-3′) and the reverse primer R685 (5′-ATCTACGCATTTCACCGCTAC-3′). The primers targeted amplification of the hypervariable regions V3 and V4 of the 16S rRNA gene. For a 100-μl PCR mixture, 5 units of Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA), 1× Pfu reaction buffer, 0.2 mM deoxynucleoside triphosphates (dNTPs) (TaKaRa, Dalian, China), 0.1 μM each barcoded primer, and 20 ng of genomic DNA template were used. PCR amplification was carried out on a thermocycle controller (MJ Research Inc., Bio-Rad) with the following program: initial denaturation at 94°C for 5 min; 26 cycles at 94°C for 30 s, 53°C for 30 s, and 72°C for 45 s; and final extension at 72°C for 6 min. The PCR products were purified using the TaKaRa Agarose Gel DNA Purification Kit (TaKaRa, Dalian, China) and quantified with the NanoDrop device. A mixture of the PCR products was prepared by mixing 200 ng of the purified 16S amplicons from each coral sample and then pyrosequenced on the Roche 454 FLX Titanium platform at the Chinese National Human Genome Centre in Shanghai, China. To evaluate intrasample variations in the bacterial communities, the 16S rRNA gene amplicons were generated using DNA from one randomly chosen replicate for each sample and pyrosequenced as described above.
Calculation of species richness and taxonomic assignment of pyrosequencing reads.
The pyrosequencing data were deposited in the NCBI Sequence Read Archive (SRA) database with the accession number SRA012656. The downstream bioinformatics analysis was performed using QIIME 1.3.0 (11). The following quality control criteria were enforced: (i) exclusion of reads with one or more ambiguous nucleotides, (ii) exclusion of reads shorter than 150 bp, (iii) exclusion of reads containing homopolymers of 6 bp and above, and (iv) application of a quality window of 50 bp with an average flowgram score of 25. Reads were assigned to their respective samples according to their barcodes and then subjected to a second round of quality control using Denoiser (58). Qualified reads were clustered using uclust (20) and then assigned to operational taxonomic units (OTUs) at 97% similarity. Representatives of the most abundant reads were selected from each OTU for subsequent analysis. Representative OTUs were de novo aligned using MUSCLE (19), and a phylogenetic tree was produced using fasttree (54). Representative OTUs were also aligned using PyNAST (10) with the Silva108 database as a reference. Successfully aligned reads were fed into ChimeraSlayer (24) to identify and discard chimeric reads. Species diversity, richness, and rarefaction curves were computed at 97% similarity as part of QIIME's alpha diversity pipeline. Beta diversity analysis was conducted after rarefying the samples at the smallest library. A step size of 100 was used, with 100 repetitions at each step. Taxonomy assignment was conducted using the Ribosomal Database Project (RDP) classifier version 2.2 (79) against Silva108 (55) with a bootstrap confidence level of 50%. The numbers of reads assigned to different genera were converted into percentages, which served as the input for Cluster3 (16). A divergence of greater than 0.5% was used to filter out genera with small differences among the samples. The remaining genera were further normalized and centered by the mean. The complete linkage method with a metric of correlation (uncentered) was used to generate a hierarchical cluster, which was then passed to the Java Treeview module in Cluster3 to generate a heat map.
Comparison of coral-associated microbial communities and their relationship with the environment.
The similarity among different microbial communities was determined by similarity matrices generated based on the phylogenetic distances among all qualified reads (i.e., UniFrac distance [46, 47]) and displayed with Jackknife-supported unweighted-pair group method using average linkages (UPGMA) clustering implemented in the QIIME pipeline.
The correlations between bacterial assemblages, coral species type, and sampling sites (i.e., environmental factors) were analyzed by ordination methods using the software Canoco (version 4.5; Microcomputer Power) (9). For both constrained and unconstrained ordination methods, the percentage abundance data of bacterial groups (at the genus level) in each library were used as the species input, and the environmental variables (both coral species type and environmental parameters) were log transformed and served as the environmental input. The significance tests of Monte Carlo permutations were used to build the optimal models of the microbe-environment relationship (40).
Nucleotide sequence accession numbers.
The 18S rRNA sequences were deposited in GenBank with accession numbers HM067604 to HM067613.
RESULTS
Environmental parameters at the study sites.
Coral samples were collected from different depths, ranging from shallow water at 8.0 m to deeper water at 19 m, at three locations along the coast of the Red Sea in Saudi Arabia (Fig. 1 and Table 1). All environmental parameters, except nitrate and total phosphorus (TP), showed a significant difference among the sites (one-way ANOVA; P < 0.05) (Table 2). Site 1 appeared to be the least turbid (with the highest visibility), yet it had the highest values for total organic carbon (TOC), total nitrogen (TN), nitrate, and ammonium, whereas site 3 was the most turbid and was characterized by a generally low nutrient content. In addition, the heavy metal contents in the sediments from different sites also varied significantly (P < 0.01) (Table 3). The sediment from site 2 had the highest copper, lead, arsenic, and mercury contents, ranging from 40 ng/g to 4 μg/g, while sediment from sites 1 and 2 contained a high level of zinc (>9 μg/g) (Table 3). In contrast, the levels of zinc, copper, lead, arsenic, and mercury were lowest in the sediment from site 3.
Table 2.
Environmental parameters at the sampling locationsa
| Site | Depth (m) | Temp (°C) | Sal (ppt) | DO (mg/liter) | pH | Turbidity (NTU) | Chl a (μg/liter) | TOC (mg/liter) | TC (mg/liter) | TN (mg/liter) | NO3 (mg/liter) | NH4 (mg/liter) | TP (μg/liter) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 15 | 26.00 ± 0.01 | 39.15 ± 0.01 | 6.64 ± 0.01 | 7.62 ± 0.01 | 0.233 ± 0.23 | 0.50 ± 0.00 | 3.591 ± 0.27 | 30.39 ± 0.22 | 0.496 ± 0.03 | 0.210 ± 0.09 | 1.256 ± 0.14 | 2.29 ± 0.001 |
| 2 | 8 | 26.79 ± 0.01 | 39.21 ± 0.01 | 6.72 ± 0.02 | 7.80 ± 0.01 | 0.833 ± 0.06 | 0.33 ± 0.06 | 2.247 ± 0.20 | 28.92 ± 1.37 | 0.192 ± 0.00 | 0.116 ± 0.01 | 1.195 ± 0.08 | 3.01 ± 0.005 |
| 3 | 19 | 25.80 ± 0.01 | 39.57 ± 0.01 | 5.70 ± 0.02 | 7.89 ± 0.02 | 1.233 ± 0.06 | 0.23 ± 0.06 | 1.767 ± 0.20 | 26.58 ± 0.18 | 0.124 ± 0.01 | 0.126 ± 0.02 | 0.888 ± 0.03 | 1.58 ± 0.003 |
Temperature (Temp), salinity (Sal), DO, pH, turbidity, and Chl a content were measured with a YSI 6600 Sonde; TOC and TC were measured with a TOC machine; and TN, nitrate, ammonium, and TP were measured with a nutrient analyzer. The data presented are means ± 1 standard deviations of 3 measurements. One-way ANOVA detected significant differences (P < 0.05) in all parameters except NO3 and TP among sites. NTU, nephelometric turbidity units.
Table 3.
Heavy metal content in the sediments collected from each sampling locationa
| Site | Cd (μg/g) | Zn (μg/g) | Cu (μg/g) | Pb (μg/g) | As (μg/g) | Hg (ng/g) |
|---|---|---|---|---|---|---|
| 1 | 0.035 ± 0.007 | 10.0 ± 0.6 | 0.69 ± 0.09 | 0.93 ± 0.15 | 1.5 ± 0.6 | 3.7 ± 0.9 |
| 2 | 0.024 ± 0.005 | 9.0 ± 0.6 | 1.44 ± 0.09 | 2.74 ± 0.29 | 4.2 ± 0.3 | 40.4 ± 9.4 |
| 3 | 0.027 ± 0.004 | 5.3 ± 0.4 | 0.65 ± 0.20 | 0.49 ± 0.06 | 1.4 ± 0.8 | 3.2 ± 0.3 |
The data presented are means ± 1 standard deviation of 3 measurements. One-way ANOVA detected significant differences (P < 0.01) in all measured heavy metals among sites.
Diversity and composition of coral-associated bacterial communities.
Quality filtering recovered a total of 43,579 reads (average number of reads per sample, 4,842; average read length, 364 bp) from pyrosequencing of the mixture of PCR amplicons of the pooled DNA. The number of OTUs and estimated species richness at a 3% dissimilarity level are listed in Fig. 2 (see Table S2 in the supplemental material). The highest number of OTUs was found in Sarcophyton sp. 2 from site 2 and the lowest in Astreopora myriophthalma from sites 1 and 3 (Fig. 2). The same species of corals from different locations showed varying degrees of bacterial diversity, and there was not a significant correlation between species diversity (i.e., the number of OTUs, diversity index, or species richness) with the coral species type or sampling site (Pearson correlation; P > 0.05).
Fig 2.
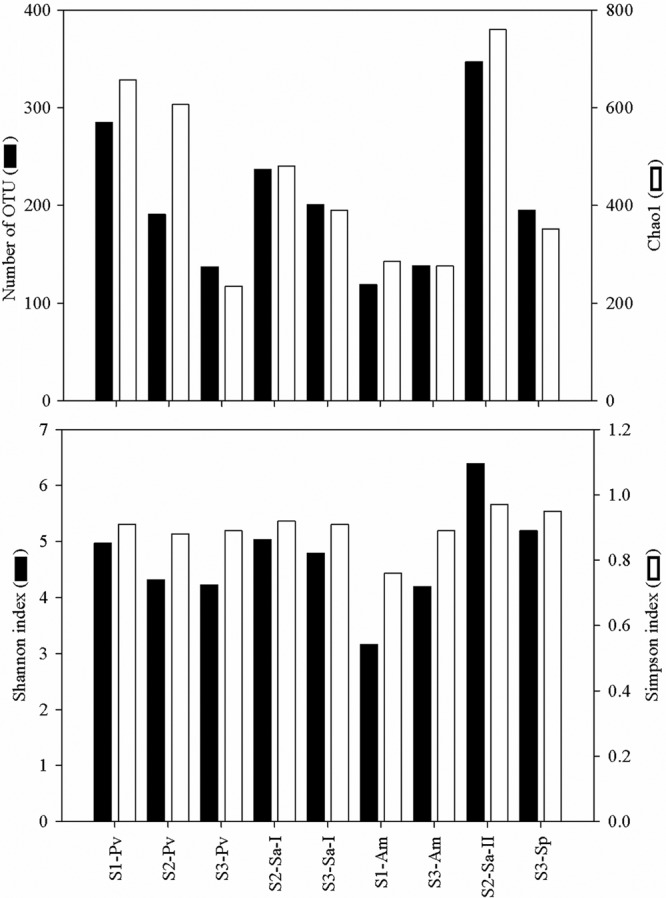
Similarity-based OTUs, species diversity (Shannon and Simpson indices), and richness estimate (Chao1) of the coral-associated bacterial communities at a dissimilarity level of 3%. The data presented are based on data sets normalized by the smallest library size (i.e., 2,165 reads). See Table 1 for sample identifiers. The number of qualified reads for each sample and nonnormalized data are listed in Table S2 in the supplemental material.
At a confidence threshold of 50%, 42,409 out of the 43,579 qualified bacterial reads (i.e., 97.3%) could be assigned to a known phylum using the QIIME analysis pipeline. Twenty-one phyla were recovered from the coral-associated bacterial communities, with an average of 96.2% of the qualified reads affiliated with seven ubiquitous phyla, including Proteobacteria, Actinobacteria, Bacteroidetes, Cyanobacteria, Chloroflexi, Deinococcus-Thermus, and Firmicutes (Fig. 3A). The proportions of these seven ubiquitous phyla varied among different coral species from different sites, yet Proteobacteria was always the most dominant and constituted 63 to 95% of the qualified bacterial reads in all of the coral samples. For the pyrosequencing run of one randomly chosen replicate, 184,978 reads (average length, 368 bp) were retrieved, and 182,142 (i.e., 98.8%) were assigned to 19 different phyla (see Fig. S3a in the supplemental material). RDP classifier revealed a similar observation that a large proportion of classified reads was affiliated with Proteobacteria, followed by Actinobacteria (see Fig. S3a in the supplemental material). Using the same threshold confidence level, the bacterial reads were further classified down to the order level, and substantial variations in the bacterial communities associated with corals from different sites were observed (Fig. 3B; see Fig. S3b in the supplemental material). For instance, corals from sites 1 and 2 were dominated by the Gammaproteobacteria, with a majority belonging to the order Enterobacteriales for both pyrosequencing runs, whereas corals from site 3 contained varying proportions of Vibrionales, Alteromonadales, and Pseudomonadales in the Gammaproteobacteria and Burkholderiales in the Betaproteobacteria. The compositions of the bacterial communities in the same coral species from disturbed and pristine areas also differed substantially, as exemplified by the comparison of P. verrucosa, A. myriophthalma, and Sarcophyton sp. 1 from sites 1 and 3 Furthermore, large variations were found in the communities in different pyrosequencing runs of samples from site 3.
Fig 3.

Taxonomic classification of bacterial reads retrieved from pooled DNA amplicons from different coral species into phylum (A) and order (B) levels using the RDP classifier. A confidence threshold of 50% was applied for classification. See Table 1 for sample identifiers.
Further classification at the genus level indicated that the bacterial communities associated with corals from sites 1 and 2, except for Sarcophyton sp. 2, were heavily loaded with Serratia, Achromobacter, Stenotrophomonas, and Pseudomonas, whereas those from site 3, except for A. myriophthalma, were dominated by Vibrio, Tenacibaculum, Photobacterium, and Alteromonas (Fig. 4). Sarcophyton sp. 2 from site 2 showed a relatively high abundance of Anoxybacillus, Marinobacter, Aestuariibacter, Chryseobacterium, Colwellia, Lutibacter, Rhodococcus, Tsukamurella, and Lactococcus. A BLAST search for the closest matches of the 10 most abundant OTUs for each coral sample also revealed dominance of Serratia and Achromobacter-like bacteria in corals from sites 1 and 2, whereas corals from site 3 contained a large number of Vibrio bacteria (see Table S3 in the supplemental material).
Fig 4.

Heat map showing the relative abundances and distributions of representative 16S rRNA gene tag sequences classified at the genus level. A divergence setting of >0.5% was used to filter out genera with small differences among the samples. The normalized data were centered by mean and clustered using the complete linkage method and a metric of correlation (uncentered). The color code indicates differences of the relative abundance from the mean, ranging from green (negative) through black (the mean) to red (positive). See Table 1 for sample identifiers.
Comparison of coral-associated bacterial communities and their relationship with the environment.
A UniFrac distance-based Jackknife cluster was computed to compare the similarities of the microbial communities among different coral species from different ecological habitats (Fig. 5). The bacterial communities associated with corals from site 3, regardless of the coral species, generally clustered together, indicating high degrees of similarity among them, and this cluster was well separated from another cluster formed by the samples from sites 1 and 2, suggesting a substantial dissimilarity of the bacterial communities in these samples compared with those associated with corals from site 3. Cluster analysis also showed that corals of the same species did not share the highest similarity in terms of the composition of associated microbes, as corals of the same species did not always form close, distinct clusters. A similar observation was made from the heat map that was generated based on taxonomic assignment at the genus level (Fig. 4).
Fig 5.
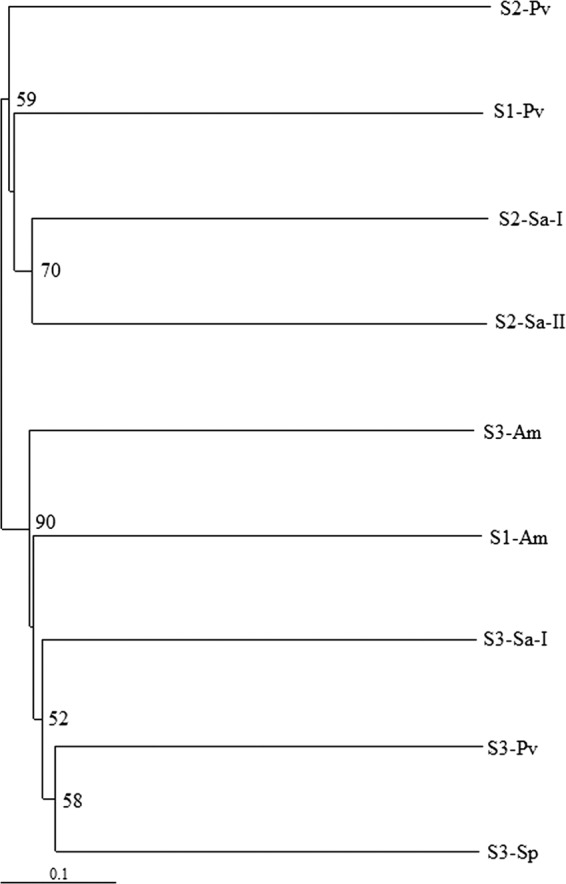
UniFrac distance-based Jackknife clustering of bacterial communities associated with different coral species from different sampling locations. The numbers at the nodes show the Jackknife support values based on 100 random resamplings. See Table 1 for sample identifiers.
Ordination methods were used to examine the relationships of bacterial community composition with coral species, sampling site, and environmental parameters. Detrended correspondence analysis (DCA) was first applied to determine the lengths of gradients, which indicated that the species data set represented a linear model. DCA showed that the measured environmental parameters correlated better with major bacterial genus composition (r = 0.874) than did the coral species (r = 0.413). Redundancy analysis (RDA) was then performed as a direct gradient analysis to study the relationship of coral species and bacterial community composition. The results indicated that 35.6% of the variance in bacterial community composition could be explained by the coral species while 15.3% and 8.8% of the variance were explained by the first and second axes, respectively (see Table S4 in the supplemental material). The first axis separated the bacterial communities in A. myriophthalma and Sarcophyton sp. 1 from the rest of the coral species, while the second axis separated those in A. myriophthalma and P. verrucosa from the others (Fig. 5). RDA showed a strong positive correlation of Chloracidobacterium and Leucothrix with A. myriophthalma but a strong negative correlation of Endozoicomonas and Synechococcus with the coral (Fig. 6a). Similarly, the genera Pseudoalteromonas, Alteromonas, and Salinimonas were positively correlated with the coral S. pistillata, while Glaciecola was positively correlated with Sarcophyton sp. 2 (Fig. 6A). RDA was also performed to study the relationship of environmental parameters with bacterial community composition. The results indicated that up to 43.3% of the variance could be explained by the environmental parameters measured in this study; a majority (35%) could be explained by the first axis and the rest (8.3%) by the second axis (see Table S4 in the supplemental material). Both the first and second axes showed good correlation with environmental data (r > 0.874) (see Table S4 in the supplemental material). Automatic forward selection was then performed to build a parsimonious model that identified salinity, followed by depth, as the most influential environmental parameters out of all those measured that significantly contributed to the variations in bacterial community composition (P < 0.05). Strong negative correlation was found for depth with TP and temperature (−0.9902 < r < −0.9887) (see Table S5 in the supplemental material), while dissolved oxygen (DO), total carbon (TC), and NH4 were highly negatively correlated with salinity (r < −0.9715) (see Table S5 in the supplemental material). A triplot illustrating the relationship between major bacterial genera and these seven environmental parameters showed that Vibrio, Pseudoalteromonas, Photobacterium, Bermanella, and Salinimonas were highly positively correlated with salinity (Fig. 6B). In contrast, Serratia, Stenotrophomonas, and Pseudomonas were negatively correlated with salinity and depth but positively correlated with TC, DO, and NH4. In addition, positive correlations were found for Achromobacter and Staphylococcus with temperature and TP (Fig. 6B). When combining the influence of environmental parameters and coral species type, up to 81.9% of the variance in the bacterial communities could be explained, and the best correlation was observed (see Table S4 in the supplemental material).
Fig 6.
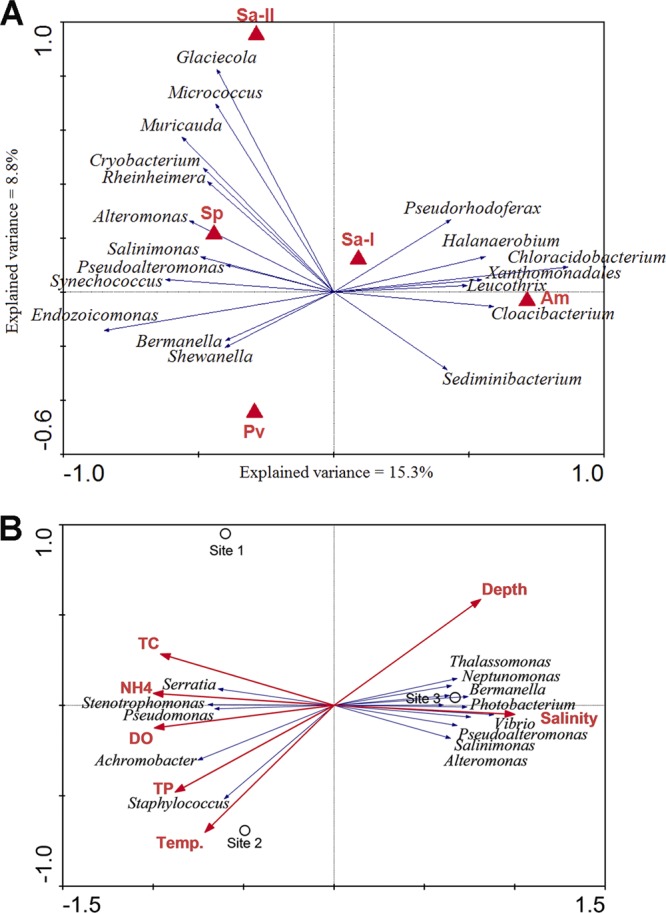
RDA ordination plots showing the relationships of coral-associated bacteria with coral species and environmental parameters at different sites. (A) Biplot showing the relationship between coral species and bacterial genus compositions. See Table 1 for sample identifiers. (B) Triplot showing the relationship between environmental parameters, sampling sites, and bacterial genus compositions. Correlations between environmental variables and the first two RDA axes are represented by the lengths and angles of the arrows (environmental-factor vectors). Automatic forward selection with Monte Carlo permutation tests was applied to build the parsimonious model, which included temperature (Temp.), salinity, depth, TC, ammonium (NH4), and TP, explaining the variance in the bacterial communities. The environmental variables were checked to minimize colinearity in the analyses.
DISCUSSION
The development of a coral-microbe association is a dynamic process possibly affected by a combination of intrinsic and external factors. Intrinsic factors may include the vertical transmission of microbial symbionts from parent to daughter cells (26, 28, 29, 71) and the selective acquisition of microbes from the surrounding environment by defined mechanisms in corals at different developmental stages (2, 4, 28), leading to coral-species-dependent microbial consortia (62, 64, 65). External factors, such as temperature, elevated nutrient levels, dissolved organic carbon load, and reduced pH, can create stressful conditions that change coral-associated microbial communities (78). In this study, we observed the combined effect of environment and species type on microbial communities associated with the corals we collected from the Red Sea.
Relationship of coral-associated microbial communities with environment and coral species.
Due to differences in depth and turbidity, the collection sites differed in light penetration, with the highest levels occurring at site 1 and the lowest at site 3. In comparison with site 3, sites 1 and 2 were relatively disturbed areas with a high nutrient content, particularly total nitrogen and phosphorus. This may be attributed to sewage discharge from anthropogenic and domestic sources along Sharm Obhur (Fig. 1). The high nutrient content, together with sufficient light supplies at these sites due to their low turbidity, possibly promoted the growth of phytoplankton, which would explain the increase in chlorophyll a (Chl a). Site 3 (Abu Madafi Reef) had the lowest nutrient content and primary productivity of all three sites, possibly due to its open-ocean setting and limited nutrient supply. Albarakati (1) found a progressive decrease in surface salinity from the head to the mouth of the sharm, possibly reflecting the dilution impact of local discharges inward from the sharm. Measurements of stable nitrogen isotopes (δ15N) in antipatharians at the middle of Sharm Obhur in June and July 2007 reflected rapid development and potential contamination by sewage discharges around the sharm over the previous 2 decades (59, 60). This may lend support to the relatively high nitrogen loads at site 2 found in the present study. Although our measurement of several environmental parameters in the seawater collected in this study is a snapshot, it may not necessarily indicate a year-round status for a given site. The high levels of heavy metals detected in the sediments from sites 1 and 2 compared with those from site 3 probably reflect long-term pollution in the sharm area. Site 3 is obviously more pristine than sites 1 and 2. Multiple sampling and measurements of water quality at different sites should improve the correlation between the environmental settings and the coral-associated microbial communities.
Many previous studies attempted to investigate the effects of environmental parameters on microbial density, diversity, and composition in a wide range of environments (15, 21, 27, 31, 32, 35, 49, 57). They generally agreed that environmental factors, particularly temperature and salinity, could shift the microbial communities there and thus supported the Bass-Becking hypothesis of “everything is everywhere, but the environment selects” (3a). However, a study investigating the bacterial community in the surface mucus layer of the reef coral Montastraea faveolata using denaturing gradient gel electrophoresis (DGGE) (23) did not find a strong correlation between bacterial community structure and any of the measured water quality parameters, including temperature, salinity, turbidity, DO, and Chl a, E. coli, fecal, nitrate, and phosphate concentrations. This is probably due to the low sensitivity and resolution of DGGE in revealing microbial diversity and community structure (51). In a recent study, which specifically surveyed the pathogenic vibrios in water, sediment, and oysters (31), the authors observed significant correlations of different Vibrio species with temperature, salinity, Chl a level, and turbidity, suggesting niche-based differences in their abundances. Given the differences in environmental settings among the sampling sites in this study, we hypothesized that different microbes associated with corals from different sites. With improved sensitivity and resolution using the tag-pyrosequencing technique, our data partially supported our hypothesis and showed that the bacterial communities associated with corals from site 3 were apparently different from those in sites 1 and 2 (Fig. 3 and 4). Vibrio, Pseudoalteromonas, Photobacterium, Bermanella, and Salinimonas were found to be more abundant in corals from site 3, where salinity was relatively high (Fig. 4). In contrast, Serratia, Stenotrophomonas, and Pseudomonas were more dominant in corals from sites 1 and 2 (Fig. 4), where salinity was comparatively low but TC, DO, and NH4 levels were high (Table 2). Their correlations with these environmental parameters were nicely supported by RDA (Fig. 6B). Furthermore, Achromobacter and Staphylococcus were more abundant in corals from site 2, where the temperature and TP were relatively high (Table 2). Similarly, these genera showed positive correlation with temperature and TP (Fig. 6B). These results suggested that the occurrence of certain genera in different corals at different sites was likely regulated by external environmental parameters.
On the other hand, previous studies also showed that coral-associated microbial communities could be coral species dependent (62, 64, 65). Our results from ordination methods partially supported this, as about 1/3 of the variance in the bacterial community could be explained by the coral species (see Table S4 in the supplemental material) and the abundance of certain genera; for instance, Chloracidobacterium was highly correlated with the coral species, particularly with A. myriophthalma (Fig. 6A). However, our results also suggested that sites (characterized by environmental parameters), rather than coral species, were the primary determining factors in shaping the bacterial communities, as the communities clustered with respect to site rather than to coral species (Fig. 4 and 5). Furthermore, ordination methods indicated that environmental parameters had better correlation with bacterial genus composition and explained more variance than did the coral species (see Table S4 in the supplemental material). Compared to the two more disturbed sites, larger variations in bacterial composition were found in corals from site 3 (Fig. 3B; see Fig. S3b in the supplemental material), where the environment was more pristine. Many of the abundant genera found in corals from this site, such as Synechococcus, Alteromonas, and Salinimonas, showed good correlation with coral species (Fig. 6A). This may suggest that the bacterial communities are more sensitive to the coral species or even to individual corals in more pristine environments. Neither the coral species nor the environmental parameters could be removed from the model, as each of them explained certain degrees of variation in bacterial community structure and combining both gave the best correlation and highest variance explained (see Table S4 in the supplemental material). Nevertheless, it should be noted that only 89.1% of the variation could be explained by the environmental factors that we assessed. The rest of the variation may be due to some other factors that we have not investigated in this study.
Closer investigation of the bacterial communities at a higher resolution revealed that corals from sites 1 and 2 contained a large number of Serratia bacteria (Fig. 4). Although this genus is ubiquitous in water, a few members have been reported to be feces-derived pathogens associated with white pox disease in the Caribbean elkhorn coral Acropora palmata (44, 53, 76). Similarly, the bacterial genus Vibrio was abundant in corals from site 3 (Fig. 4). This genus consists of a huge number of species with a wide range of bioactivity. Although the majority of the members of the genus are harmless to corals, some previous studies have reported that certain Vibrio species can induce coral bleaching (37, 38) and are pathogenic (75). Some of them might be related to black band disease or yellow blotch/band disease in corals (3, 5, 14). Although there is no record of disease outbreak in the studied locations so far and the corals collected in this study did not show any signs of disease or bleaching, a deeper phylogenetic study might help identify Serratia and Vibrio at the species level and determine the potential threat to their hosts.
Coral-associated microbial communities revealed by tag pyrosequencing compared with other studies.
The number of ribotypes detected in a single species of corals in our study ranged from 119 to 631, with richness estimates of up to 1,000 species (Fig. 2; see Table S2 in the supplemental material). In a similar study that pyrosequenced the V6 hypervariable region, up to 2,000 bacterial OTUs with species estimates of up to 4,000 were detected from seven corals collected from the Caribbean Sea (74). The tag-pyrosequencing approach reveals a far better-resolved representation of microbial communities associated with corals, with extremely wide coverage, than do the traditional cloning and sequencing techniques, which typically recover fewer than 50 bacterial ribotypes from a single coral species (52, 65, 80, 82). In comparison with the results of Sunagawa et al. (74), this study revealed lower bacterial diversity in corals from the Red Sea than in those from the Caribbean Sea. This difference may be due to differences in PCR primer selection, OTU calculation, sequencing depth, coral species, and environmental factors between the two studies. Future studies involving deeper sequencing of multiple individuals of the same coral species from different environments should help further resolve the roles of host and environmental factors in shaping microbial communities in corals.
Altogether, 21 bacterial phyla were detected in our corals, many of which have been reported in corals around the world, ranging from tropical and subtropical to Antarctic environments (43, 52, 65, 80, 81, 82). We recovered all of the major phyla previously reported in corals, including Proteobacteria (particularly Gammaproteobacteria), Actinobacteria, Cyanobacteria, and Firmicutes. Additionally, this is the first report of a consistent occurrence of Deinococcus-Thermus and Chloroflexi in all of the coral species investigated in this study (Fig. 3A; see Fig. S3a in the supplemental material). The consistent occurrence and ecological function of these groups in our Red Sea corals warrant further confirmation and investigation. It has been reported that a large number of bacteria populate the mucus of corals (18, 35, 81), many of which are thought to be commensal associates. However, these microbes were excluded from this study, as we were unable to differentiate the true associates (considered symbionts) from the transients (contaminants) in the water column. Further study of microbial communities in the surrounding seawater and mucus may enable us to study their interrelationships with those in tissues and to enrich our knowledge of the diversity of coral-associated microbes.
Conclusion.
In this study, the bacterial communities associated with three different species of corals from disturbed areas were more similar to each other (more homogeneous) than those from pristine areas, where the communities appeared to be more coral species dependent. Salinity and sampling depth, which were highly correlated with temperature, were found to be the most influential environmental parameters among those measured in driving the variations observed in the bacterial community compositions. In particular, these factors exerted a strong influence on the genera Vibrio, Pseudoalteromonas, Serratia, Stenotrophomonas, Pseudomonas, and Achromobacter, while the genera Chloracidobacterium and Endozoicomonas appeared to be more sensitive to the coral species. We must emphasize that generalizations about how coral-associated microbial communities are regulated and respond to the host and environment based on our observations are tenuous, because not all coral species were found in all sampling locations and only a limited number of coral species were included. It would be ideal to sample the same coral species in an undisturbed area with similar depth and geographic setting for comparison. Nonetheless, the natural distribution of coral populations is beyond our control, although every attempt was made to sample as many coral species as possible in the vicinity. In addition, our observations were based on a one-time sampling, which did not address the possible influence of temporal factors on microbial communities. Some environmental parameters, such as temperature and salinity, can fluctuate drastically over time, and differences in microbial populations could be a result of factors other than those we have analyzed. A larger-scale sampling, preferably global, that maximizes the number of host species, sampling sites, and sampling times for comparison and long-term measurements of environmental conditions is definitely needed to untangle the relationship between corals and associated microbes and to make generalization possible. Furthermore, although amplicon pyrosequencing has been widely used to analyze microbial community structure, caution must be exercised in experimental design and data analysis and interpretation due to the potential problems of this sequencing technique, including random sequencing, low reproducibility, and overestimation of the true diversity (22, 36, 83). However, our study did suggest a highly complex interaction between environmental parameters and coral species in the bacterial communities in our Red Sea corals and emphasized the need for considering both, and perhaps other factors, when assessing the specificity of coral-associated microbial communities in other species and habitats in future studies.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to H. C. Chung, Y. H. Wong, M. Li, J. P. Ren, and D. Lau from the Hong Kong University of Science and Technology and to the technical team from King Abdullah University of Science and Technology for providing technical help during sample collection. We also thank H. C. Chung, K. Cheng, K. Pan, and Y. K. Tam for help with the nutrient and heavy metal analyses. We greatly appreciate the constructive discussion on ordination analysis with C. ter Braak from Wageningen University and Research Centre. We thank Virginia Unkefer and Soumaya Belkharchouche for English editing.
This publication was based on work supported by the National Basic Research Program of China (973 Program, no. 2012CB417304), an award (no. SA-C0040/UK-C001) made by King Abdullah University of Science and Technology (KAUST), and the SKLMP seed collaborative research fund (CITYU12SC01) (P.-Y.Q.).
Footnotes
Published ahead of print 3 August 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Albarakati AMA. 2009. Water exchange of Sharm Obhur, Jeddah, Red Sea. J. King Abdulaziz Univ. Mar. Sci. 20:49–58 [Google Scholar]
- 2. Apprill A, Marlow HQ, Martindale MQ, Rappé MS. 2009. The onset of microbial associations in the coral Pocillopora meandrina. ISME J. 3:685–699 [DOI] [PubMed] [Google Scholar]
- 3. Arotsker L, et al. 2009. Vibrio sp. as a potentially important member of the Black Band Disease (BBD) consortium in Favia sp. corals. FEMS Microbiol. Ecol. 70:515–524 [DOI] [PubMed] [Google Scholar]
- 3a. Baas-Becking LGM. 1934. Geobiologie of inleiding tot de milieukunde. W. P. Van Stockum & Zoon, The Hague, Netherlands [Google Scholar]
- 4. Benayahu Y, Achituv Y, Berner T. 1988. Embryogenesis and acquisition of algal symbionts by planulae of Xenia umbellata (Octocorallia: Alcyonacea). Mar. Biol. 100:93–101 [Google Scholar]
- 5. Ben-Haim Y, et al. 2003. Vibrio coralliilyticus sp. nov., a temperature-dependent pathogen of the coral Pocillopora damicornis. Int. J. Syst. Evol. Microbiol. 53:309–315 [DOI] [PubMed] [Google Scholar]
- 6. Binladen J, et al. 2007. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One 2:e197 doi:10.1371/journal.pone.0000197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bourne D, Iida Y, Uthicke S, Smith-Kuene C. 2008. Changes in coral-associated microbial communities during a bleaching event. ISME J. 2:350–363 [DOI] [PubMed] [Google Scholar]
- 8. Bourne DG, Munn CB. 2005. Diversity of bacteria associated with the coral Pocillipora damicornis from the Great Barrier Reef. Environ. Microbiol. 7:1162–1174 [DOI] [PubMed] [Google Scholar]
- 9. Braakter CJF, Šmilauer P. 2002. CANOCO reference manual and CanoDraw for Windows user's guide: software for canonical community ordination (version 4.5). Microcomputer Power, Ithaca, NY [Google Scholar]
- 10. Caporaso JG, et al. 2009. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caporaso JG, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castillo I, Lodeiros C, Nunez M, Campos I. 2001. In vitro evaluation of antibacterial substances produced by bacteria isolated from different marine organisms. Rev. Biol. Trop. 49:1213–1221 [PubMed] [Google Scholar]
- 13. Ceh J, van Keulen M, Bourne DG. 2011. Coral associated bacterial communities on Ningaloo Reef, Western Australia. FEMS Microbiol. Ecol. 75:134–144 [DOI] [PubMed] [Google Scholar]
- 14. Cervino JM, et al. 2004. Relationship of Vibrio species infection and elevated temperatures to yellow blotch/band disease in Caribbean corals. Appl. Environ. Microbiol. 70:6855–6864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chiu JMY, Thiyagarajan V, Pechenik JA, Qian PY. 2007. The influence of temperature and salinity on microbial film development and metamorphosis of the prosobranch gastropod Crepidula onyx. Mar. Biol. 151:1417–1431 [Google Scholar]
- 16. de Hoon MJL, Imoto S, Nolan J, Miyano S. 2004. Open source clustering software. Bioinformatics 20:1453–1454 [DOI] [PubMed] [Google Scholar]
- 17. DiSalvo LH. 1969. Isolation of bacteria from the corallum of Porites lobata (Vaughn) and its possible significance. Am. Zool. 9:735–740 [Google Scholar]
- 18. Ducklow HW, Mitchel R. 1979. Bacterial populations and adaptations in the mucus layers on living corals. Limnol. Oceanogr. 24:715–725 [Google Scholar]
- 19. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461 [DOI] [PubMed] [Google Scholar]
- 21. Edlund A, Soule T, Sjöling S, Jansson JK. 2006. Microbial community structure in polluted Baltic Sea sediments. Environ. Microbiol. 8:223–232 [DOI] [PubMed] [Google Scholar]
- 22. Gomez-Alvarez V, Teal TK, Schmidt TM. 2009. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 3:1314–1317 [DOI] [PubMed] [Google Scholar]
- 23. Guppy R, Bythell JC. 2006. Environmental effects on bacterial diversity in the surface mucus layer of the reef coral Montastraea faveolata. Mar. Ecol. Prog. Ser. 328:133–142 [Google Scholar]
- 24. Haas BJ, et al. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21:494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harder T, Lau SCK, Dobretsov S, Fang TK, Qian PY. 2003. A distinctive epibiotic bacterial community on the soft coral Dendrohephthya sp. and antibacterial activity of coral tissue extracts suggest a chemical mechanism against bacteria epibiosis. FEMS Microbiol. Ecol. 43:337–347 [DOI] [PubMed] [Google Scholar]
- 26. Harrison PL, Wallace CC. 1990. Reproduction, dispersal and recruitment of scleractinian corals, p 133–207 In Dubinsky Z. (ed), Coral reefs, ecosystem of the world. Elsevier, Amsterdam, Netherlands [Google Scholar]
- 27. Henriques IS, et al. 2006. Seasonal and spatial variability of free-living bacterial community composition along an estuarine gradient (Ria de Aveiro, Portugal). Estuarine Coastal Shelf Sci. 68:139–148 [Google Scholar]
- 28. Hirose M, Kinzie RA, III, Hidaka M. 2000. Early development of zooxanthella-containing eggs of the corals Pocillopora verrucosa and P. eydouxi with special reference to the distribution of zooxanthellae. Biol. Bull. 199:68–75 [DOI] [PubMed] [Google Scholar]
- 29. Hirose M, Yamamoto H, Nonaka M. 2008. Metamorphosis and acquisition of symbiotic algae in planula larvae and primary polyps of Acropora spp. Coral Reefs 27:247–254 [Google Scholar]
- 30. Hong MJ, Yu YT, Chen CA, Chiang PW, Tang SL. 2009. Influence of species specificity and other factors on bacteria associated with the coral Stylophora pistillata in Taiwan. Appl. Environ. Microbiol. 75:7797–7806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson CN, et al. 2010. Relationships between environmental factors and pathogenic vibrios in the northern Gulf of Mexico. Appl. Environ. Microbiol. 76:7076–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaspar CW, Tamplin ML. 1993. Effects of temperature and salinity on the survival of Vibrio vulnificus in seawater and shellfish. Appl. Environ. Microbiol. 59:2425–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kellogg CA. 2004. Tropical Archaea: diversity associated with the surface microlayer of corals. Mar. Ecol. Prog. Ser. 273:81–88 [Google Scholar]
- 34. Kimes NE, Van Nostrand JD, Weil E, Zhou J, Morris PJ. 2010. Microbial functional structure of Montastraea faveolata, an important Caribbean reef-building coral, differs between healthy and yellow-band diseased colonies. Environ. Microbiol. 12:541–556 [DOI] [PubMed] [Google Scholar]
- 35. Koren O, Rosenberg E. 2006. Bacteria associated with mucus and tissues of the coral Oculina patagonica in summer and winter. Appl. Environ. Microbiol. 72:5254–5259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kunin V, Engelbrektson Am Ochman H, Hugenholtz P. 2010. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12:118–123 [DOI] [PubMed] [Google Scholar]
- 37. Kushmaro A, Rosenberg E, Fine M, Loya Y. 1997. Bleaching of the coral Oculina patagonica by Vibrio AK-1. Mar. Ecol. Prog. Ser. 147:159–165 [Google Scholar]
- 38. Kushmaro A, Banin E, Loya Y, Stackebrandt E, Rosenberg E. 2001. Vibrio shiloi sp. nov., the causative agent of bleaching of the coral Oculina patagonica. Int. J. Syst. Evol. Microbiol. 51:1383–1388 [DOI] [PubMed] [Google Scholar]
- 39. Lee OO, Chui PY, Wong YH, Pawlik JR, Qian PY. 2009. Evidence for vertical transmission of bacterial symbionts from adult to embryo in the Caribbean sponge Svenzea zeai. Appl. Environ. Microbiol. 75:6147–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lepš J, Šmilauer P. 2003. Multivariate analysis of ecological data using CANOCO. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 41. Lesser MP, Mazel CH, Gorbunov MY, Falkowski PG. 2004. Discovery of symbiotic nitrogen-fixing cyanobacteria in corals. Science 305:997–1000 [DOI] [PubMed] [Google Scholar]
- 42. Lieske E, Myers RF. 2004. Coral reef guide; Red Sea. Harper Collins, London, United Kingdom [Google Scholar]
- 43. Lins-de-Barros MM, et al. 2010. Archaea, bacteria and algal plastids associated with the reef-building corals Siderastrea stellata and Mussismilia hispida from Búzios, South Atlantic Ocean, Brazil. Microb. Ecol. 59:523–532 [DOI] [PubMed] [Google Scholar]
- 44. Lipp EK, et al. 2002. Preliminary evidence for human fecal contamination in corals of the Florida Keys, U. S. A. Mar. Pollut. Bull. 44:666–670 [DOI] [PubMed] [Google Scholar]
- 45. Littman RA, Willis BL, Pfeffer C, Bourne DG. 2009. Diversities of coral-associated bacteria differ with location, but not species, for three acroprid corals on the Great Barrier Reef. FEMS Microbiol. Ecol. 68:152–163 [DOI] [PubMed] [Google Scholar]
- 46. Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lozupone C, Hamady M, Knight R. 2006. UniFrac: an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Margulies M, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Motes ML, et al. 1998. Influence of water temperature and salinity on Vibrio vulnificus in Northern Gulf and Atlantic Coast oysters (Crassostrea virginica). Appl. Environ. Microbiol. 64:1459–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mouchka ME, Hewson I, Harvell CD. 2010. Coral-associated bacterial assemblages: current knowledge and the potential for climate-driven impacts. Integr. Comp. Biol. 50:662–674 [DOI] [PubMed] [Google Scholar]
- 51. Muyzer G, Smalla K. 1998. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 73:127–141 [DOI] [PubMed] [Google Scholar]
- 52. Neulinger SC, Järnegren Ludvigsen JM, Lochte K, Dullo WC. 2008. Phenotype-specific bacterial communities in the cold-water coral Lophelia pertusa (Scleractinia) and their implications for the coral's nutrition, health, and distribution. Appl. Environ. Microbiol. 74:7272–7285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Patterson KL, et al. 2002. The etiology of white pox, a lethal disease of the Caribbean elkhorn coral, Acropora palmate. Proc. Natl. Acad. Sci. 99:8725–8730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Price MN, Dehal PS, Arkin AP. 2009. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26:1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pruesse E, et al. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Raina JB, Tapiolas D, Willis BL, Bourne DG. 2009. Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl. Environ. Microbiol. 75:3492–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Randa MA, Polz MF, Lim E. 2004. Effects of temperature and salinity on Vibrio vulnificus population dynamics as assessed by quantitative PCR. Appl. Environ. Microbiol. 70:5469–5476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Reeder J, Knight R. 2010. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat. Methods 7:668–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Research Planning Inc., Baird and Associates 2008. Jeddah coastal assessment. Report prepared for the Presidency of Meteorology and Environment, Kingdom of Saudi Arabia, p 311 Research Planning Inc., Baird and Associates, Saudi Arabia [Google Scholar]
- 60. Risk MJ, Sherwood OA, Nairn R, Gibbons C. 2009. Tracking the record of sewage discharge off Jeddah, Saudi Arabia, since 1950, using stable isotope records from antipatharians. Mar. Ecol. Prog. Ser. 397:219–226 [Google Scholar]
- 61. Ritchie KB, Smith GW. 1995. Preferential carbon utilization by surface bacterial communities from water mass, normal, and white-band diseased Acropora cervicornis. Mol. Mar. Biol. Biotechnol. 4:345–352 [Google Scholar]
- 62. Ritchie KB, Smith GW. 2004. Microbial communities of coral surface mucopolysaccharide layers, p 259–264 In Rosenberg E, Loya Y. (ed). Coral health and disease. Springer-Verlag, New York, NY [Google Scholar]
- 63. Rohwer F, Kelley S. 2004. Culture independent analysis of coral-associated microbes, p 265–277 In Rosenberg E, Loya Y. (ed). Coral health and disease. Springer-Verlag, New York, NY [Google Scholar]
- 64. Rohwer F, Breitbart M, Jara J, Azam F, Knowlton N. 2001. Diversity of bacteria associated with the Caribbean coral Montastraea franksi. Coral Reefs 20:85–91 [Google Scholar]
- 65. Rohwer F, Seguritan V, Azam F, Knowlton N. 2002. Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243:1–10 [Google Scholar]
- 66. Ronaghi M, Uhlen M, Nyren P. 1998. A sequencing method based on real-time pyrophosphate. Science 281:363–365 [DOI] [PubMed] [Google Scholar]
- 67. Rosenberg E, Koren O, Reshef L, Efrony R, Zilber-Rosenberg I. 2007. The role of microorganisms in coral health, disease, and evolution. Nat. Rev. Microbiol. 5:355–362 [DOI] [PubMed] [Google Scholar]
- 68. Rothberg JM, Leamon JH. 2008. The development and impact of 454 sequencing. Nat. Biotechnol. 26:1117–1124 [DOI] [PubMed] [Google Scholar]
- 69. Santavy DL. 1995. The diversity of microorganisms associated with marine invertebrates and their roles in the maintenance of ecosystems, p 211–229 In Allsopp D, Colwell R, Hawksworth DL. (ed), Microbial diversity and ecosystem function. CAB International, Wallingford, United Kingdom [Google Scholar]
- 70. Schloss PD, Hay AG, Wilson DB, Gossett JM, Walker LP. 2005. Quantifying bacterial population dynamics in compost using 16S rRNA gene probes. Appl. Microbiol. Biotechnol. 66:457–463 [DOI] [PubMed] [Google Scholar]
- 71. Sharp KH, Distel D, Paul VJ. 2012. Diversity and dynamic of bacterial communities in early life stages of the Caribbean coral Porites astreoides. ISME J. 6:790–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shashar N, Cohen Y, Loya Y, Sar N. 1994. Nitrogen-fixation (acetylene-reduction) in stony corals: evidence for coral-bacteria interactions. Mar. Ecol. Prog. Ser. 111:259–264 [Google Scholar]
- 73. Sogin ML, et al. 2006. Microbial diversity in the deep sea and the underexplored ‘rare biosphere.’ Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sunagawa S, Woodley CM, Medina M. 2010. Threatened corals provide underexplored microbial habitats. PLoS One 5:e9554 doi:10.1371/journal.pone.0009554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sussman M, Willis BL, Victor S, Bourne DG. 2008. Coral pathogens identified for White Syndrome (WS) epizootics in the Indo-Pacific. PLoS One 6:e2393 doi:10.1371/journal.pone.0002393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sutherland KP, et al. 2010. Human sewage identified as likely source of white pox disease of the threatened Caribbean elkhorn coral, Acropora palmata. Environ. Microbiol. 12:1122–1131 [DOI] [PubMed] [Google Scholar]
- 77. U.S. Environmental Protection Agency 1994. Method 3051. Microwave assistant acid digestion of sediments, sludges, soils and oils. U.S. Environmental Protection Agency, Washington, DC [Google Scholar]
- 78. Vega Thurber R, et al. 2009. Metagenomic analysis of stressed coral holobionts. Environ. Microbiol. 11:2148–2163 [DOI] [PubMed] [Google Scholar]
- 79. Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Webster NS, Bourne D. 2007. Bacterial community structure associated with the Antarctic soft coral, Alconium antarcticum. FEMS Microbiol. Ecol. 59:81–94 [DOI] [PubMed] [Google Scholar]
- 81. Wegley L, Edwards R, Rodriguez-Brito B, Liu H, Rohwer F. 2007. Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ. Microbiol. 9:2707–2719 [DOI] [PubMed] [Google Scholar]
- 82. Yakimov MM, et al. 2006. Phylogenetic survey of metabolically active microbial communities associated with the deep-sea Lophelia pertusa from the Apulian plateau, Central Mediterranean Sea. Deep Sea Res. 53:62–75 [Google Scholar]
- 83. Zhou JZ, et al. 2011. Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 5:1303–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.