

Abstract
Counterselection systems facilitate marker-free genetic modifications in microbes by enabling positive selections for both the introduction of a marker gene into the microbe and elimination of the marker from the microbe. Here we report a counterselection system for Geobacillus kaustophilus HTA426, established through simultaneous disruption of the pyrF and pyrR genes. The pyrF gene, essential for pyrimidine biosynthesis and metabolization of 5-fluoroorotic acid (5-FOA) to toxic metabolites, was disrupted by homologous recombination. The resultant MK54 strain (ΔpyrF) was auxotrophic for uracil and resistant to 5-FOA. MK54 complemented with pyrF was prototrophic for uracil but insensitive to 5-FOA in the presence of uracil. To confer 5-FOA sensitivity, the pyrR gene encoding an attenuator to repress pyrimidine biosynthesis by sensing uracil derivatives was disrupted. The resultant MK72 strain (ΔpyrF ΔpyrR) was auxotrophic for uracil and resistant to 5-FOA. MK72 complemented with pyrF was prototrophic for uracil and 5-FOA sensitive even in the presence of uracil. The results suggested that pyrF could serve as a counterselection marker in MK72, which was demonstrated by efficient marker-free integrations of heterologous β-galactosidase and α-amylase genes. The integrated genes were functionally expressed in G. kaustophilus and conferred new functions on the thermophile. This report describes the first establishment of a pyrF-based counterselection system in a Bacillus-related bacterium, along with the first demonstration of homologous recombination and heterologous gene expression in G. kaustophilus. Our results also suggest a new strategy for establishment of counterselection systems.
INTRODUCTION
The genus Geobacillus comprises aerobic or facultatively anaerobic, Gram-positive, thermophilic bacilli that were reclassified from the genus Bacillus in 2001 (24). Members of this genus have been isolated from a wide range of environments, including temperate soils, as well as natural and artificial hot environments (4, 21, 35). This implies great environmental adaptability, which is supported by the remarkable properties of isolated Geobacillus species, such as ethanol tolerance (10), arsenate resistance (7), and the ability to degrade long-chain alkanes (35) and herbicides (21). Because of these properties, along with their thermophilicity and catabolic versatility, Geobacillus spp. have attracted interest as high-temperature bioprocessing tools, exemplified by ethanol production using genetically engineered Geobacillus thermoglucosidasius (6, 32). High-temperature bioprocesses have many advantages compared with temperate bioprocesses, including reduced risk of contamination, low energy expenditure for agitation and cooling, and easy removal of volatile products (36).
Geobacillus kaustophilus HTA426, isolated from deep sea sediments of the Mariana Trench (29), can grow aerobically between 42°C and 74°C, with an optimum growth temperature of 60°C (30, 31). This thermophile can grow even in media containing more than 3% NaCl (28–30). Its growth is as rapid as that of Escherichia coli and Bacillus subtilis. Its low nutrient requirements and high ability to utilize various carbon sources have also been observed (28). The published genome sequence of G. kaustophilus HTA426 (31) and our abundant knowledge of related bacilli, such as B. subtilis, enable us to predict gene functions in strain HTA426. Thus, strain HTA426 has great potential as a thermophilic host for various high-temperature bioprocesses and as a model for biological studies of this genus. However, this strain has not yet been utilized effectively, primarily because of inadequate genetic tools.
We have undertaken to develop genetic tools for the effective utilization of strain HTA426. We previously reported plasmid transformation of this strain using conjugative transfer from E. coli (28). In the present study, we have established a pyrF-based counterselection system, which enables positive selections for both introduction of a pyrF marker into the microbe and elimination of the marker, thereby facilitating marker-free genetic modifications. The pyrF gene (ura3 in eukaryotes) encodes orotidine 5′-phosphate decarboxylase, which is involved in de novo biosynthesis of pyrimidine-related metabolites, such as UMP, UDP, and UTP (Fig. 1). In Saccharomyces cerevisiae, ura3 deficiency causes starvation for these essential metabolites, although this can be circumvented by uracil supplementation because uracil is converted to UMP by uracil phosphoribosyltransferase. ura3 is also responsible for the toxicity of 5-fluoroorotic acid (5-FOA). One reason for this is that 5-fluorouridine 5′-monophosphate produced from 5-FOA by Ura3 is further metabolized into 5-fluorodeoxyuridine 5′-monophosphate, a potent inhibitor of thymidylate synthetase (2). Therefore, ura3 deficiency confers uracil auxotrophy and 5-FOA resistance, permitting counterselection using a ura3 marker in ura3-deficient microbes (3). Following its use for counterselection in S. cerevisiae, pyrF- or ura3-based counterselections have been demonstrated in many microbes (5, 11, 19, 20, 25, 27, 33). However, there are no reports of pyrF-based counterselections in Bacillus-related bacteria, possibly because of their resistance to 5-FOA (26).
Fig 1.

Proposed pathway of orotic acid metabolism in G. kaustophilus HTA426, based on the B. subtilis pathway. Orotic acid is metabolized to UMP by PyrE and PyrF. These enzymes also convert 5-FOA to 5-fluorouridine monophosphate, which is further metabolized to the toxic metabolite 5-fluorodeoxyuridine 5′-monophosphate. pyrR encodes an mRNA-binding attenuator that negatively regulates pyr expression by sensing UMP or UTP (34). UMP is also produced from uracil by uracil phosphoribosyltransferase.
Here we report the first establishment of a pyrF-based counterselection in a Bacillus-related bacterium, along with the first demonstration of homologous recombination and heterologous gene expression in G. kaustophilus. This was achieved through disruptions of pyrF and also of pyrR, which encodes the negative regulator of pyr expression (34). Our results not only increase the potential of strain HTA426 for biological studies and bioprocessing applications, but also suggest a new strategy for establishing counterselection systems in pyrR-possessing microbes.
MATERIALS AND METHODS
Bacterial strains, culture conditions, plasmids, and primers.
Table 1 lists the strains and plasmids used in this study. Table 2 lists the primers used. E. coli strains were grown at 37°C in Luria-Bertani (LB) medium supplemented with the appropriate antibiotics. G. kaustophilus strains were grown at 60°C overnight in LB medium or in minimal medium (MM) comprising 0.3 g/liter K2SO4, 2.5 g/liter Na2HPO4 · 12H2O, 1 g/liter NH4Cl, 0.1% (vol/vol) trace element solution (1), 1 g/liter Casamino Acids (Difco), 0.4 g/liter MgSO4, 3 mg/liter MnCl2 · 4H2O, 5 mg/liter CaCl2 · 2H2O, 7 mg/liter FeCl3 · 6H2O, 10 mM Tris-HCl (pH 7.5), and 10 g/liter d-glucose. Kanamycin (5 mg/liter), uracil (10 mg/liter), and 5-FOA (50 mg/liter) were added as required.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| G. kaustophilus | ||
| HTA426 | Wild type | JCM 12893 |
| MK54 | ΔpyrF | This study (Fig. 3) |
| MK54c | Mutant MK54 that integrates pGAM46 at GK0707 locus | This study |
| MK72 | ΔpyrF ΔpyrR | This study (Fig. 5) |
| MK72c | Mutant MK72 that integrates pGAM46 at GK0707 locus | This study |
| MK95 | Derivative of mutant MK72; GK0707::PsigA-bgaB | This study (Fig. 6) |
| MK96 | Derivative of mutant MK72; GK0707::bgaB | This study (Fig. 6) |
| MK109 | Derivative of mutant MK72; GK0707::PsigA-amyE | This study (Fig. 6) |
| MK304 | Derivative of mutant MK72; GK0707::amyE | This study (Fig. 6) |
| G. stearothermophilus | ||
| IAM11011 | Source of bgaB | ATCC 8005 |
| CU21 | Source of amyE | ATCC 12980 |
| E. coli BR408 | Donor strain for conjugative plasmid transfer | 28 |
| Plasmids | ||
| pUC19 | Cloning vector; Ampr | TaKaRa Bio (Fig. 2) |
| pCR4Blunt-TOPO | Cloning vector; Ampr Kmr | Invitrogen |
| pCR4B-TK101 | pCR4Blunt-TOPO carrying TK101 marker | This study |
| pCR4B-pyrF | pCR4Blunt-TOPO carrying pyrF marker | This study |
| pGKE11T | Derivative of pUC19; oriT Ampr | This study (Fig. 2) |
| pTK19 | Derivative of pGKE11T, oriT, Ampr, TK101 marker | This study (Fig. 2) |
| pΔpyrF | pTK19 carrying ΔpyrF fragment | This study (Fig. 2) |
| pΔpyrR | pTK19 carrying ΔpyrR fragment | This study (Fig. 2) |
| pGAM46 | Derivative of pGKE11T, GK0707 fragments, oriT, Ampr, pyrF marker | This study (Fig. 2) |
| pGAM46-bgaB | pGAM46 carrying bgaB between SphI and BamHI sites | This study |
| pGAM46-amyE | pGAM46 carrying amyE between SphI and BamHI sites | This study |
| pGAM47 | pGAM46 carrying PsigA between HindIII and SphI sites | This study |
| pGAM47-bgaB | pGAM47 carrying bgaB between SphI and BamHI sites | This study |
| pGAM47-amyE | pGAM47 carrying amyE between SphI and BamHI sites | This study |
Ampr and Kmr are genes coding for resistance to ampicillin and kanamycin, respectively. oriT is the conjugative transfer origin from pRK2013 (9). PsigA is the sigA promoter from G. kaustophilus HTA426. The TK101 marker represents the TK101 gene encoding thermostable kanamycin nucleotidyltransferase (18) under the control of the sigA promoter. The pyrF marker is the pyrF gene from G. kaustophilus HTA426 under the control of the sigA promoter.
Table 2.
Primers used in this study
| Primer | Sequence (5′→3′) |
|---|---|
| tk101F | GATACGGGGTTGTTCGTATGAGAATAGTGAATG |
| tk101R | AGGATCCTCATCGTTCAAAATGGTATG |
| sigAF1 | AGGATCCCTTCGCCTCATCCGCACGATTTC |
| sigAR1 | CATTCACTATTCTCATACGAACAACCCCGTATC |
| sigAF2 | AAGATCTCTTCGCCTCATCCGCACGATTTC |
| sigAR2 | GAACGGCGTGTGCATACGAACAACCCCGTATC |
| sigAF3 | GAAGCTTCTTCGCCTCATCCGCACGATTTC |
| sigAR3 | GCCGCATGCGAACAACCCCGTATCGTATT |
| 1155F | GATACGGGGTTGTTCGTATGCACACGCCGTTC |
| 1155R | AAGATCTCTAAGTGGGGGTAGTTGAC |
| oriTF | GCCACATGTCCGCCTTTTCCTCAATCGCTC |
| oriTR | GCCACATGTAATGAAATAAGATCACTACC |
| mfbF | GTATCAGCTCACTCAATTGCGGTGATCAAGATCTCCACAGAATCAGGGG |
| mfbR | CCCCTGATTCTGTGGAGATCTTGATCACCGCAATTGAGTGAGCTGATAC |
| 1155upF | GCCGAATTCCTTTGCCGACATCGGCTAC |
| 1155upR | GGCGGATCCATCAAGCGCGACAATGAAC |
| 1155dwF | GGCGGATCCCGTGCGCTTGGTTCTGAC |
| 1155dwR | GGCGCATGCCCGTTCGCTAACAACAGCTG |
| 1147upF | GCCCTGCAGTAAGACACGATCACATTTCACATTG |
| 1147upR | GGCGTCGACGGACGCCCCTTCAATTTG |
| 1147dwF | GCCGTCGACGGCATCGATCAAGTCTCC |
| 1147dwR | GGCGGATCCATCCATCGGAATGCCGACCGCATG |
| 0707upF | GGACGTCCGCCGGCGGAACGGAC |
| 0707upR | GGACGTCATATTGTTAAACCGGTCGACC |
| 0707dwF | GGGATCCGACAAACGACCAGAACGTG |
| 0707dwR | GGGATCCTCCAATAAAAAGCCGCGCAG |
| bgaF | GCCGCATGCAAGTGTTATCCTCAATTTGTTAC |
| bgaR | CGGATCCCTAGTGGTGGTGGTGGTGGTGAACCTTCCCGGCTTCATCATG |
| amyF | GCATGCTAACGTTTCACCGCATC |
| amyR | AGATCTAGATCAAGGCCATGCCACCAAC |
| 2400F | AACGACCAGCTGCCAAAAAACAAGGAACTG |
| 3800R | TCTTTCCTGCGTTATCCCCTGATTCTGTGG |
| −20F | TCCGAATGCATAGGAAGGAGCTGGAAGACC |
| 700R | TTTTGCGTCAGCCCCTTTTCGCTTGCGTTG |
Plasmid introduction into G. kaustophilus.
Plasmids were introduced into G. kaustophilus by conjugative transfer from E. coli BR408, as described previously (28). This method allows the transfer of oriT-containing plasmids from E. coli donors to G. kaustophilus recipients. Briefly, E. coli donors and G. kaustophilus recipients were grown in LB medium and mixed. Cells were collected and incubated on LB plates at 37°C overnight. The resultant cells were spread on the appropriate media and incubated at 60°C to isolate transconjugants.
Construction of plasmids pCR4B-TK101 and pCR4B-pyrF.
An upstream region of dnaG to sigA (GK2483 to GK2482) that encodes housekeeping proteins (14) was amplified from the HTA426 chromosome using primers sigAF1 and sigAR1. This region was used as the sigA promoter described below. The TK101 gene, encoding thermostable kanamycin nucleotidyltransferase (18), was amplified from pSTE33 (23) using primers tk101F and tk101R. Two amplified fragments were combined by overlap extension PCR (15) and cloned into pCR4Blunt-TOPO to give pCR4B-TK101 containing the sigA promoter-TK101 sequence flanked by BamHI sites.
To obtain pCR4B-pyrF, the sigA promoter was amplified using primers sigAF2 and sigAR2. The pyrF (GK1155) gene was amplified using primers 1155F and 1155R. The two fragments were combined and cloned into pCR4Blunt-TOPO, resulting in pCR4B-pyrF. This plasmid contained sigA promoter-pyrF sequence flanked by BglII sites.
Construction of plasmids pGKE11T, pTK19, pΔpyrF, and pΔpyrR.
Figure 2 is a schematic representation of the plasmid construction procedure. The oriT region (essential for conjugative transfer) was amplified from pRK2013 (9) using primers oriTF and oriTR and cloned into the PciI site of pUC19. To obtain pGKE11T, MunI, FbaI, and BglII sites were introduced into the resulting plasmid using the QuikChange site-directed mutagenesis kit (Stratagene) and primers mfbF and mfbR. The TK101 marker was excised from pCR4B-TK101 with BamHI and cloned into the BglII site of pGKE11T to yield pTK19.
Fig 2.

Construction of plasmids pGKE11T, pGAM46, pTK19, pΔpyrF, and pΔpyrR. pGKE11T was constructed from pUC19 by introduction of oriT and restriction sites. pGAM46 was constructed from pGKE11T by introduction of the pyrF marker and the GK0707 upstream (GK0707′) and downstream (′GK0707) fragments. pTK19 was constructed by introduction of the TK101 marker into pGKE11T and was used for construction of pΔpyrF and pΔpyrR. pΔpyrF contains pyrF upstream (pyrF up) and downstream (pyrF dw) regions to form the ΔpyrF sequence. pΔpyrR contains pyrR upstream (pyrR up) and downstream (pyrR dw) regions to form the ΔpyrR sequence. The ampicillin resistance gene (Ampr), the replicon from pUC19 (ori), and the conjugative transfer origin from pRK2013 (oriT) are shown, along with the relevant restriction sites. “MCS” denotes the multiple cloning sites of pUC19.
pΔpyrF was constructed for a pyrF in-frame deletion in strain HTA426. The pyrF upstream region (2.0 kb) was amplified using primers 1155upF and 1155upR and trimmed with EcoRI and BamHI. The downstream region (2.0 kb) was amplified using the primers 1155dwF and 1155dwR and trimmed with BamHI and SphI. The upstream fragment was cloned between the EcoRI and BamHI sites of pTK19, and the downstream fragment was cloned between the BamHI and SphI sites, to yield pΔpyrF. This plasmid contained a defective gene with the sequence 5′-GGATCC-3′ replacing pyrF codons 11 to 202.
pΔpyrR was constructed for a pyrR in-frame deletion. The pyrR (GK1147) upstream region (1.3 kb) was amplified using primers 1147upF and 1147upR and trimmed with PstI and SalI. The downstream region (2.0 kb) was amplified using primers 1147dwF and 1147dwR and trimmed with SalI and BamHI. The upstream fragment was cloned between the PstI and SalI sites of pTK19, and the downstream fragment was cloned between the SalI and BamHI sites, to yield pΔpyrR. This plasmid contained a defective pyrR gene lacking codons 62 to 168.
Construction of pGAM plasmids.
pGAM plasmids were constructed for marker-free integrations of heterologous genes at the GK0707 locus. The GK0707 upstream region (1.2 kb) was amplified using primers 0707upF and 0707upR and trimmed with AatII. The GK0707 downstream region (1.2 kb) was amplified using primers 0707dwF and 0707dwR and trimmed with BamHI. The upstream and downstream fragments were cloned into the AatII and FbaI sites of pGKE11T, respectively, in the correct direction. The pyrF marker was excised from pCR4B-pyrF with BglII and cloned into the BglII site of the resulting plasmid, to yield pGAM46 (Fig. 2).
The sigA promoter was amplified using primers sigAF3 and sigAR3. The fragment was trimmed with HindIII and SphI and cloned into the HindIII and SphI sites of pGAM46 to yield pGAM47. The bgaB gene (GenBank accession no. M13466) (16) was amplified from the Geobacillus stearothermophilus IAM11011 chromosome using primers bgaF and bgaR. After being cloned into pCR4Blunt-TOPO, the bgaB sequence was excised with SphI and BamHI and subcloned between the SphI and BamHI sites of pGAM46 and pGAM47, to yield pGAM46-bgaB and pGAM47-bgaB, respectively.
The amyE gene (GenBank accession no. M11450) (22) was amplified from the G. stearothermophilus CU21 chromosome using primers amyF and amyR. After being cloned into pCR4Blunt-TOPO, the amyE sequence was excised with SphI and BglII and cloned between the SphI and BamHI sites of pGAM46 and pGAM47 to yield pGAM46-amyE and pGAM47-amyE, respectively.
In-frame deletions of pyrF and pyrR.
pΔpyrF was introduced into strain HTA426 to obtain kanamycin-resistant transconjugants that had integrated pΔpyrF into the chromosome as a result of the first crossover. A transconjugant was subcultured in kanamycin-free LB medium for the second crossover to remove pΔpyrF from the chromosome. After four sequential subcultures, cells were colonized on kanamycin-free LB plates and analyzed to isolate kanamycin-sensitive clones. PCR analysis of their chromosomes revealed some ΔpyrF mutants, one of which was designated mutant strain MK54.
pΔpyrR was introduced into the mutant MK54 to obtain kanamycin-resistant transconjugants. Following a procedure similar to that for MK54 construction, pyrR in MK54 was disrupted by reciprocal crossovers. One of the mutants obtained was designated mutant strain MK72.
Southern hybridization.
Chromosomal DNA was digested and separated on a 0.8% (wt/vol) agarose gel by electrophoresis. DNA was transferred to a nylon membrane and hybridized with digoxigenin (DIG)-labeled DNA probes that were synthesized using a PCR DIG probe synthesis kit (Roche). Hybridized DNA was detected by the chromogenic method using a DIG nucleic acid detection kit (Roche).
Transcription analysis.
Total RNA was isolated from G. kaustophilus cells according to a method used for B. subtilis (17). The purified RNA was separated by electrophoresis, transferred to a nylon membrane, and hybridized with digoxigenin-labeled RNA probes synthesized using a DIG RNA labeling kit (Roche). The hybridized RNA was detected by the chemiluminescence method using a DIG luminescence detection kit (Roche).
Marker-free integration of heterologous genes.
Gene integrations were performed by pyrF-based counterselection using pGAM plasmids. The plasmid was introduced into the mutant MK72 cells by conjugative transfer. Cells were grown on MM plates to obtain uracil-prototrophic transconjugants that had integrated the pGAM plasmid at the GK0707 locus. A transconjugant was subcultured twice in LB medium for the second crossover and then once in MM containing 1 mg/liter uracil and 5-FOA to reduce false positives (see below). Aliquots (102 to 103 cells) were incubated on MM plates containing 10 mg/liter uracil and 5-FOA. Chromosomes were isolated from grown colonies and analyzed for the mutant genotype by PCR.
Sequencing analysis of pyrE and pyrF regions.
During pyrF-based counterselection, a significant number of 5-FOA-resistant, uracil-prototrophic, false positives were observed. To analyze spontaneous mutations in pyrE and pyrF genes in these clones, pyrF marker (sigA promoter-pyrF) and pyrE (ΔpyrF-pyrE) regions were amplified by PCR using primers 2400F and 3800R and −20F and 700R, respectively. The amplified fragments were cloned into pCR4Blunt-TOPO and sequenced.
In vitro β-galactosidase assay.
G. kaustophilus was cultured in LB medium until the late logarithmic phase. Harvested cells were sonicated in buffer (50 mM sodium phosphate, pH 6.0) and centrifuged to remove cell debris. β-Galactosidase activities in crude extracts were determined using p-nitrophenyl-β-d-galactopyranoside as the chromogenic substrate. The reaction mixture contained 50 mM sodium phosphate (pH 6.0) and 2 mM substrate in 100 μl. Reactions were performed at 60°C for 30 min and terminated by adding 100 μl ice-cold 2 M sodium carbonate. The amount of p-nitrophenol released was determined by measuring the absorbance at 405 nm with reference to an experimentally derived standard curve. One unit was defined as the amount of enzyme that released 1 pmol p-nitrophenol per s. Proteins were quantified by the Bradford method using a protein assay kit (Bio-Rad) with bovine serum albumin as the standard. Results are expressed as means ± standard deviations (SD) from four independent experiments.
In vitro α-amylase assay.
G. kaustophilus was cultured in LB medium for 24 h. Harvested cells were sonicated in buffer [100 mM 3-(N-morpholino)propanesulfonic acid sodium, pH 6.9] and centrifuged. α-Amylase activities in crude extracts and the culture supernatant were determined using an EnzChek Ultra amylase assay kit (Invitrogen) according to the manufacturer's instructions. The amount of fluorescent substance released from starch substrates was determined by measuring fluorescence (excitation at 485 nm and detection at 535 nm) with reference to an experimentally derived standard curve. One unit was defined as the amount of enzyme that released 1 pmol of fluorescent substance per s at 60°C. Results are expressed as the means ± SD from four independent experiments.
RESULTS
pyrF disruption in strain HTA426.
Strain HTA426 harbors pyr genes that are homologous to those of B. subtilis (Fig. 3A), implying that these genes are involved in de novo biosynthesis of pyrimidine-related metabolites. The pyrF (GK1155) gene was disrupted by a 0.6-kb in-frame deletion using pΔpyrF. pΔpyrF was introduced into strain HTA426 by conjugative transfer from E. coli BR408. The conjugation using 108 recipient cells yielded approximately 102 kanamycin-resistant transconjugants. Because pΔpyrF did not contain a functional replicon for G. kaustophilus, the plasmid should have been integrated into the chromosome through homologous recombination (first crossover) at either the pyrF upstream or downstream regions. After a transconjugant was subcultured under nonselective conditions for the second crossover, cells were analyzed for kanamycin sensitivity. Eight kanamycin-sensitive clones were identified among the 1,040 analyzed clones. PCR analysis showed that four of the eight clones were revertants with the wild-type genotype; however, the other four clones, including MK54, were ΔpyrF mutants. Southern analysis of MK54 confirmed the correct 0.6-kb deletion in pyrF (Fig. 3B). Mutant strain MK54 was auxotrophic for uracil and 5-FOA resistance, whereas MK54 complemented with pyrF (MK54c) was prototrophic for uracil (Fig. 3C). However, mutant strain MK54c and strain HTA426, as well as mutant strain MK54, could grow on MM plates containing uracil and 5-FOA (Fig. 4).
Fig 3.
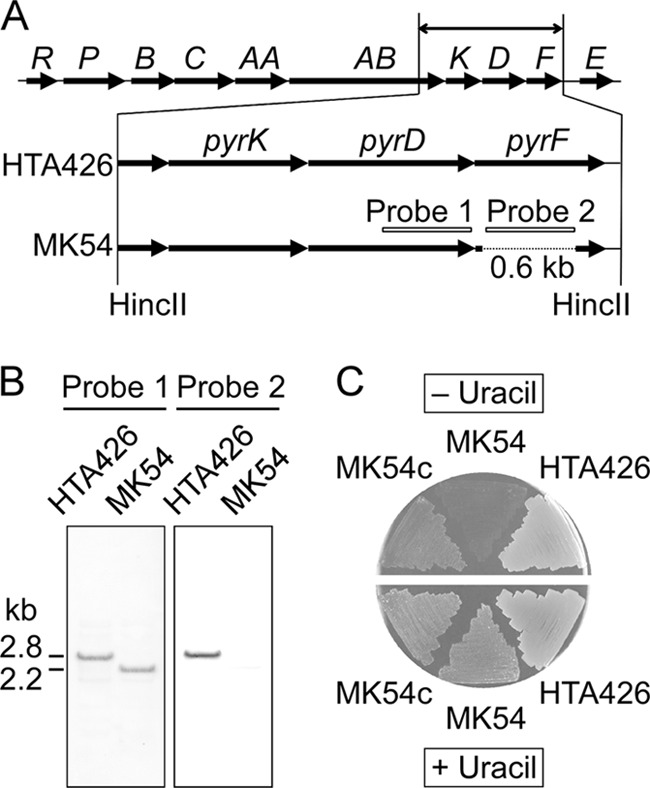
pyrF deletion in strain HTA426. (A) Gene organization of the pyr cluster (GK1147 to GK1157) in G. kaustophilus HTA426 and schematic diagram of construction of mutant strain MK54. The 0.6-kb region of pyrF was deleted by reciprocal crossovers. Probes 1 and 2 were used for Southern hybridization. (B) Southern hybridization to verify the correct deletion. Chromosomal DNA was digested with HincII. (C) Analysis of uracil auxotrophy. Strain HTA426, mutant strain MK54, and pyrF-complemented MK54 (MK54c) were incubated on MM plates with and without 10 mg/liter uracil.
Fig 4.
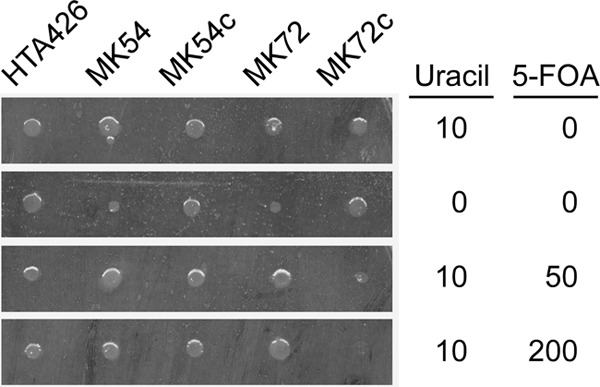
5-FOA sensitivity of strain HTA426 and mutant strains MK54, pyrF-complemented MK54 (MK54c), MK72, and pyrF-complemented MK72 (MK72c). Overnight cultures (105 cells) were applied as spots and incubated for 24 h on MM plates containing uracil and 5-FOA at the indicated concentrations (mg/liter).
pyrR disruption in mutant strain MK54.
In B. subtilis, pyrR encodes an mRNA-binding regulatory protein that negatively regulates pyr expression by sensing UMP or UTP (Fig. 1) (34). The pyrR gene in mutant strain MK54 was disrupted by a 0.3-kb in-frame deletion using pΔpyrR (Fig. 5A). The plasmid was introduced with a conjugation efficiency of approximately 1 × 10−6 recipient−1. After a transconjugant that had integrated pΔpyrR into the chromosome was cultured under nonselective conditions, 600 clones were analyzed to obtain 10 kanamycin-sensitive clones. PCR analysis showed that eight of the 10 clones had the MK54 genotype, and two clones, including MK72, were ΔpyrR mutants. Southern analysis of mutant strain MK72 confirmed the correct 0.3-kb deletion in pyrR (Fig. 5B). Mutant strain MK72 was auxotrophic for uracil and 5-FOA resistant (Fig. 4). Mutant strain MK72 complemented with pyrF (MK72c) was prototrophic for uracil. Most importantly, MK72c was unable to grow on MM plates containing uracil and 5-FOA.
Fig 5.
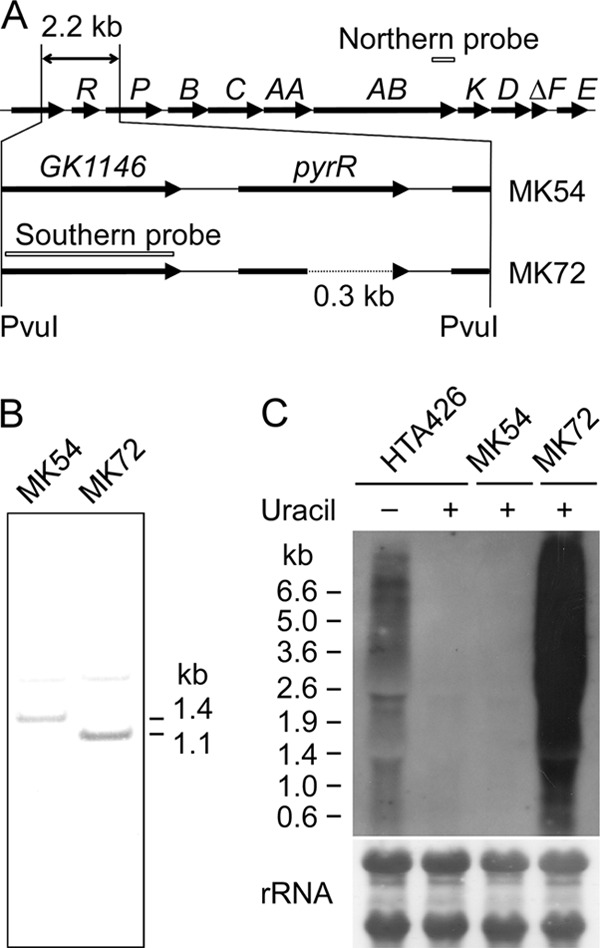
pyrR deletion in mutant strain MK54. (A) Schematic diagram of the construction of mutant strain MK72 from mutant strain MK54. The 0.3-kb region in pyrR was deleted by reciprocal crossovers. Probes used for Southern hybridization and northern analysis are indicated. (B) Southern hybridization to verify the correct deletion. Chromosomal DNA was digested with PvuI. (C) Northern analysis of pyr transcription in strain HTA426 and the mutant strains MK54 and MK72. Cells were cultured in MM with (+) or without (−) 10 mg/liter uracil.
Transcription of pyr genes.
Northern analysis (Fig. 5C) showed that pyr transcription in strain HTA426 was promoted in medium lacking uracil but repressed in a uracil-rich medium. Similar repression was observed in mutant strain MK54. However, pyr genes were transcribed in mutant strain MK72 even in uracil-rich medium.
Marker-free integration of bgaB.
pGAM47-bgaB was introduced into mutant strain MK72 to obtain uracil-prototrophic transconjugants that had integrated the plasmid at the GK0707 locus of the chromosome. One transconjugant was subcultured three times in LB medium and then incubated on MM plates containing uracil and 5-FOA. More than 20 colonies were obtained from a plate spread with approximately 103 cells. High-density incubation (>105 cells/plate) did not give rise to any apparent colonies. This may be attributed to the accumulation of toxic metabolites produced from 5-FOA by background cells. After colony isolation, eight colonies were analyzed for uracil auxotrophy. Four clones showed uracil auxotrophy, indicating pyrF elimination, although the other four clones were false positives that were uracil prototrophs (see below). PCR analysis revealed that two of the four positive clones had the MK72 genotype and that the other two clones had integrated bgaB at the GK0707 locus (Fig. 6A). A mutant was designated mutant strain MK95 and analyzed further. Similarly, mutant strain MK96 was constructed using pGAM46-bgaB. This construction also yielded 50% false positives.
Fig 6.
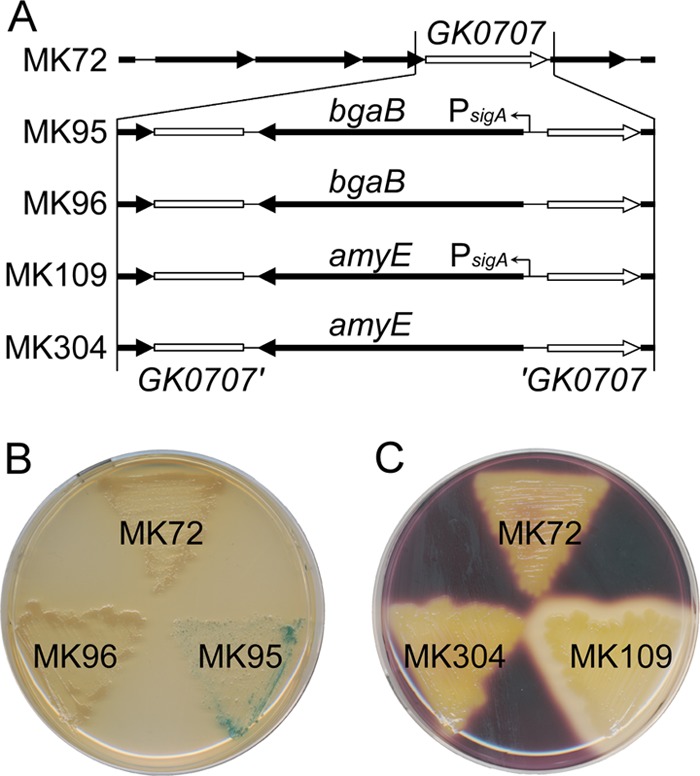
Marker-free integrations of heterologous genes and their expression. (A) Gene organization surrounding GK0707 and a schematic diagram of construction of mutant strains MK95, MK96, MK109, and MK304. Mutant strains MK95 and MK109 had integrated bgaB and amyE, respectively, under the control of the sigA promoter (PsigA). Mutant strains MK96 and MK304 had integrated bgaB and amyE, respectively, without the promoter. (B) β-Galactosidase assays of G. kaustophilus mutants. Cells were incubated for 48 h on an LB plate containing 200 mg/liter X-Gal. (C) α-Amylase assays of G. kaustophilus mutants. Cells were incubated for 24 h on an LB plate containing 1% (wt/vol) soluble starch. Residual starch was stained with I2-KI solution.
Characterization of false positives.
False positives observed during mutant strain MK95 construction (see above) were prototrophic for uracil, 5-FOA resistant in the presence of uracil, and 5-FOA sensitive in the absence of uracil. They were sensitive to 5-fluorouracil in the presence or absence of uracil. PCR analysis showed that these false positives harbored the pyrF marker. Among eight false positives, one had a nonsynonymous change in the pyrF sequence (C→T at position 215 in the pyrF open reading frame [ORF], resulting in PyrF A72V), and another one had a synonymous change (T→A at position 12), but the other six clones had no mutations. No mutations were observed in the pyrE regions of the eight clones.
Marker-free integration of amyE.
pGAM47-amyE was introduced into mutant strain MK72. Following a procedure similar to that used for the bgaB integrations, a uracil-prototrophic transconjugant that had integrated pGAM47-amyE was subcultured in LB medium three times and screened for clones from which pyrF had been deleted. We analyzed 24 clones resistant to 5-FOA for uracil auxotrophy. However, all clones were false positives exhibiting uracil prototrophy.
Because the false positives were 5-FOA sensitive in the absence of uracil, we assumed that false positives frequently occurred under uracil-rich conditions and may be reduced under uracil-restricted conditions. Thus, a uracil-prototrophic transconjugant was subcultured twice in LB medium and then in MM containing a small amount of uracil (1 mg/liter) and 5-FOA. Grown cells were incubated on MM plates containing 10 mg/liter uracil and 5-FOA and analyzed for uracil auxotrophy without colony purification. All 48 analyzed colonies were uracil auxotrophs. Of these, eight clones were analyzed by PCR. Three clones were revertants to the MK72 genotype, and another five were mutants that had integrated amyE in the GK0707 locus. One mutant was designated mutant strain MK109 (Fig. 6A). By a similar procedure, mutant strain MK304 was constructed using pGAM46-amyE.
Effect of subculture conditions on pyrF elimination and generation of false positives.
To analyze the frequencies of pyrF elimination and false-positive generation, four uracil-prototrophic transconjugants that had integrated pGAM47-bgaB were again cultured in LB medium followed by MM containing 1 mg/liter uracil and 5-FOA (procedure A in Table 3). 5-FOA-resistant clones generated after subculture in LB medium accounted for <1% of the total cells, and approximately 63% of these were false positives. The proportions of 5-FOA-resistant clones and false positives were significantly increased and reduced, respectively, after subculture in MM with 1 mg/liter uracil and 5-FOA. Approximately 61% of the clones from which pyrF had been deleted were revertants to the MK72 genotype. We also examined subculture in MM with 10 mg/liter uracil (procedure B in Table 3). Procedure B yielded 5-FOA-resistant clones faster and more frequently than procedure A, although the ratio did not increase after further subculture in MM with 1 mg/liter uracil and 5-FOA. The frequencies of false positives and revertants were comparable in procedures A and B.
Table 3.
Effect of subculture conditions on pyrF elimination and generation of false positives
| Subculture | Medium | Frequency of occurrence (%)a |
||
|---|---|---|---|---|
| 5-FOA resistantb | False positivec | Revertantd | ||
| Procedure A | ||||
| 1 | LB | 0.01 ± 0.00 | ||
| 2 | LB | 0.45 ± 0.42 | ||
| 3 | LB | 0.29 ± 0.28 | 63 ± 5 | |
| 4 | +5-FOAe | 59 ± 53 | 7.8 ± 6.0 | 61 ± 17 |
| Procedure B | ||||
| 1 | MMf | 4.5 ± 0.9 | ||
| 2 | MM | 5.5 ± 3.9 | ||
| 3 | MM | 5.5 ± 2.9 | 86 ± 9 | |
| 4 | +5-FOA | 9.4 ± 5.6 | 8.1 ± 13 | 68 ± 12 |
The values shown are means ± SD from four independent experiments using transconjugants that had integrated pGAM47-bgaB.
The values shown are the number of colonies growing from subculture aliquots on MM plates containing 10 mg/liter uracil and 5-FOA compared with the number on LB plates (i.e., 5-FOA-resistant cells per total cells).
The values shown are the number of clones growing on MM plates without uracil per 48 5-FOA-resistant clones (i.e., uracil-prototrophic cells per 5-FOA-resistant cells).
The values shown are the number of clones exhibiting white colonies on LB plates with 200 mg/liter X-Gal per 32 uracil-auxotrophic clones generated (i.e., MK72 revertant cells per total cells from which pyrF had been eliminated).
MM containing 1 mg/liter uracil and 5-FOA.
MM containing 10 mg/liter uracil.
bgaB expression.
bgaB encoding thermostable β-galactosidase was used to examine heterologous gene expression in G. kaustophilus. Genes homologous to bgaB have not been identified in the HTA426 genome. bgaB was integrated into mutant strain MK72 to generate the mutant strains MK95 and MK96 (Fig. 6A). Their expression was analyzed by incubation on LB plates with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). Mutant strain MK95 produced blue pigment on the plate, although growth was slightly inhibited, whereas mutant strains MK72 and MK96 did not produce pigment (Fig. 6B). These results suggested that the heterologous gene was functionally expressed under the control of the sigA promoter in mutant strain MK95. bgaB expression was further confirmed by an in vitro β-galactosidase assay. The specific activity of crude extract from mutant strain MK95 was 74 ± 17 U/mg protein. Cultivation in MM had a negligible effect on specific activity (69 ± 5 U/mg protein), implying the functionality of the sigA promoter under multiple conditions. No activity was detected in the culture supernatant of mutant strain MK95 or in crude extracts of mutant strains MK72 or MK96 (<0.01 U/mg protein).
amyE expression.
amyE encoding a heterologous α-amylase was also used for expression analysis. This gene was integrated into mutant strain MK72 with and without the sigA promoter, to generate mutant strains MK109 and MK304, respectively. Expression was analyzed by soluble starch degradation on LB plates (Fig. 6C). Although putative α-amylase genes are found in the HTA426 genome, mutant strains MK72 and MK304 showed no α-amylase activity. However, mutant strain MK109 degraded soluble starch, indicating α-amylase production. An in vitro assay was used to determine α-amylase localization. The total activity in the culture supernatant was 1,500 ± 250 U per 100-ml culture, while that in the crude extracts was 13 ± 1 U. Mutant strain MK72 had negligible activity in both the culture supernatant and crude extracts (<10 U). These results suggested that amyE was functionally expressed and that the product was secreted.
DISCUSSION
The gene products of pyrF and pyrE are essential for de novo biosynthesis of pyrimidine-related metabolites and for conversion of 5-FOA to toxic metabolites (Fig. 1); thus, pyrF deficiency confers uracil auxotrophy and 5-FOA resistance to microbes. This property enables a pyrF-based counterselection system that has been demonstrated in many microbes (3, 5, 11, 19, 20, 25, 27, 33). In order to establish a counterselection system for strain HTA426, the pyrF gene was disrupted using pΔpyrF. This plasmid was efficiently integrated in the chromosome, demonstrating homologous recombination in G. kaustophilus, and was then eliminated along with the pyrF deletion to yield mutant strain MK54, which was auxotrophic for uracil (Fig. 3C). A pyrF-complemented strain (MK54c) was prototrophic for uracil; however, MK54c and MK54 were able to grow on MM plates containing 5-FOA and uracil (Fig. 4). A similar result has been described in B. subtilis (26). The results showed that pyrF could serve as a genetic marker, but not as a counterselection marker in mutant strain MK54.
Both strains HTA426 and MK54c were unable to grow on MM plates containing 5-FOA without uracil (data not shown). This suggested the possibility that G. kaustophilus may not be resistant to 5-FOA, but rather the 5-FOA metabolism was repressed in the presence of uracil due to repression of pyr expression. In fact, pyr transcription in strain HTA426 and mutant strain MK54 was repressed in the presence of uracil (Fig. 5C), supporting this hypothesis. Since it is known that in B. subtilis, the pyrR product negatively regulates pyr expression by sensing UMP and UTP (Fig. 1) (34), we focused on pyrR as a key factor in the apparent 5-FOA resistance of G. kaustophilus and constructed mutant strain MK72 (ΔpyrF ΔpyrR). As expected, pyr transcription in mutant strain MK72 was insensitive to uracil (Fig. 5C). A pyrF-complemented strain of MK72 (MK72c) showed 5-FOA sensitivity even in the presence of uracil (Fig. 4). These results suggested that pyrF could be used as a counterselection marker in mutant strain MK72.
Counterselection in mutant strain MK72 was demonstrated by marker-free integrations of heterologous genes using pGAM plasmids. Integration and elimination of the plasmid were positively selected by uracil prototrophy and 5-FOA resistance, respectively. Although the counterselection produced significant numbers of false positives, these were greatly reduced by additional subculturing in MM with restricted uracil and 5-FOA following subculture in LB media. Detailed analysis indicated that the false positives were reduced to <10% by this procedure (Table 3). This counterselection system was very effective. It enabled us to obtain mutant strains MK95, MK96, MK109, and MK304 by analyzing only a few clones, whereas the obtaining of mutant strains MK54 and MK72 required analysis of more than 1,000 and 600 clones, respectively. Thus, we have established a pyrF-based counterselection system for G. kaustophilus HTA426 through pyrF and pyrR disruptions. This is the first report of a pyrF-based counterselection in Bacillus-related bacteria, although counterselections based on other strategies have been reported in B. subtilis (8, 26). Our results suggest that this approach may be applied to the establishment of pyrF-based counterselection systems in the many other organisms harboring pyrR, including Desulfotomaculum, Listeria, Clostridium, Enterococcus, and Bacillus-related bacteria.
False positives observed during counterselection were sensitive to 5-fluorouracil. Therefore, it was likely that metabolism from 5-fluorouridine 5′-monophosphate to toxic metabolites was functional in false positives. Grogan et al. (12, 13) reported that 5-FOA resulted in a low frequency of spontaneous mutations of pyr genes in Sulfolobus acidocaldarius. Some of the spontaneous mutants had a frameshift mutation in the pyrE gene and were uracil prototrophic and 5-FOA resistant, suggesting that false positives observed in this study may arise from pyrE mutations. We therefore analyzed the pyrF and pyrE regions of false positives; however, mutations observed in these regions were negligible. Thus, the 5-FOA resistance mechanism of the false positives is unclear. One possibility is that G. kaustophilus may have a minor mechanism for repressing pyrimidine biosynthesis and/or incorporation under uracil-rich conditions independently of pyrR. This idea is consistent with the fact that false positives are 5-FOA sensitive in the absence of uracil and are reduced in restricted uracil.
In addition to counterselection, this study demonstrates heterologous gene expression in G. kaustophilus (Fig. 6). The bgaB and amyE genes were functionally expressed in mutant strains MK95 and MK109, respectively, under the control of the sigA promoter. The amyE product was secreted into the culture supernatant, confirming the protein-secreting capability of this strain. These results raise the possibility that various thermostable proteins can be produced intracellularly or extracellularly in G. kaustophilus. The results of this study will facilitate genetic modification of G. kaustophilus HTA426, including gene disruptions and gene overexpression, and therefore may contribute to the effective utilization of this strain as a thermophilic host for various applications and as a model for biological studies of the genus.
ACKNOWLEDGMENTS
This work was supported by Special Coordination Funds for Promoting Science and Technology under the project Creation of Innovation Centers for Advanced Interdisciplinary Research Areas (Innovative Bioproduction Kobe), MEXT, Japan, and, in part, by KAKENHI (22310130).
Footnotes
Published ahead of print 10 August 2012
REFERENCES
- 1. Amartey SA, Leak DJ, Hartley BS. 1991. Development and optimization of a defined medium for aerobic growth of Bacillus stearothermophilus LLD-15. Biotechnol. Lett. 13:621–626 [Google Scholar]
- 2. Bisson LF, Thorner J. 1981. Thymidylate synthetase from Saccharomyces cerevisiae: purification and enzymatic properties. J. Biol. Chem. 256:2456–2462 [PubMed] [Google Scholar]
- 3. Boeke JD, LaCroute F, Fink GR. 1984. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197:345–346 [DOI] [PubMed] [Google Scholar]
- 4. Burgess SA, Lindsay D, Flint SH. 2010. Thermophilic bacilli and their importance in dairy processing. Int. J. Food Microbiol. 144:215–225 [DOI] [PubMed] [Google Scholar]
- 5. Cava F, Hidalgo A, Berenguer J. 2009. Thermus thermophilus as biological model. Extremophiles 13:213–231 [DOI] [PubMed] [Google Scholar]
- 6. Cripps RE, et al. 2009. Metabolic engineering of Geobacillus thermoglucosidasius for high yield ethanol production. Metab. Eng. 11:398–408 [DOI] [PubMed] [Google Scholar]
- 7. Cuebas M, Sannino D, Bini E. 2011. Isolation and characterization of an arsenic resistant Geobacillus kaustophilus strain from geothermal soils. J. Basic Microbiol. 51:364–371 [DOI] [PubMed] [Google Scholar]
- 8. Fabret C, Ehrlich SD, Noirot P. 2002. A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol. Microbiol. 46:25–36 [DOI] [PubMed] [Google Scholar]
- 9. Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. U. S. A. 76:1648–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fong JCN, et al. 2006. Isolation and characterization of two novel ethanol-tolerant facultative-anaerobic thermophilic bacteria strains from waste compost. Extremophiles 10:363–372 [DOI] [PubMed] [Google Scholar]
- 11. Galvão TC, de Lorenzo V. 2005. Adaptation of the yeast URA3 selection system to gram-negative bacteria and generation of a ΔbetCDE Pseudomonas putida strain. Appl. Environ. Microbiol. 71:883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grogan DW, Carver GT, Drake JW. 2001. Genetic fidelity under harsh conditions: analysis of spontaneous mutation in the thermoacidophilic archaeon Sulfolobus acidocaldarius. Proc. Natl. Acad. Sci. U. S. A. 98:7928–7933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grogan DW, Gunsalus RP. 1993. Sulfolobus acidocaldarius synthesizes UMP via a standard de novo pathway: results of a biochemical-genetic study. J. Bacteriol. 175:1500–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haldenwang WG. 1995. The sigma factors of Bacillus subtilis. Microbiol. Rev. 59:1–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hirata H, Fukazawa T, Negoro S, Okada H. 1986. Structure of a β-galactosidase gene of Bacillus stearothermophilus. J. Bacteriol. 166:722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Igo MM, Losick R. 1986. Regulation of a promoter that is utilized by minor forms of RNA polymerase holoenzyme in Bacillus subtilis. J. Mol. Biol. 191:615–624 [DOI] [PubMed] [Google Scholar]
- 18. Liao H, McKenzie T, Hageman R. 1986. Isolation of a thermostable enzyme variant by cloning and selection in a thermophile. Proc. Natl. Acad. Sci. U. S. A. 83:576–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lipscomb GL, et al. 2011. Natural competence in the hyperthermophilic archaeon Pyrococcus furiosus facilitates genetic manipulation: construction of markerless deletions of genes encoding the two cytoplasmic hydrogenases. Appl. Environ. Microbiol. 77:2232–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu H, Han J, Liu X, Zhou J, Xiang H. 2011. Development of pyrF-based gene knockout systems for genome-wide manipulation of the archaea Haloferax mediterranei and Haloarcula hispanica. J. Genet. Genomics 38:261–269 [DOI] [PubMed] [Google Scholar]
- 21. McMullan G, et al. 2004. Habitat, applications and genomics of the aerobic, thermophilic genus Geobacillus. Biochem. Soc. Trans. 32:214–217 [DOI] [PubMed] [Google Scholar]
- 22. Nakajima R, Imanaka T, Aiba S. 1985. Nucleotide sequence of the Bacillus stearothermophilus α-amylase gene. J. Bacteriol. 163:401–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Narumi I, et al. 1993. Construction of a new shuttle vector pSTE33 and its stabilities in Bacillus stearothermophilus, Bacillus subtilis, and Escherichia coli. Biotechnol. Lett. 15:815–820 [Google Scholar]
- 24. Nazina TN, et al. 2001. Taxonomic study of aerobic thermophilic bacilli: descriptions of Geobacillus subterraneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermoglucosidasius and Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G. thermocatenulatus, G. thermoleovorans, G. kaustophilus, G. thermoglucosidasius and G. thermodenitrificans. Int. J. Syst. Evol. Microbiol. 51:433–446 [DOI] [PubMed] [Google Scholar]
- 25. Sato T, Fukui T, Atomi H, Imanaka T. 2005. Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl. Environ. Microbiol. 71:3889–3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sekowska A, Danchin A. 1999. Identification of yrrU as the methylthioadenosine nucleosidase gene in Bacillus subtilis. DNA Res. 6:255–264 [DOI] [PubMed] [Google Scholar]
- 27. Shaw AJ, Covalla SF, Hogsett DA, Herring CD. 2011. Marker removal system for Thermoanaerobacterium saccharolyticum and development of a markerless ethanologen. Appl. Environ. Microbiol. 77:2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suzuki H, Yoshida K. 2012. Genetic transformation of Geobacillus kaustophilus HTA426 by conjugative transfer of host-mimicking plasmids. J. Microbiol. Biotechnol. 22:1279–1287 [DOI] [PubMed] [Google Scholar]
- 29. Takami H, Inoue A, Fuji F, Horikoshi K. 1997. Microbial flora in the deepest sea mud of the Mariana Trench. FEMS Microbiol. Lett. 152:279–285 [DOI] [PubMed] [Google Scholar]
- 30. Takami H, Nishi S, Lu J, Shinamura S, Takaki Y. 2004. Genomic characterization of thermophilic Geobacillus species isolated from the deepest sea mud of the Mariana Trench. Extremophiles 8:351–356 [DOI] [PubMed] [Google Scholar]
- 31. Takami H, et al. 2004. Thermoadaptation trait revealed by the genome sequence of thermophilic Geobacillus kaustophilus. Nucleic Acids Res. 32:6292–6303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taylor MP, et al. 2009. Thermophilic ethanologenesis: future prospects for second-generation bioethanol production. Trends Biotechnol. 27:398–405 [DOI] [PubMed] [Google Scholar]
- 33. Tripathi SA, et al. 2010. Development of pyrF-based genetic system for targeted gene deletion in Clostridium thermocellum and creation of a pta mutant. Appl. Environ. Microbiol. 76:6591–6599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turnbough CL, Switzer RL. 2008. Regulation of pyrimidine biosynthetic gene expression in bacteria: repression without repressors. Microbiol. Mol. Biol. Rev. 72:266–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang L, et al. 2006. Isolation and characterization of a novel thermophilic Bacillus strain degrading long-chain n-alkanes. Extremophiles 10:347–356 [DOI] [PubMed] [Google Scholar]
- 36. Wiegel J, Ljungdahl LG. 1986. The importance of thermophilic bacteria in biotechnology. Crit. Rev. Biotechnol. 3:39–108 [Google Scholar]