

Abstract
Foot-and-mouth disease virus (FMDV) nonstructural protein 3A plays important roles in virus replication, virulence, and host range. In other picornaviruses, homodimerization of 3A has been shown to be relevant for its biological activity. In this work, FMDV 3A homodimerization was evidenced by an in situ protein fluorescent ligation assay. A molecular model of the FMDV 3A protein, derived from the nuclear magnetic resonance (NMR) structure of the poliovirus 3A protein, predicted a hydrophobic interface spanning residues 25 to 44 as the main determinant for 3A dimerization. Replacements L38E and L41E, involving charge acquisition at residues predicted to contribute to the hydrophobic interface, reduced the dimerization signal in the protein ligation assay and prevented the detection of dimer/multimer species in both transiently expressed 3A proteins and in synthetic peptides reproducing the N terminus of 3A. These replacements also led to production of infective viruses that replaced the acidic residues introduced (E) by nonpolar amino acids, indicating that preservation of the hydrophobic interface is essential for virus replication. Replacements that favored (Q44R) or impaired (Q44D) the polar interactions predicted between residues Q44 and D32 did not abolish dimer formation of transiently expressed 3A, indicating that these interactions are not critical for 3A dimerization. Nevertheless, while Q44R led to recovery of viruses that maintained the mutation, Q44D resulted in selection of infective viruses with substitution D44E with acidic charge but with structural features similar to those of the parental virus, suggesting that Q44 is involved in functions other than 3A dimerization.
INTRODUCTION
Foot-and-mouth disease virus (FMDV) is the prototypic member of the aphthovirus genus within the family Picornaviridae (5, 24, 52) and the etiological agent of a devastating disease of livestock (34). The viral particle is composed of a protein capsid that contains a positive-sense RNA molecule of about 8,500 nucleotides that is infectious and encodes a single polyprotein, which is processed in infected cells by cis- and trans-acting viral proteases (55) to yield different polypeptide precursors and the mature viral proteins (9, 62). The viral genome encodes four structural capsid proteins (VP1 to VP4) and seven nonstructural (NS) proteins, the leader Lb/ab protease, and proteins encoded in the P2 (2B and 2C) and P3 (3A, 3B, 3C, and 3D) regions (9).
Replication of picornaviruses occurs associated to cell endomembranes that are recruited during viral infection (25). NS proteins are involved in crucial aspects of the viral cycle and pathogenesis, such as rearrangements of intracellular membranes required for endomembrane recruitment and the lysis of host cells (1, 12, 14, 18, 73). Protein 3A is an example of this multifunctional role; in poliovirus (PV), the interaction between the RNA replication complex and intracellular membranes appears to be accomplished by proteins 3A and 2C, which have membrane-binding properties (11, 60). When expressed as a recombinant protein in transfected cells, PV 3A cofractionates with endoplasmic reticulum markers (66), and its single transient expression can disrupt the secretory apparatus (23) and decrease major histocompatibility complex (MHC) class I expression (22). On the other hand, 3AB presumably anchors 3B in intracellular membranes originated de novo during the early steps of RNA replication, where uridylylated 3B primes the synthesis of nascent viral RNAs (2, 37, 68, 69). PV 3AB has a nonspecific RNA-binding activity and associates with the cloverleaf structure in the 5′ end of viral RNA and with the 3CD precursor to form a ribonucleoprotein complex required for PV RNA synthesis (32, 74, 76).
While FMDV shows considerable functional and structural analogies with PV and other picornaviruses, some differences have been reported, such as its resistance to the Golgi disruption exerted by brefeldin A, the different pattern of membrane reorganization induced in infected cells (39, 43), and the ability of 2B and 2BC, instead of 3A, to inhibit the secretory pathway in cultured cells (42), likely contributing to the evasion of innate and acquired immune responses (58). In addition, FMDV is the only picornavirus encoding 3 copies of 3B protein, required for both optimal replication in cell culture (26) and for virulence in natural hosts (50). All 3 copies have been shown to undergo uridylylation in vitro (44). In FMDV, 3A protein has been reported to play a role on viral host range, as a single amino acid replacement (Q44R) conferred FMDV the ability to cause vesicular lesions in guinea pigs (48). As 2B and 2C, FMDV nonstructural protein 3A contains a hydrophobic domain that in other picornaviruses mediates its targeting to intracellular membranes (19), which could be responsible for the location of the replication complex within a membrane context (20, 23, 27, 69). The C-terminal fragment of 3A (from the C terminus of the hydrophobic domain) is considerably longer in FMDV (77 amino acids [aa]) than in other picornaviruses (i.e., 7 aa for PV), and deletions and mutations in 3A are known to contribute both to viral attenuation in cattle (8) and to decreased replication rates in bovine epithelial cells (49). However, little is known on the interactions of 3A with other viral and cellular proteins, and no structural data are available for this protein.
Homodimer formation has been revealed by the NMR structure determined for a soluble version lacking the trans-membrane domain (aa 1 to 59) of PV 3A (65). Each monomer has a structured region consisting of two amphipathic α-helices (aa 23 to 29 and 32 to 41) separated by a 180° loop that forms a helical hairpin, flanked by nonstructured N and C regions. The N terminus of 3A from PV also contains a conserved patch of anionic residues at the top of the dimer structure, in the loop between the two α-helices, as well as three solvent-exposed charged residues (E38, K39, and K40) that may be important for viral replication (65). A role for 3A homodimer formation in both RNA replication and inhibition of cellular protein transport has also been reported for coxsackievirus (CV) B3. In this case, while the general organization of the CVB3 dimer was similar to that of PV, the establishment of salt bridges between residues D24 and K41 was found critical for dimer stability; using an optimized PV 3A structure, these salt bridges were also found in equivalent PV residues (D23 and K40) (72).
To gain insight into the structure-function relationship of FMDV 3A protein, we devised a molecular model for the N-terminal region of this protein, using as the template the structure reported for PV 3A. This model predicted hydrophobic interactions between residues at two α-helices in each monomer as the main homodimerization determinant. Here, we show that amino acid replacements L38E and L41E, located at the predicted hydrophobic dimerization interface, and expected to contribute to dimer stability, decrease 3A dimerization in cells transiently expressing 3A, and abolish dimer/multimer formation in peptides reproducing the N terminus of 3A. Replacements L38E and L41E significantly reduced the homodimerization signal detected for transiently expressed 3A by means of an in situ proximity ligation assay (63). In addition, replacements L38E and L41E were detrimental for virus growth, leading to selection of viruses that for mutants L38E and L41E restored the hydrophobicity of the residues, suggesting that 3A dimer formation plays a relevant role in FMDV replication. On the other hand, replacement Q44R that favors or replacement Q44E that impairs the polar interactions that, according to the model, Q44 could establish with residue D32 of the opposite monomer did not abolish dimer formation of transiently expressed 3A, indicating that these polar interactions are not critical for 3A dimerization. Nevertheless, while Q44R led to infectious virus recovery, Q44D resulted in the selection of infective viruses with substitution D44E with acidic charge but with structural features similar to those of the parental virus, suggesting that residue Q44, despite not being essential for 3A dimerization, is involved in biological functions relevant for virus multiplication.
MATERIALS AND METHODS
Modeling procedures of FMDV 3A proteins.
Three-dimensional (3D) models of FMDV 3A protein dimer (primary UniProtKB/TrEMBL accession code Q9DJC8) and 3A mutants Q44R, Q44D, M29R, L38E, and L41E were generated using comparative modeling procedures. As a structural template, the soluble domain of human PV 3A protein was used, attending to its hypothetical structural similarity to the target sequence (Protein Data Bank code 1NG7) (65). Best representative conformer in the NMR spectroscopy ensemble was selected as a source of coordinates for the FMDV 3A wild type and subsequent models. All models were built using the SWISS-MODEL server (31, 51, 59), available at http://swissmodel.expasy.org//SWISS-MODEL.html, and their structural quality was checked using the analysis programs provided by the same server (Anolea/Gromos). Global model quality estimation was performed in terms of QMEAN4 raw score (10). To optimize geometries, models were energy minimized using the GROMOS 43B1 force field implemented in DeepView (31), using 500 steps of steepest descent minimization followed by 500 steps of conjugate-gradient minimization. Figures were generated using the Pymol Molecular Graphics System (Schrödinger, LLC). Structurally based sequence alignment of wild-type model templates and other 3A proteins was refined with T-COFFEE (46).
Peptides.
The N-terminal region of 3A, spanning residues I1 to F52, as well as four analogues with the M29R, L38E, L41E, and L38E-L41E replacements (N-ter peptides; see Table 1), were made in C-terminal carboxamide form on Rink amide ChemMatrix resin in a Prelude synthesizer (Protein Technologies, Tucson, AZ). Standard 9-fluorenylmethoxy carbonyl (Fmoc) protocols at a 0.05-mmol scale were used, with 5-fold molar amounts of Fmoc-amino acids and O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate and 10-fold molar amounts of N,N-diisopropylethylamine, with systematic double coupling for all residues. After deprotection and cleavage with trifluoroacetic acid-water-triisopropylsilane (95:2.5:2.5 [vol/vol], 90 min, room temperature), peptides were precipitated with chilled diethyl ether, redissolved in water, lyophilized, and purified by preparative reverse-phase high-performance liquid chromatography (HPLC) to a minimum purity of 95% by analytical HPLC. Their identities were confirmed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry in a Voyager DE-STR instrument (Applied Biosystems), using α-cyano-4-hydroxycinnamic acid matrix and linear mode acquisition (Table 1).
Table 1.
Synthetic peptide sequences
| Wild type or mutation | Amino acid sequencea | Avg mol wt (calculated/found) |
|---|---|---|
| Wild type | ISIPSQKSVLYFLIEKGQHEAAIEFFEGMVHDSIKEELRPLIQQTSFVKRAF | 6,036/6,035 |
| L38E | ISIPSQKSVLYFLIEKGQHEAAIEFFEGMVHDSIKEEERPLIQQTSFVKRAF | 6,051/6,052 |
| L41E | ISIPSQKSVLYFLIEKGQHEAAIEFFEGMVHDSIKEELRPEIQQTSFVKRAF | 6,051/6,052 |
| L38L41E | ISIPSQKSVLYFLIEKGQHEAAIEFFEGMVHDSIKEEERPEIQQTSFVKRAF | 6,068/6,069 |
| M29R | ISIPSQKSVLYFLIEKGQHEAAIEFFEGRVHDSIKEELRPLIQQTSFVKRAF | 6,061/6,063 |
Sequences correspond to residues 1 to 52 of the 3A protein. Bold, underlined letters refer to replacements relative to the 3A sequence of FMDV C-S8c1.
For SDS-PAGE analysis, aliquots containing 1.87 μg and 0.187 μg of each peptide were prepared from 5-mg/ml stock solutions of the lyophilized peptides in Milli-Q water, mixed with Laemmli sample buffer containing 2% SDS and 100 mM dithiothreitol (DTT), and incubated for 25 min at room temperature. Samples were resolved on 12% SDS-PAGE gels, stained with Coomassie blue, transferred to nitrocellulose membranes, and immunoblotted with 3A-specific rabbit polyclonal antibody (Ab) and a goat anti-rabbit horseradish peroxidase-coupled secondary Ab (GE Healthcare). Samples were subsequently developed using a chemiluminescence kit (Perkin-Elmer).
Cells and viruses.
Vero, IBRS-2, and BHK-21 cells were grown at 37°C and maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco-BRL), supplemented with 5% fetal bovine serum (Gibco-BRL), 1 μg/ml streptomycin, and 1 μg/ml penicillin. A viral stock from a type C FMDV C-S8c1 (61) isolate was produced by amplification in BHK-21 cells. Procedures for infections and virus titration in semisolid agar medium were as described previously (54).
Antibodies and reagents.
Monoclonal Ab (MAb) 2C2 to nonstructural protein 3A (21) was used in the proximity ligation assay. Rabbit polyclonal anti-β-tubulin 196 was used in Western blot analysis (4). Anti-rabbit IgGs secondary Ab coupled to Alexa Fluor (AF) 647, used in immunofluorescence assays, was from Molecular Probes (Invitrogen). Polyclonal Ab 443 against the N terminus of 3A protein was produced by immunizing two New Zealand White rabbits with a synthetic peptide spanning 3A residues F26 to Q43 (FEGMVHDSIKEELRPLIQ) coupled to Cys-KLH. To generate polyclonal Ab 346 against the C terminus of 3A, plasmid pRSET-C (Invitrogen) was used for the inducible isopropyl-β-d-thiogalactopyranoside (IPTG) expression in Escherichia coli BL21(DE3)pLysS of a His-tagged polypeptide spanning 3A residues 81 to 153 fused to 3BBB, whose sequence was amplified from the pMT28 clone with primers 5′-TAGGCGCTAGCAGACAGAAGATGGTG-3′ and 5′-GCAGATCTTTACTCAGTGACAATCAA-3′. The restriction enzymes NheI and BglII (New England Biolabs) were used for cloning. The His-tagged protein was purified in an Ni column (Probond resin; Invitrogen) with a pH 4.5 elution buffer, and two New Zealand White rabbits were immunized with the purified protein. Rabbit immunizations were as described previously (54).
Plasmids.
To assess their effect on FMDV infectivity, 3A mutations were introduced in plasmid pMT28 encoding the type C FMDV isolate C-S8c1 full-length sequence (29). Substitutions of selected amino acids were performed by site-directed mutagenesis (41), using the primers shown in Table 2. To this end, a 3A-containing fragment was amplified from pMT28 using primer 3A-F and the corresponding mutated reverse primer (-R) or primer 3A-R and the corresponding mutated forward primer (-F) at a 1 μM concentration, in a reaction, including the Expand high-fidelity PCR system BioTaq (Bioline) polymerase (1.25 U), plus Pfu (Biotools) polymerase (0.15U), 2 mM MgCl2, and 1 mM deoxynucleoside triphosphate (dNTPs) (Roche). After amplification, each PCR fragment was purified and an overlap extension PCR was performed using outer primers 3A-F and 3A-R. The resulting amplicons were digested with BglII and RsrII and cloned into pMT28 digested with the same restriction enzymes, as described previously (40). The correct orientation and sequence of the resulting plasmids were confirmed by sequencing with primers 3A-F and 3A-R (Table 2), as well as primers 3A-1 and 3ABB-2 (30). For transient expression, plasmid pRSV3Awt (30) and derivatives of pRSV/L in which the luciferase gene was replaced by the 3A coding sequence of the different mutants described above were used. To this end, 3A-containing sequences were amplified from the corresponding pMT28 derivatives using oligonucleotides 3A-1 and 3A-2 (30), which included restriction sites for cloning into pRSV (HindIII and KpnI).
Table 2.
Oligonucleotides used for construction of mutants in the 3A protein
| Oligonucleotidea | Sequence (5′→3′)b | Positionb |
|---|---|---|
| 3AF | GAAGATCTCCGACTCGCTCTCCAG | 4199 |
| 3AR | ATCGGACCGGCGTAAGGTCCCTC | 5839 |
| L38E-F | CATTAAGGAGGAAGAGCGGCCCCTCATC | 5397 |
| L38E-R | GATGAGGGGCCGCTCTTCCTCCTTAATG | 5424 |
| L41E-F | GAACTCCGGCCCGAGATCCAACAAAC | 5407 |
| L41E-R | GTTTGTTGGATCTCGGGCCGGAGTTC | 5432 |
| L38EL41E-F | TTAGGAGGAAGAGCGGCCCGAGATCCAA | 5398 |
| L38EL41E-R | TTGGATCTCGGGCCGCTCTTCCTCCTTA | 5437 |
| Q44R-F | CCTCATCCAACGAACTTCATTTGTGAAACGT | 5418 |
| Q44R-R | CACAAATGAAGTTCGTTGGATGAGGGGC | 5442 |
| Q44D-F | CCTCATCCAAGATACTTCATTTGTGAAACGTG | 5418 |
| Q44D-R | CACAAATGAAGTATCTTGGATGAGGGGC | 5442 |
| M29R-F | CTTTGAGGGCCGAGTACACGACTCC | 5373 |
| M29R-R | GGAGTCGTGTACTCGGCCCTCAAAG | 5397 |
Forward and reverse primers are indicated by F and R, respectively.
Sequences in bold and nucleotide numbering correspond to those of FMDV C-S8c1 (GenBank no. AJ133357), and substituted nucleotides are in italics. Restriction sites are indicated, and their nucleotides are underlined.
RNA synthesis, transfection, and infectivity.
For RNA synthesis, plasmids were linearized with NdeI (New England BioLabs) and in vitro transcribed using SP6 RNA polymerase (Promega). After transcription, the reaction mixture was treated with RQ1 DNase (1U/μg RNA; Promega). Then, RNA was extracted with phenol-chloroform and precipitated with ethanol. The RNA integrity and concentration were determined by electrophoresis on agarose gels. In vitro-transcribed RNAs were transfected into BHK-21 cells using the Lipofectin reagent (Invitrogen), as described previously (40). Cells were maintained at 37°C in Dulbecco's modified Eagle's medium (DMEM)-supplemented fetal bovine serum (FBS). To assess RNA infectivity, at 4 h posttransfection (hpt) cells were maintained in semisolid DMEM-0.5% agar supplemented with 5% FBS. At 24 hpt, the viral titer was determined by plaque assay (54).
Viral RNA extraction, cDNA synthesis, and DNA sequencing.
Viral RNA was extracted from supernatants of cell cultures or tissue homogenates from suckling mice (7), using TRI reagent (Sigma), as described by the manufacturer. cDNA was synthesized by reverse transcription of viral RNA using M-MuLV reverse transcriptase (Roche) and primers 3A-F and 3A-R. cDNA was amplified by PCR using the same primers and BioTaq DNA polymerase (Bioline) supplemented with 10% of Expand high-fidelity polymerase (Biotools) for proofreading activity. PCR products were purified with Wizard SV gel and PCR clean-up system (Promega) and their sequences determined by automatic DNA sequencing. DNA sequences, spanning the nucleotides corresponding to residues 105 of 2B to 5 of 3B2 viral proteins, were confirmed by at least two independent sequencing reactions using the primers indicated above. Nucleotide positions correspond to those previously described for the FMDV C-S8c1 isolate (GenBank no. AJ133357).
In vitro translation.
Rabbit lysate reticulocytes (Promega) were incubated with 1 μg of transcribed RNAs and 14.3 μCi of [35S]methionine and cysteine (Amersham) for 1 h at 30°C and treated with 50 μg of RNase A for 15 min at room temperature, as described previously (56). Translated proteins were analyzed by 10% SDS-PAGE.
In situ PLA.
Samples were processed as described by the manufacturer (OLINK Bioscience). Briefly, IBRS cells were grown on glass coverslips and transfected with 1 μg of pRSV3Awt and derivatives using Lipofectamine (Invitrogen) as described by the manufacturer or infected with FMDV C-S8c1 at a multiplicity of infection (MOI) of 5 PFU/cell. At 24 hpt, or 3 h postinfection (hpi), monolayers were washed with phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde for 15 min at room temperature, blocked, and permeabilized with PBTG buffer (0.1% Triton X-100, 1% bovine serum albumin [BSA], and 1 M glycine in PBS) for 15 min at room temperature. Primary Ab was prepared using the Probemaker kit (OLINK, Bioscience) by using the manufacturer's recommendations: 1 mg/ml of monoclonal Ab (IgG class) 2C2, affinity purified through a protein G column, was independently conjugated to each of a pair of oligonucleotides to generate plus and minus proximity ligation assay (PLA) probes. Cells on coverslips were incubated with conjugated primary antibodies diluted in PBS with 1% BSA for 1 h at room temperature and then washed twice with buffer A (0.01 M Tris, 0.15 M NaCl, and 0.05% Tween 20) for 5 min. The signal development (ligation, amplification, and hybridization) was performed according to the manufacturer's instructions in PLA Duolink II. Briefly, a ligation-ligase solution was added to each sample, and slides were incubated in a preheated humidity chamber for 30 min at 37°C and washed twice in buffer A for 2 min with gentle agitation. Then, the amplification-polymerase solution, including the fluorescent probe (λexc of 554 nm), was added and slides were incubated in the preheated humidity chamber for 100 min at 37°C. Finally, the slides were washed three times in buffer B (0.2 M Tris, 0.1 M NaCl [pH 7.5]) for 10 min with agitation and protected from light. In the case of transiently transfected cells, samples were further incubated with primary polyclonal Ab 346 and anti-rabbit IgG secondary antibodies coupled to Alexa Fluor 647 (Invitrogen) to detect 3A protein. Slides were mounted with a coverslip using a minimal volume of Duolink II mounting medium with DAPI (4′,6-diamidino-2-phenylindole). Cells were observed with a confocal LSM710 vertical (Bio-Rad/Zeiss) microscope. As reported for dimerization detection by PLA (45), fluorescence was quantified using the ImageJ software (analyze particles plug-in). The percentage of the fluorescence intensity, relative to that determined for cells expressing 3Awt protein, was plotted for each mutant protein ± standard error.
Western blot analysis.
Vero cells were grown on 35-mm tissue culture plates and transfected as described above with 1 μg of different plasmids. At 24 hpt, the cells were scraped on ice into NP-40 lysis buffer (10 mM EGTA, 2.5 mM MgCl2, 1% NP-40, 20 mM HEPES [pH 7.4]) and sonicated. Equal amounts of total protein of each sample, estimated by Bradford (13), were mixed with Laemmli sample buffer and boiled. Samples were separated by 12% SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was blocked, and proteins were detected by incubation with the corresponding 3A-specific polyclonal rabbit Ab as described above.
Mouse experiments.
The infectivity of the FMDV 3A mutants in suckling mice was tested as described previously (7). Briefly, in vitro-transcribed viral RNA (from 102 to 105 ng) mixed with 20 μl of Lipofectin (Invitrogen) in a final volume of 100 μl in PBS was inoculated intraperitoneally in litters (4 to 5 mice per RNA) of Swiss newborn mice. Dead animals were scored up to 11 days after infection. Mice showing severe signs of disease (tremors ataxia, paralysis of the hind limbs) were euthanized. All animals in this study were handled in the BSL-3 facilities at CISA-INIA (Madrid, Spain), in strict accordance with the guidelines of the European Community 86/609/CEE. The protocol was approved by the Committee on the Ethics of Animal Experiments of INIA (permit number CBS 2008/016).
Data analysis.
One-way analysis of variance was performed with statistical package SPSS 19.0 (SPSS, Inc.) for Windows. Data are presented as means ± standard errors.
RESULTS
Homology model of the 3A protein and dimerization interface.
To provide basic insights into the structure-function relationships of the FMDV 3A protein, 3D structural models of its N-terminal region (residues 1 to 94) were generated by bioinformatic procedures, using the analogous structure of the soluble domain of human poliovirus 3A protein (65) as the template. As a result, compatible ungapped models were rendered for residues 15 to 48 of the FMDV 3A protein, in correspondence to the structured region in solution from the experimental template. Quality QMEAN4 raw scores for the model of wild-type protein monomers in this region were 0.622 (Z score = 0.09) and 0.621 (Z score = 0.09), respectively. These values are comprised within the range of quality standards for accepted homology-based structure models (10). The tertiary structure of each monomer is organized mainly on the structural basis of two α-helices (α1, residues 25 to 33, and α2, residues 37 to 44) connected by a 3-residue loop (Fig. 1A and B), hypothetically responsible for the dimerization process. Consensus from diverse methods of transmembrane domain predictions (6, 33, 70) points to the region comprised between residues 59 to 76 as putatively responsible for 3A protein anchorage to membrane (Fig. 1A). General inspection of a minimized 3D structure of the wild-type FMDV 3A homodimer model showed a quaternary organization of the N-terminal region protein similar to that of PV 3A protein obtained by NMR spectroscopy (65). The dimerization interface in FMDV 3A would include all residues in the 25-to-44 stretch of the two antiparallel monomers. As in PV 3A, a number of hydrophobic contacts between α1 and α2 helices could provide physical stability to the dimer. Such interactions would involve residues F25, F26, M29, V30, L38, L41, and I42, in both monomers. Genotypes carrying mutations introducing similar electrostatic repulsive interactions in the hydrophobic core could be unable to be functionally competent. In addition, gradations of that biological effect would be also related to the differential local accumulation of heterogeneous repulsive interactions. Thus, mutations L38E, L41E, and M29R within the hydrophobic core could disrupt the binding interaction in the region by creating repulsions from facing negatively (or positively) charged residues (Fig. 1C). On the other hand, polar contacts such as that of residues D32 and Q44 (Fig. 1C), flanking the C-terminal ends of α1 and α2 helices and exposed to the solvent, could also modulate the interactions between both monomers. Mutation Q44R, found to mediate FMDV adaptation to the guinea pig (48), could significantly modify the electrostatic surface of the region, favoring the formation of salt bridges between both R44 and D32 residues and eventually increasing dimer stability.
Fig 1.

(A) Top, structure-derived multiple sequence alignment of the soluble domain and vicinities of diverse 3A proteins of enteroviruses and FMDVs (Uniprot IDs are given in parenthesis): 1NG7, poliovirus type 1 strain Mahoney (P03300), sequence used as the template for homology modeling procedures (65); PV1, poliovirus type 1, CHN-Guangdong/92-2 isolate (Q9E912); CVB3 (P03313); CVA11 (Q7T7P4); CVA20 (Q7T7N8); FMDV type C, C-S8c1 isolate, (Q9E2G4); FMDV type O (P03305); FMDV type A (P49303); FMDV type Asia 1 (Q7TDB3). Positions in the alignment are colored according to sequence conservation by average BLOSUM 62 score (similar residues according to BLOSUM 62 score are colored as the most conserved one using standard Belvu colors: <0.50, white; 0.51 to 1.50, light gray; 1.51 to 3.00, medium blue; >3.00, cyan) (http://sonnhammer.sbc.su.se/Belvu.html) (64). Positions of D32 and Q44 residues in the FMDV-3A modeled sequence are indicated by magenta arrows. Position of hydrophobic residues M29, L38, and L41 are marked by green arrows. (B) General view of the 3D model of wild-type FMDV 3A protein dimer. Position of selected residues hypothetically forming the binding interface and involved in FMDV 3A dimerization is indicated. Secondary structure elements α1 and α2 are also indicated. (C) 3D models of analyzed mutants at the dimerization binding face. From left to right and top to bottom: M29R, L38E, L41E, and the relative positions of D32 and Q44 residues (mutants Q44D and Q44R).
Based on these predictions, residues at positions 29, 38, and 41, highly conserved among FMDVs of different serotypes (16), were selected to be replaced by a negatively charged E residue, which would interfere in the hydrophobic interactions predicted in this 3A region. On the other hand, two replacements at residue 44 were analyzed: Q44R and Q44D, predicted to influence dimer stability.
Substitutions at the predicted hydrophobic interface affect 3A dimerization.
Identification of protein bands potentially corresponding to 3A dimers in FMDV-infected cells is impaired by the presence of protein precursors from the 3ABBB region (30). Therefore, we first addressed the effect of the substitutions selected on FMDV 3A protein transiently expressed in cultured cells. In cells transfected with plasmid pRSV3Awt, a major protein band of the expected mobility was observed by Western blotting using a 3A-specific Ab (Fig. 2A). Additional lower-mobility bands suggestive of 3A dimer/multimer formation were also observed. When cells were transfected with pRSV derivatives expressing 3A with mutations M29R, L38E, or L41E or double-mutant L38EL41E, expected to destabilize dimer formation, no bands corresponding to putative dimers were observed (Fig. 2A). Dimerization was also observed in cells expressing mutant proteins Q44D and Q44R, with the intensity of the dimer band in both cases slightly lower than that of 3Awt.
Fig 2.
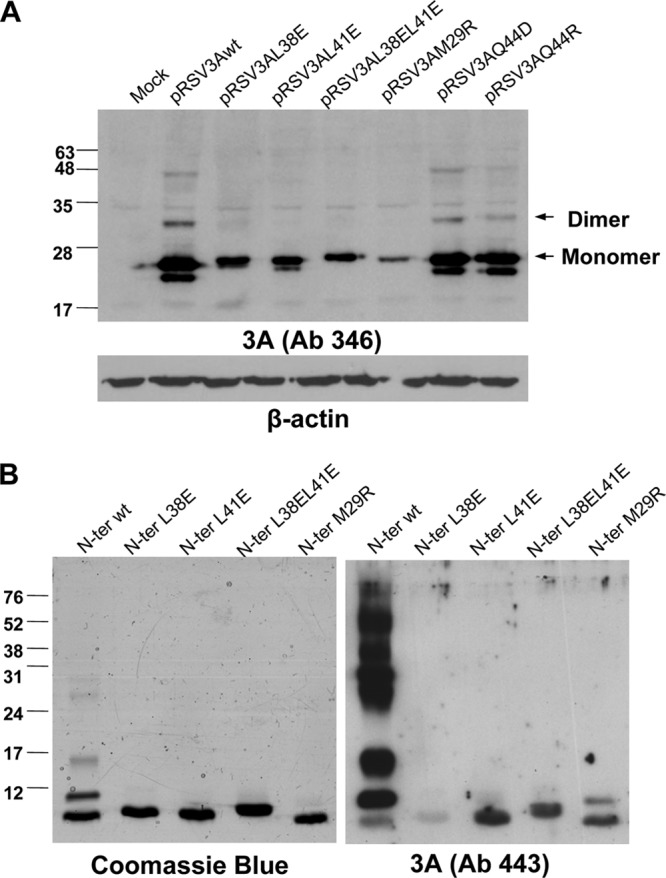
Analysis of 3A proteins and peptides, including mutations potentially affecting dimerization. (A) Expression in eukaryotic cells of 3A proteins. Immunoblot of lysates of Vero cells transfected with 1 μg of each of the plasmids indicated. Proteins were resolved on an 12% SDS-PAGE gel, transferred to a membrane, and incubated with polyclonal Ab 346 generated against the C terminus of 3A. The protein bands whose migrations correspond to the monomeric and the dimeric forms of 3A are indicated with arrows. Membranes were reprobed with a MAb against β-tubulin, as a control for protein loading. (B) Analysis of synthetic peptides from the N terminus of the 3A wild-type protein and L38E, L41E, L38E/L41E, and M29R mutants. Each peptide (1.25 μg) was resolved on two 12% SDS-PAGE parallel gels, one of which was stained with Coomassie blue. The second gel was transferred to nitrocellulose membrane and blotted with polyclonal Ab 443 produced against the N terminus of 3A. The migrations and sizes of the molecular weight markers are indicated on the left.
To further characterize 3A homodimerization, we set up an in situ protein ligation assay previously used to visualize protein-protein interaction in the cell by fluorescence microscopy (3, 28, 45). The strategy, detailed in Materials and Methods and summarized in Fig. 3A, is based on coupling a monoclonal Ab that recognizes a single epitope on each 3A molecule to two different oligonucleotide probes, + and −, which subsequently hybridize to oligonucleotides included in a ligation solution. Should the pair of antibodies locate close enough (<40 nm), their attached probes would ligate, forming a circle; amplification of this circle upon addition of fluorescently labeled nucleotides allows visualization of each single interaction by fluorescence microscopy (71). The monoclonal Ab selected for this assay was directed against an epitope on the C-terminal region of 3A, distant from the N-terminal region analyzed to avoid loss of recognition due to the mutations introduced. The assay included a biological control of noninfected or transfected cells and two technical controls lacking one of the probes each. As shown in Fig. 3B, when cells were FMDV infected and incubated with probes 2C2+ and 2C2−, a dimer fluorescent signal was observed. Fluorescence was also noticed in cells transfected with pRSV3Awt. Significant reductions in the fluorescence intensity, of about 40% of that of 3Awt, were detected in cells transfected with mutant plasmids pRSV3AL38E and pRSV3AL41E (Fig. 3C and D). When plasmid pRSV3AL38E-L41E bearing the two substitutions was analyzed, the signal reduction observed was higher, at about 80% (Fig. 3C). These results support the capacity of 3A to form homodimers and suggest that replacements L38E and L41E impaired dimer formation.
Fig 3.

Proximity ligation assay to confirm homodimer protein complexes. (A) The scheme summarizes the procedure: (i) conjugation of primary monoclonal Ab 2C2, with PLA probes + (red) and − (blue); (ii) incubation of the sample where dimerized proteins are represented in green and cherry, with the two conjugates of monoclonal Ab 2C2, probes + and −; (iii) hybridization and ligation reaction with oligonucleotides complementary to the PLA probes; (iv) final rolling-circle amplification and detection of the amplified products with the nucleotide fluorescent probe (orange balls). (B) Confocal fluorescence microscopy, from left to right: (i) PLA reaction in cells infected with FMDV (MOI of 5 PFU/ml); negative controls of infected cells incubated with (ii) 2C2-probe + or (iii) 2C2-probe −; and (iv) PLA reaction in mock-infected cells with probes + and −. (C) Plot of percentage of fluorescence intensity, relative to that of cells transfected with pRSV3Awt, in cells transfected with pRSV3AL38E, pRSV3AL41E, and pRSV3AL38EL41E. Raw data were quantified using the ImageJ program, n ≥ 40 cells scored in the experiment. Mean values and standard errors are represented. Statistically significant differences, relative to pRSV3Awt, are indicated by * (P ≤ 0.05) or ** (P ≤ 0.005). (D) PLA reaction (orange) in cells transfected with the plasmids in panel C (24 hpt) and incubated with rabbit polyclonal Ab 346 (red) detected with Alexa Fluor 647 anti-rabbit Ab. Nuclei were stained with DAPI. Scale bar, 20 μm.
Replacements M29R, L38E, and L41E also prevent dimer formation by synthetic peptides spanning the 3A dimerization interface.
To study the potential of the N terminus of 3A to form dimers and multimers, peptides spanning residues I1 to F52 (termed N peptides) were synthesized by solid-phase procedures, as described in Materials and Methods. The electrophoretic mobility of peptide N-wt, spanning the sequence corresponding to the parental C-S8c1 FMDV, revealed by Western blotting with an Ab against the N terminus of 3A protein, showed a band of a size about that corresponding to the monomeric peptide, as well as additional bands corresponding to higher-order forms (Fig. 2B). A pattern of bands similar to that observed by Western blotting was revealed by mass staining; in this case, the monomeric form was the major band, indicating that the Ab used preferentially recognized dimer/multimeric peptide forms. The inverse relationship observed between the higher-order forms revealed by the Ab recognition or by mass staining could be related to an increase of Ab recognition due to epitope multimerization. On the other hand, only monomeric bands were detected by mass staining of mutant peptides, which showed slight differences in mobility, as reported to occur among 48-mer peptides differing in single residues (53). In addition, the Ab staining showed a dramatic decrease of higher-order forms, N-M29R being the only peptide for which a faint dimeric band was observed. The poor Ab recognition of peptide N-L38E could be explained by either an altered conformation adopted by this peptide or by a possible implication of the mutated residue in the peptide-Ab interaction.
Thus, the overall results indicate that (i) FMDV 3A protein can form dimers and multimers, (ii) its N-terminal fragment is involved in this process, and (iii) replacement of the hydrophobic residue L at positions 38 and 41 by a charged amino acid E diminishes dimerization.
Mutations that abolish hydrophobic interactions at residues L38 and L41 of the 3A protein are detrimental for virus infectivity.
To assess the effect of replacements L38E and L41E located at the predicted hydrophobic dimerization interface on the FMDV life cycle, the infectivity of mutant RNAs transcribed from plasmids pMT28L38E and pMT28L41E carrying these substitutions was compared with that of the corresponding parental C-S8c1 RNA derived from plasmid pMT28. To this end, plaque formation was determined after 24 h of transfection with different amounts of RNA (from 1 to 103 ng) of BHK-21 cell monolayers maintained in semisolid medium. No infectious virus was recovered after transfection of cells with up to 1 μg of RNA transcribed from pMT28L38E or pMT28L41E (data not shown), indicating that RNAs with these mutations showed an infectivity at least five log lower than that of the RNA control pMT28. When transfected cells were incubated in liquid medium, a delay in the recovery of infectious virus, relative to transcript pMT28, was observed for transcripts pMT28L38E and pMT28L41E (no infective virus was found at 24 hpt, and limited titers of about 1 × 102 and 10 PFU/ml were detected at 72 hpt, respectively) (Fig. 4). Upon two additional passages of the transfection supernatant, the viral titers recovered from transcripts pMT28L38E and pMT28 were similar, while those recovered from transcript pMT28L41E were about 1 log lower (Fig. 4). Emergence of infectious virus was concomitant with the imposition in the viral populations of replacements for each of the mutant RNAs analyzed—E38V and E41A—that restored the hydrophobicity of the residues, as determined by sequencing of the viral RNA at the second cell passage given to the transfection medium (Table 3). Selection of mutant E38V was observed in the two additional transfections of RNA from pMT28L38E performed. On the other hand, infectious virus was recovered in only one of the three additional transfections carried out with RNA from pMT28L41E, in which replacement E41A was found imposed on the viral populations (Table 3). These results indicate that replacement of hydrophobic residues by charged residues at positions 38 and 41 of 3A drastically reduces viral multiplication, leading to selection of substitutions to nonpolar V and A residues that restore hydrophobicity and favor dimer stability.
Fig 4.
Effect of 3A mutations on FMDV infectivity. (A) Viral titers of viruses harboring different 3A mutations recovered after transfection, first or second passage on BHK-21 cells, were determined at the time of complete CPE, as follows: 24 hpt for pMT28 and pMT28Q44R or 72 hpt for pMT28L38E, pMT28L41E, and pMT28Q44E; 24 hpi for pMT28 and pMT28Q44R or 48 hpi for pMT28L38E, pMT28L41E, and pMT28Q44D after the first passage; 24 hpi for all the viruses after the second passage. Data are the averages from three independent transfections; one out of the three experiments for pMT28-L41E RNA failed to yield infectious virus.
Table 3.
Replacements selected from RNAs with mutations in 3A
| Residuea | Substitution introduced | Sequence recoveredb |
|---|---|---|
| L38(CTC) | E38(GAG) | V38(GTG)c |
| L41(CTC) | E41(GAG) | A41(GCG)d |
| Q44(CAA) | R44(CGA) | R44(CGA)c |
| Q44(CAA) | D44(GAT) | E44(GAA)c |
Amino acid residue and position in 3A are indicated relative to the C-S8c1 sequence. The corresponding codon is given in parenthesis.
Replacement selected in the virus recovered upon RNA transfection of BHK-21 cells (see Materials and Methods for details). In each case, the substitution shown was the only mutation found in the RNA region sequenced.
Sequence of the viral population recovered from the 3 independent experiments performed.
Sequence of the viral population recovered from 2 of the 4 independent experiments performed.
To study whether the point mutations introduced in 3A could alter the translation of the viral polyprotein, in vitro-transcribed RNAs from pMT2-L38E and pMT28L41E were in vitro translated in a rabbit reticulocytes lysate. The protein pattern obtained for the different mutants was similar to that of pMT28 control RNA (data not shown).
Mutation Q44D is detrimental for virus infectivity.
To study the effect of electrostatic charge acquisition at residue Q44 on FMDV life cycle, the infectivity of mutants with replacements Q44R and Q44D, which resulted in a slightly lower dimer detection in transiently expressed 3A mutants (Fig. 2A), was analyzed. An infectivity of about 197 PFU/ng of RNA was determined for transcript pMTQ44R, a value slightly higher than that observed for the control RNA from pMT28 (data not shown). When transfected cells were incubated in liquid medium, RNA from mutant Q44R produced a cytopathic effect at a time similar to that of pMT28, comparable viral titers were found in the culture medium at 24 hpt and upon the two additional passages of the transfection supernatant (Fig. 4), and the viruses recovered maintained the substitution over 3 additional cell passages. Similar results were obtained from two additional transfections performed with pMTQ44R RNA. These results confirm that replacement Q44R does not exert a detrimental effect on FMDV infectivity in cultured cells.
On the contrary, an infectivity of <1 PFU/μg was found for RNA from pMTQ44D, a delay in the recovery of infectious virus was observed after its transfection (no infective virus was found at 24 hpt, and limited titers of about <10 PFU/ml were detected at 72 hpt), and the viral titers recovered after two passages of the supernatant of cells transfected were about 1 log lower than those recovered from pMT28 RNA. Emergence of infectious virus was associated with the imposition of replacement D44E, which was also observed in three additional transfections of RNA from pMT28-Q44D (Table 3).
On the other hand, in vitro translation of RNAs from pMT28Q44R and pMT28Q44D showed a protein pattern similar to that of pMT28 control RNA (data not shown). Thus, while replacement Q44R leads to viruses with infectivity similar to that of the parental C-S8c1 virus, introduction of an acidic residue at position 44 (replacement Q44D) imposes selection of an E residue that maintains the negative charge but acquires a structure similar to that of the parental Q.
Mutations at residues L38, L41, and Q44 of 3A protein abolish infectivity in suckling mice.
To assess the effect of the mutations introduced in 3A on the replication of the virus in an animal model, RNAs transcribed from pMT28L38E, pMT28L41E, pMT28Q44R, and pMT28Q44D were injected into suckling mice, and the emergence of clinical signs and instances of death were followed for 10 days. RNA from pMT28 was used as a control. An amount of 100 ng RNA from pMT28 killed 4 of the 5 animals injected, a lethality value similar to that reported for other FMDV RNAs derived from full-length infectious clones (7). RNA from pMT28-Q44R was also lethal for suckling mice, as 100 ng of RNA killed 3 out of 4 injected animals, and sequencing of the 3A coding region of RNA extracted from dead mice showed the presence of replacement Q44R, confirming the capacity of the virus carrying this mutation to infect suckling mice (47). Conversely, RNAs from pMT28Q44D, pMT28L38E, and pMT28L41E were not lethal at doses of RNA up to 105 ng (Table 4), suggesting that these replacements allow viral replication in cultured cells, enabling selection of revertant mutants, and abolish viral multiplication in an animal model, such as suckling mice.
Table 4.
RNA infectivity in suckling mice of FMDV 3A mutants
| RNA | RNA dose (ng) | No. of dead mice/total no. of mice |
|---|---|---|
| pMT28 | 102 | 4/5 |
| 103 | 5/5 | |
| pMT28L38E | 105 | 0/5 |
| pMT28L41E | 105 | 0/5 |
| pMT28Q44D | 105 | 0/4 |
| pMT28Q44R | 102 | 3/4 |
| 103 | 4/4 |
DISCUSSION
The self-association of proteins to form dimers and higher-order oligomers is a common biological phenomenon. Recent structural and biophysical studies show that protein dimerization or oligomerization is a key factor in the regulation of different protein functions (38), including proteins relevant for virus replication (36). In this line, dimerization/multimerization has been shown to be relevant for the biological role of nonstructural proteins of different picornaviruses (15, 17, 57, 77), including FMDV (67). In this study, we describe experiments aimed to gain insight on the structure-function relationship of FMDV 3A protein, a nonstructural protein relevant for virus replication, virulence, and host range for which the molecular mechanisms that mediate its biological activity are poorly understood. Previous attempts to obtain structural data from E. coli-expressed 3A and peptides corresponding to the N terminus of 3A showed an aggregation tendency that impaired subsequent analyses. In this work, using the NMR structure of PV 3A as a template, a molecular model for the N-terminal 94 residues of FMDV 3A was derived. The model shows that 3A contains two α helices (α1, residues 25 to 33, and α2, residues 37 to 44) and that, as in PV and CVB3 3A structures, a number of hydrophobic contacts in helices α1 and α2 could provide physical stability to the dimer. In addition, in FMDV 3A, the two alpha-helices are connected by a 3-residue loop (I34, K35, and E36) that conforms a patch of charged residues. In the equivalent loop positions, a patch of negatively charged residues is present in PV (D29 and E32) (65) and in CVB3 (D30 and E32) (72) 3A proteins.
According to the model, the analysis of the transiently expressed FMDV 3A protein suggested that 3A forms dimers/multimers as shown by Western blotting analyses (Fig. 2A). Protein oligomerization even in the presence of SDS, has been previously described for PV 3AB (35, 74). This homodimerization of 3A was also evidenced by an in situ protein ligation assay designed to visualize protein-protein interactions in the cell by fluorescence microscopy (Fig. 3). The contribution of the N-terminal fragment of 3A to the dimerization was detected using the N-wt synthetic peptide, spanning residues 1 to 52 of 3A, whose mobility in SDS-PAGE revealed by mass staining showed a major band of a size about that corresponding to the monomeric peptide, as well as bands corresponding to higher-order oligomers (Fig. 2B).
Based on the model and on sequence conservation among FMDV isolates, hydrophobic interactions between residues at the helical regions of both monomers were expected to be the main dimerization determinant. Indeed, replacements L38E, L41E, and M29R, involving charge acquisition at residues predicted to contribute to the hydrophobic interface, abolished formation of dimer bands in transiently expressed 3A (Fig. 2A). Moreover, the single replacements L38E and L41E and, to a higher extent, the double replacement L38EL41E showed a reduced fluorescence signal in the proximity ligation assay (Fig. 2B), while expression of 3A was evidenced by conventional immunofluorescence with a different anti-3A antibody (Ab 346). A similar decrease in the dimerization signal in a double hybrid system has been reported for CVB3 3A carrying a double mutation (by alanine replacement) at residues L25 and L26 (72). On the other hand, peptides from the N terminus of 3A with substitutions L38E, L41E, M29R, or L38EL41E showed a dramatic reduction of the electrophoretic bands of a size higher than that corresponding to their monomeric forms. Overall, these results suggest that, as reported for CVB3, preservation of a cluster of hydrophobic interactions in this region is essential for 3A dimer stability.
When FMDV RNA with replacement L38E was transfected in BHK-21 cells, a delay, relative to cells transfected with parental C-S8c1 RNA, in both the emergence of cytopathic effect and the recovery of infectious virus was observed in three independent experiments. Viruses recovered from the second passage in cultured cells of the transfection medium displayed replacement E38V that resembled the nonpolar (L) residue of the parental C-S8c1 virus. On the other hand, infectious virus could be recovered in two of the four independent transfection experiments performed with RNA carrying replacement L41E, and the sequencing of the viral populations obtained displayed the replacement E41A, which again restored the nonpolar nature of this position. The lower viral titers recovered from transfections with L41E RNA, relative to those produced by L38E RNA, as well as the lack of recovery of infectious virus from two of the transfections with RNA L41E, suggest that this replacement could affect FMDV replication more severely than L38E. While direct reversion to the parental residue L at E38 and E44 residues requires at least 2 nucleotide substitutions, single transversions mediate replacements E38V (A5411T) and E41A (A5420C) (Table 3), making their selection more likely. These results indicate that the presence of nonpolar hydrophobic residues at positions 38 and 41 of 3A is essential for virus replication and that no second-site suppressor mutations at other 3A residues, unlike as reported for coxsackievirus RNA with replacements at analogous 3A hydrophobic residues (72), are frequently selected during the limited replication of RNA mutants L38E and L41E. Thus, taken together, our results suggest that the conservation of hydrophobic interactions at the predicted dimerization interface is required for efficient FMDV replication in cultured cells.
In CVB3, polar interactions, other than the hydrophobic contacts such as those contributed by residues L25 and L26, have been shown to participate in 3A dimer stability; thus, the establishment of salt bridges between residues D24 and K41 was found critical for dimer stability, RNA replication, and inhibition of protein transport (72). K41 is part of a cluster of charged residues located at the C terminus of CVB 3A α-helix 2. A similar cluster of charged, solvent exposed residues is found in PV 3A α-helix 2, and mutations at these residues (E38, K39, and K40) yield nonviable viruses, indicating the biological relevance of these polar, charged residues located at the α-helix 2 (75). In contrast, the equivalent positions at the C terminus of FMDV 3A α-helix 2 are occupied by a cluster of polar but not charged residues: Q43, Q44, and T45. According to our model, residues D32 and Q44 of FMDV 3A, located flanking the C-terminal extremes of helices α1 and α2 and exposed to the solvent, could establish polar interactions between both monomers. Replacement Q44R, which mediates adaptation to the guinea pig (48), could allow formation of an intermolecular salt bridge with D32 with the consequent dimer stabilization. Conversely, replacement Q44D would impair polar interactions with D32. Despite these predictions, neither of these substitutions produced a marked effect on the dimer formation of transiently expressed 3A, suggesting that the polar interactions between Q44 and D32 are not critical for 3A dimerization, albeit they could modulate dimer stability. A similar observation was reported for CVB3 3A in which replacements to A at polar residues S28 and Y37, predicted to establish an intermolecular hydrogen bond, did not affect dimerization and protein transport, while only replacement Y37A impaired virus replication, leading to recovery of a second-site suppressor mutation (72).
When the effect of electrostatic charge acquisition at position 44 on the infectiveness of FMDV RNA was analyzed, transfected RNA with replacement Q44R produced a cytopathic effect and infectious virus in a manner similar to that of the parental RNA pMT28, and the viruses recovered maintained the substitution over 3 additional cell passages, indicating that this change is not detrimental for virus replication in cultured cells. This result is consistent with the ability of the guinea pig-adapted virus carrying replacement Q44R to kill suckling mice and to cause acute disease in the pig (47). On the contrary, in cells transfected with RNA carrying the replacement Q44D, a delay in the production of infectious virus was observed and the recovered virus displayed replacement D44E. In this case, substitution of D by E did not restore a polar, uncharged side chain, maintaining a milder negative charge and structural features similar to those of the parental Q. Direct reversion to the parental residue Q requires 2 nucleotide substitutions, while a single transversion mediates replacement D44E (T5430A) (Table 3). Residue Q44 is rather conserved among FMDVs, and replacements at these residue have been found in guinea pig-adapted isolates (16). Our results suggest that, despite the fact that replacements at Q44 do not substantially affect the capacity of transiently expressed 3A to form dimers, this residue is relevant for virus replication. Thus, while replacement Q44R retains infectivity in cultured cells at levels similar to that of the parental C-S8c1, Q44D leads to the recovery of the D44E mutant.
Despite the fact that replication impairment introduced by mutations L38E, L41E, and Q44D permitted selection of revertant mutants able to grow in cultured cells, RNAs carrying these substitutions caused no death or clinical signs when inoculated in suckling mice, opposite to what was observed for RNA from C-S8c1 (pMT28) and from mutant Q44R. These results indicate that, as previously reported (7), in vivo multiplication frequently imposes different constrains for virus replication and disease emergence compared to those found in cultured cells.
Although further work is required to assess the contribution of dimerization inhibition on the detrimental effect of the mutants studied, our results suggest that, despite the unique characteristics of FMDV 3A protein among picornaviruses, FMDV requires 3A dimerization for efficient replication. Our data support that the hydrophobic interactions established between the α-helices of both monomers are the main determinant for dimerization and its impairment is detrimental for virus multiplication. On the other hand, mutations affecting polar interactions between residues at the α-helices can affect FMDV replication, without abolishing 3A dimerization.
ACKNOWLEDGMENTS
We thank E. Domingo and C. Escarmís for infectious clone pMT28. We are indebted to B. Borrego for advice and help with animal experiments and to M. A. Martín-Acebes and M. Sáiz for discussions and critical reading of the manuscript.
Work at the F.S. laboratory was supported by grants from Ministerio de Ciencia e Innovación (MICINN, Spain): BIO2008-0447-C03-01, CSD2006-0007, and BIO2011-24351. Work at the D.A. laboratory was supported by MICINN grants BIO2008-0447-C03-01 and SAF2011-24899 and by Generalitat de Catalunya (SGR2009-0492). Work at the P.G.-P. laboratory was supported by MICINN grants SAF2007-61926, IPT2011-0964-900000, and SAF2011-13156-E and by the European Commission through grants FP7 HEALTH-F3-2009-223431 (EU project “Divinocell”) and FP7 HEALTH-2011-278603 (EU project “Dorian”). Support from the Fundación Ramón Areces and the Centro de Computación Científica CCC-UAM (computational support) is also acknowledged. Work at Biomol-Informatics was partially financed by the European Social Fund. Work at the L.K. laboratory was supported by MICINN grants PI 201120E007 and PI 201120E089 and by MSC grant FIS 2010-PI10/00594.
Footnotes
Published ahead of print 11 July 2012
REFERENCES
- 1. Andino R, Boddeker N, Silvera D, Gamarnik AV. 1999. Intracellular determinants of picornavirus replication. Trends Microbiol. 7:76–82 [DOI] [PubMed] [Google Scholar]
- 2. Andino R, Rieckhof GE, Achacoso PL, Baltimore D. 1993. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 12:3587–3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antonio LS, et al. 2011. P2X4 receptors interact with both P2X2 and P2X7 receptors in the form of homotrimers. Br. J. Pharmacol. 163:1069–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Armas-Portela R, Parrales MA, Albar JP, Martinez AC, Avila J. 1999. Distribution and characteristics of betaII tubulin-enriched microtubules in interphase cells. Exp. Cell Res. 248:372–380 [DOI] [PubMed] [Google Scholar]
- 5. Bachrach HL. 1977. Foot-and-mouth disease virus, properties, molecular biology and immunogenicity, p 3–32 In Romberger JA. (ed), Beltsville symposia in agricultural research, vol I. Virology in agriculture. Allanheld, Osmund, Monclair, NJ [Google Scholar]
- 6. Bagos PG, Liakopoulos TD, Hamodrakas SJ. 2006. Algorithms for incorporating prior topological information in HMMs: application to transmembrane proteins. BMC Bioinformatics 7:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baranowski E, Molina N, Nunez JI, Sobrino F, Saiz M. 2003. Recovery of infectious foot-and-mouth disease virus from suckling mice after direct inoculation with in vitro-transcribed RNA. J. Virol. 77:11290–11295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beard CW, Mason PW. 2000. Genetic determinants of altered virulence of Taiwanese foot-and-mouth disease virus. J. Virol. 74:987–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belsham GJ. 2005. Translation and replication of FMDV RNA. Curr. Top. Microbiol. Immunol. 288:43–70 [DOI] [PubMed] [Google Scholar]
- 10. Benkert R, Templin T, Schim SM, Doorenbos AZ, Bell SE. 2011. Testing a multigroup model of culturally competent behaviors among underrepresented nurse practitioners. Res. Nurs. Health 34:327–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bienz K, Egger D, Pasamontes L. 1987. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 160:220–226 [DOI] [PubMed] [Google Scholar]
- 12. Bienz K, Egger D, Rasser Y, Bossart W. 1983. Intracellular distribution of poliovirus proteins and the induction of virus-specific cytoplasmic structures. Virology 131:39–48 [DOI] [PubMed] [Google Scholar]
- 13. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 14. Buenz EJ, Howe CL. 2006. Picornaviruses and cell death. Trends Microbiol. 14:28–36 [DOI] [PubMed] [Google Scholar]
- 15. Cameron CE, Suk Oh H, Moustafa IM. 2010. Expanding knowledge of P3 proteins in the poliovirus lifecycle. Future Microbiol. 5:867–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carrillo C, et al. 2005. Comparative genomics of foot-and-mouth disease virus. J. Virol. 79:6487–6504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cuconati A, Xiang W, Lahser F, Pfister T, Wimmer E. 1998. A protein linkage map of the P2 nonstructural proteins of poliovirus. J. Virol. 72:1297–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cho MW, Teterina N, Egger D, Bienz K, Ehrenfeld E. 1994. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202:129–145 [DOI] [PubMed] [Google Scholar]
- 19. Choe SS, Kirkegaard K. 2004. Intracellular topology and epitope shielding of poliovirus 3A protein. J. Virol. 78:5973–5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Datta U, Dasgupta A. 1994. Expression and subcellular localization of poliovirus VPg-precursor protein 3AB in eukaryotic cells: evidence for glycosylation in vitro. J. Virol. 68:4468–4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Diego M, Brocchi E, Mackay D, De Simone F. 1997. The non-structural polyprotein 3ABC of foot-and-mouth disease virus as a diagnostic antigen in ELISA to differentiate infected from vaccinated cattle. Arch. Virol. 142:2021–2033 [DOI] [PubMed] [Google Scholar]
- 22. Deitz SB, Dodd DA, Cooper S, Parham P, Kirkegaard K. 2000. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. U. S. A. 97:13790–13795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doedens JR, Giddings TH, Jr, Kirkegaard K. 1997. Inhibition of endoplasmic reticulum-to-Golgi traffic by poliovirus protein 3A: genetic and ultrastructural analysis. J. Virol. 71:9054–9064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Domingo E, et al. 1990. Genetic variability and antigenic diversity of foot-and-mouth disease virus, p 233–266 In Murphy FA, Van Regenmortel MHV. (ed), Applied virology research, vol 2 Plenum Publishing Corporation, New York, NY [Google Scholar]
- 25. Egger D, Teterina N, Ehrenfeld E, Bienz K. 2000. Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J. Virol. 74:6570–6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Falk MM, et al. 1990. Foot-and-mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3. J. Virol. 64:748–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fujita K, et al. 2007. Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry 46:5185–5199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gajadhar A, Guha A. 2009. A proximity ligation assay using transiently transfected, epitope-tagged proteins: application for in situ detection of dimerized receptor tyrosine kinases. Biotechniques 48:145–152 [DOI] [PubMed] [Google Scholar]
- 29. García-Arriaza J, Manrubia SC, Toja M, Domingo E, Escarmís C. 2004. Evolutionary transition toward defective RNAs that are infectious by complementation. J. Virol. 78:11678–11685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garcia-Briones M, et al. 2006. Differential distribution of non-structural proteins of foot-and-mouth disease virus in BHK-21 cells. Virology 349:409–421 [DOI] [PubMed] [Google Scholar]
- 31. Guex N, Diemand A, Peitsch MC. 1999. Protein modelling for all. Trends Biochem. Sci. 24:364–367 [DOI] [PubMed] [Google Scholar]
- 32. Hope DA, Diamond SE, Kirkegaard K. 1997. Genetic dissection of interaction between poliovirus 3D polymerase and viral protein 3AB. J. Virol. 71:9490–9498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kall L, Krogh A, Sonnhammer EL. 2007. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius Web server. Nucleic Acids Res. 35:W429–W432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Knowles NJ, Samuel AR, Davies PR, Kitching RP, Donaldson AI. 2001. Outbreak of foot-and-mouth disease virus serotype O in the UK caused by a pandemic strain. Vet. Rec. 148:258–259 [PubMed] [Google Scholar]
- 35. Lama J, Paul AV, Harris KS, Wimmer E. 1994. Properties of purified recombinant poliovirus protein 3aB as substrate for viral proteinases and as co-factor for RNA polymerase 3Dpol. J. Biol. Chem. 269:66–70 [PubMed] [Google Scholar]
- 36. Li S, et al. 2009. Dimerization of hepatitis E virus capsid protein E2s domain is essential for virus-host interaction. PLoS Pathog. 5:e1000537 doi:10.1371/journal.ppat.1000537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lyle JM, et al. 2002. Similar structural basis for membrane localization and protein priming by an RNA-dependent RNA polymerase. J. Biol. Chem. 277:16324–16331 [DOI] [PubMed] [Google Scholar]
- 38. Marianayagam NJ, Sunde M, Matthews JM. 2004. The power of two: protein dimerization in biology. Trends Biochem. Sci. 29:618–625 [DOI] [PubMed] [Google Scholar]
- 39. Martin-Acebes MA, et al. 2008. Subcellular distribution of swine vesicular disease virus proteins and alterations induced in infected cells: a comparative study with foot-and-mouth disease virus and vesicular stomatitis virus. Virology 374:432–443 [DOI] [PubMed] [Google Scholar]
- 40. Martin-Acebes MA, Rincon V, Armas-Portela R, Mateu MG, Sobrino F. 2010. A single amino acid substitution in the capsid of foot-and-mouth disease virus can increase acid lability and confer resistance to acid-dependent uncoating inhibition. J. Virol. 84:2902–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin-Acebes MA, Vazquez-Calvo A, Rincon V, Mateu MG, Sobrino F. 2011. A single amino acid substitution in the capsid of foot-and-mouth disease virus can increase acid resistance. J. Virol. 85:2733–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moffat K, et al. 2005. Effects of foot-and-mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum-to-Golgi transport. J. Virol. 79:4382–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Monaghan P, Cook H, Jackson T, Ryan M, Wileman T. 2004. The ultrastructure of the developing replication site in foot-and-mouth disease virus-infected BHK-38 cells. J. Gen. Virol. 85:933–946 [DOI] [PubMed] [Google Scholar]
- 44. Nayak A, Goodfellow IG, Belsham GJ. 2005. Factors required for the uridylylation of the foot-and-mouth disease virus 3B1, 3B2, and 3B3 peptides by the RNA-dependent RNA polymerase (3Dpol) in vitro. J. Virol. 79:7698–7706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nilsson I, et al. 2010. VEGF receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J. 29:1377–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217 [DOI] [PubMed] [Google Scholar]
- 47. Nunez JI, et al. 2007. Guinea pig-adapted foot-and-mouth disease virus with altered receptor recognition can productively infect a natural host. J. Virol. 81:8497–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nuñez JI, et al. 2001. A single amino acid substitution in nonstructural protein 3A can mediate adaptation of foot-and-mouth disease virus to the guinea pig. J. Virol. 75:3977–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. O'Donnell VK, Pacheco JM, Henry TM, Mason PW. 2001. Subcellular distribution of the foot-and-mouth disease virus 3A protein in cells infected with viruses encoding wild-type and bovine-attenuated forms of 3A. Virology 287:151–162 [DOI] [PubMed] [Google Scholar]
- 50. Pacheco JM, Henry TM, O'Donnell VK, Gregory JB, Mason PW. 2003. Role of nonstructural proteins 3A and 3B in host range and pathogenicity of foot-and-mouth disease virus. J. Virol. 77:13017–13027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peitsch MC. 1996. ProMod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem. Soc. Trans. 24:274–279 [DOI] [PubMed] [Google Scholar]
- 52. Pereira HG. 1981. Foot-and-mouth disease, p 333–363 In Gibbs EPJ. (ed), Virus diseases of food animals. Academic Press Inc., London, United Kingdom [Google Scholar]
- 53. Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM. 2009. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 106:1760–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rosas MF, et al. 2008. Susceptibility to viral infection is enhanced by stable expression of 3A or 3AB proteins from foot-and-mouth disease virus. Virology 380:34–45 [DOI] [PubMed] [Google Scholar]
- 55. Ryan MD, et al. 2004. Foot-and-mouth disease virus proteinases. In Sobrino F, Domingo E. (ed), Foot and mouth disease: current perspectives. Horizon Bioscience, Norfolk, United Kingdom [Google Scholar]
- 56. Saiz M, Gomez S, Martinez-Salas E, Sobrino F. 2001. Deletion or substitution of the aphthovirus 3′ NCR abrogates infectivity and virus replication. J. Gen. Virol. 82:93–101 [DOI] [PubMed] [Google Scholar]
- 57. Samuilova O, Krogerus C, Poyry T, Hyypia T. 2004. Specific interaction between human parechovirus nonstructural 2A protein and viral RNA. J. Biol. Chem. 279:37822–37831 [DOI] [PubMed] [Google Scholar]
- 58. Sanz-Parra A, Sobrino F, Ley V. 1998. Infection with foot-and-mouth disease virus results in a rapid reduction of MHC class I surface expression. J. Gen. Virol. 79(Pt 3):433–436 [DOI] [PubMed] [Google Scholar]
- 59. Schwede T, Kopp J, Guex N, Peitsch MC. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Semler BL, Anderson CW, Hanecak R, Dorner LF, Wimmer E. 1982. A membrane-associated precursor to poliovirus VPg identified by immunoprecipitation with antibodies directed against a synthetic heptapeptide. Cell 28:405–412 [DOI] [PubMed] [Google Scholar]
- 61. Sobrino F, Dávila M, Ortín J, Domingo E. 1983. Multiple genetic variants arise in the course of replication of foot-and-mouth disease virus in cell culture. Virology 128:310–318 [DOI] [PubMed] [Google Scholar]
- 62. Sobrino F, et al. 2001. Foot-and-mouth disease virus: a long known virus, but a current threat. Vet. Res. 32:1–30 [DOI] [PubMed] [Google Scholar]
- 63. Soderberg O, et al. 2006. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3:995–1000 [DOI] [PubMed] [Google Scholar]
- 64. Sonnhammer EL, Hollich V. 2005. Scoredist: a simple and robust protein sequence distance estimator. BMC Bioinformatics 6:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Strauss DM, Glustrom LW, Wuttke DS. 2003. Towards an understanding of the poliovirus replication complex: the solution structure of the soluble domain of the poliovirus 3A protein. J. Mol. Biol. 330:225–234 [DOI] [PubMed] [Google Scholar]
- 66. Suhy DA, Giddings TH, Jr, Kirkegaard K. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74:8953–8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sweeney TR, et al. 2010. Foot-and-mouth disease virus 2C is a hexameric AAA+ protein with a coordinated ATP hydrolysis mechanism. J. Biol. Chem. 285:24347–24359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Takegami T, Kuhn RJ, Anderson CW, Wimmer E. 1983. Membrane-dependent uridylylation of the genome-linked protein VPg of poliovirus. Proc. Natl. Acad. Sci. U. S. A. 80:7447–7451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Towner JS, Ho TV, Semler BL. 1996. Determinants of membrane association for poliovirus protein 3AB. J. Biol. Chem. 271:26810–26818 [DOI] [PubMed] [Google Scholar]
- 70. Tusnady GE, Simon I. 2001. Topology of membrane proteins. J. Chem. Infect. Comput. Sci. 41:364–368 [DOI] [PubMed] [Google Scholar]
- 71. Weibrecht I, et al. 2010. Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev. Proteomics 7:401–409 [DOI] [PubMed] [Google Scholar]
- 72. Wessels E, et al. 2006. Structure-function analysis of the coxsackievirus protein 3A: identification of residues important for dimerization, viral RNA replication, and transport inhibition. J. Biol. Chem. 281:28232–28243 [DOI] [PubMed] [Google Scholar]
- 73. Whitton JL, Cornell CT, Feuer R. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3:765–776 [DOI] [PubMed] [Google Scholar]
- 74. Xiang W, Cuconati A, Hope D, Kirkegaard K, Wimmer E. 1998. Complete protein linkage map of poliovirus P3 proteins: interaction of polymerase 3Dpol with VPg and with genetic variants of 3AB. J. Virol. 72:6732–6741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xiang W, Cuconati A, Paul AV, Cao X, Wimmer E. 1995. Molecular dissection of the multifunctional poliovirus RNA-binding protein 3AB. RNA 1:892–904 [PMC free article] [PubMed] [Google Scholar]
- 76. Xiang W, Harris KS, Alexander L, Wimmer E. 1995. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J. Virol. 69:3658–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zell R, Seitz S, Henke A, Munder T, Wutzler P. 2005. Linkage map of protein-protein interactions of porcine teschovirus. J. Gen. Virol. 86:2763–2768 [DOI] [PubMed] [Google Scholar]