

Abstract
Epstein-Barr virus (EBV) BamHI-A rightward frame 1 (BARF1) is considered a major viral oncogene in epithelial cells and has immune-modulating properties. However, in B cells and lymphomas, BARF1 expression is restricted to the viral lytic replication cycle. In this report, the transcriptional regulation of BARF1 during lytic replication is unraveled. Bisulfite sequencing of various cell lines indicated a high level of methylation of the BARF1 gene control region. A BARF1 promoter luciferase reporter construct was created using a CpG-free vector, enabling true assessment of promoter methylation. Induction of the EBV lytic cycle is mediated by the immediate-early proteins BZLF1 (Z) and BRLF1 (R). R was found to activate expression of the BARF1 promoter up to 250-fold independently of Z and unaffected by BARF1 promoter methylation. Chromatin immunoprecipitation (ChIP), electrophoretic mobility shift assay (EMSA), and specific mutagenesis of the R-responsive elements (RREs) demonstrated direct binding of R to RREs between nucleotides −554 and −327 relative to the BARF1 transcriptional ATG start site. The kinetics of BARF1 expression upon transactivation by R showed that BARF1 mRNA was expressed within 6 h in the context of the viral genome. In conclusion, expression of the BARF1 protein during lytic replication is regulated by direct binding of R to multiple RREs in the gene control region and is independent of the promoter methylation status. The early kinetics of BARF1 upon transactivation by R confirm its status as an early gene and emphasize the necessity of early immune modulation during lytic reactivation.
INTRODUCTION
Epstein-Barr virus (EBV) infects 90% of the world population and persists in the host for life. It causes a relatively mild primary disease if acquired early in life and infectious mononucleosis if acquired after adolescence. EBV has dual tropism in vivo, infecting B lymphocytes and stratified epithelium (52, 57), as reflected by its association with several lymphomas and carcinomas (33, 42, 50, 66). Being a gamma herpesvirus, EBV can infect cells in either latent or lytic form. Lytic replication, required for progeny virus production, occurs in epithelial cells and following differentiation of B cells into plasma cells (32, 35, 52). In latency, EBV expresses only a few genes necessary for viral genome persistence, altering signal transduction and cell cycle control and causing apoptosis inhibition and modulation of immune recognition.
The BamHI-A rightward frame 1 (BARF1) protein is highly and selectively expressed in carcinomas such as nasopharyngeal carcinoma (NPC) and gastric carcinoma (GC) and is considered a major viral oncogene in epithelial cells (13, 55, 58, 64, 67). BARF1 may drive carcinogenesis by immortalizing and transforming epithelial cells of different origins and by upregulating anti-apoptotic Bcl-2, enabling cell survival under inappropriate conditions (56, 63). In addition, secreted hexameric BARF1 inhibits macrophage colony-stimulating factor (M-CSF), thus manipulating myeloid cell growth and functions (12, 26a, 59). In B cells and lymphomas, however, BARF1 expression is restrained to the viral lytic replication cycle (24, 47).
Lytic replication is mediated by the virally encoded DNA polymerase using the oriLyt replication origin and results in the release of infectious viral particles (28, 32). The switch from latent to lytic Epstein-Barr virus (EBV) infection is mediated by the viral immediate-early (IE) proteins BZLF1 (Z, Zta, ZEBRA, EB1) and BRLF1 (R, Rta). Z and R are transcription factors which autostimulate their own expression, reciprocally activate each other, and cooperatively induce expression of all early lytic viral proteins, allowing the virus to replicate (28, 32, 39). Z is a 245-amino-acid (aa) bZip family protein homologous to c-Jun and c-Fos, together forming the AP1 transcription factor. It contains three functional domains: a transactivation (TA) domain (aa 1 to 167), a DNA binding domain (aa 178 to 196), and a dimerization domain (aa 197 to 221) (49). Z binds to the consensus AP1 motif as well as atypical AP1-like motifs known as Z-responsive elements (ZREs) (10, 17, 19, 20).
R is a 605-aa protein with homologues among the gamma herpesviruses (14, 40, 65). It contains a combined N-terminal DNA binding and dimerization domain (aa 1 to 232), and the TA (transactivator) domain is found in the C-terminal region (41). Rta homodimerizes in the absence of DNA. R activates some promoters through a direct binding to specific DNA sequences, known as R-responsive elements (RRE) (21, 26, 27, 31), but other promoters are activated by indirect mechanisms (1, 9, 38, 51). R activates the BZLF1 promoter indirectly through effects on cellular transcription factors (c-Jun and ATF-2) which bind to a cyclic AMP-responsive element (CRE) motif (1, 11, 22). RREs are GC-rich motifs of which the consensus recognition sequence is gNcc-N9-ggNg, where “N9” is a 9-nucleotide linker. The sequences of both the central nucleotides and, to a lesser extent, the flanking sequences contribute to the binding affinity and transcriptional activation by R (11). R promoter activation is inhibited by direct binding of the EBV LF2 protein which mediates its translocation from the nucleus (25). R also directly interacts with the histone acetyl transferase CREB-binding protein (CBP) (60), with Oct-1 (54), and with RanBPM, promoting R sumoylation (8).
Viral gene expression is, in addition to regulation by transcription factors, controlled by epigenetic modulation. The linear EBV genome in virions is not methylated. However, in latently infected cells the majority of the EBV genome becomes highly methylated. EBV uses controlled methylation of its genome initially to prevent production of viral progeny after initial infection, which would kill its host, and to suppress the expression of immunodominant latent viral antigens shortly after host cell immortalization (2, 5, 16, 32, 46). DNA methylation, which plays a critical role in modulating the expression of both cellular and viral genes, induces transcriptional repression by multiple mechanisms, including prevention of transcription factor binding to DNA and the recruitment of histone deacetylase (HDAC) complexes (6). The patterns of EBV genome methylation are specific, and selected viral promoters, such as Qp and both EBER genes and flanking sequences, appear never to be methylated (45, 53, 62). For the (down)regulation of these unmethylated regions, mechanisms other than methylation have previously been proposed (61, 62). Z has an enhanced ability to bind to methylated promoters (4, 15), and methylation is required for the virus to enter the lytic phase (29, 30). R preferentially activates unmethylated lytic promoters; however, methylation does not inhibit DNA binding (C. K. Wille, presented at the 36th International Herpesvirus Workshop, Gdansk, Poland, 24 to 28 July 2011).
BARF1 is considered an early lytic gene (39), but detailed information about its transcriptional activation in the lytic cycle is absent. The control region of BARF1 largely overlaps that of BALF2. The BALF2 coding sequence is on the minus strand and its ATG start site only 734 nucleotides apart from BARF1. The methylation status of the BARF1 gene control region in various cell lines was investigated, showing a high level of methylated CpG islands. So far, it was unknown how BARF1 expression is regulated during the lytic stage. An independent study analyzing R DNA binding using chromatin immunoprecipitation (ChIP) sequencing indicated that R binds to the bidirectional BARF1/BALF2 promoter region, but no detailed mapping was provided and BARF1 gene responsiveness to R was not further analyzed (26). In this study, the regulation of BARF1 in the lytic cycle by Z or by R or by Z and R in combination was investigated. We demonstrate that the BARF1 gene is transactivated by R and not by the major lytic switch protein Z, independently of methylation status, and show direct binding of R to multiple identified RRE sites. Site-directed mutagenesis of RRE sites showed RRE 2 located between −516 and −498 and RRE 3 located between −426 and −409 relative to the BARF1 transcriptional ATG start site to be the major activating sites. These results revealed a new mechanism for the regulation of BARF1 expression.
MATERIALS AND METHODS
Cell culture.
Hone-1, an EBV-negative human nasopharyngeal carcinoma (NPC) cell line, SNU-719, a naturally derived EBV-infected gastric carcinoma cell line (48), and the P3HR1-derived cell line HH514 (a gift of G. Miller) were maintained in RPMI 1640 medium. HeLa cells, 293 cells, and C666.1 cells, a NPC cell line consistently harboring EBV (a gift from D. Thorley-Lawson), were maintained in Dulbecco's modified Eagle's medium (DMEM). C666.1 cells were cultured in fibronectin-coated flasks (Sigma-Aldrich, Buchs, Switzerland). AGS cells were maintained in Ham's F-12 medium. EBV-infected 293 cells, 293 cells infected with a Z knockout EBV mutant, EBV-positive AGS cells (gifts from H.-J. Delecluse), and 293 cells infected with Z stop and R stop EBV have been described previously (18, 54) and were maintained under conditions of hygromycin (Roche, Basel, Switzerland) (100 μg/ml) selection. CNE-2 Akata cells (a gift from K. W. Lo), a NPC cell line superinfected with the Akata strain of EBV, were maintained in RPMI 1640 under conditions of G418 (Invitrogen, Carlsbad, CA) (400 μg/ml) selection. All media contained 10% fetal calf serum (FCS), 100 U/ml sodium penicillin, 100 μg/ml streptomycin sulfate, and 2 mM l-glutamine. HH514 was induced by using 20 ng of 12-tetradecanoylphorbol-13-acetate (TPA) per ml and 3 mM sodium butyrate (NaB) as described previously (43).
Bisulfite sequencing PCR (BSP).
Tumor material from C15 and C17 human NPC xenografted in mice was a kind gift from P. Busson (7). Genomic DNA was isolated from cells and tumor material using silica-based extraction (Basic kit; bioMérieux, Craponne, France). An EZ DNA methylation kit (Zymo Research, Orange, CA) was used for bisulfate treatment of 500 ng DNA, after which the area of interest was amplified using AmpliTaq Gold (Roche). The following primer sequences designed to anneal to conversed DNA, and flanking the two largest methylation islands, were used (GenBank accession no. NC007605): region 1 (164414 to 164615) forward, AGTTAGTTAGGTTGGTTAGGGTTTA; region 1 reverse, CTCAAAATAATACTATACTACACAATAATA; region 2 (164550 to 164792) forward, GTTTTTGTGGTTATTTAGGTAGTTT; region 2 reverse, CCTTTACCAACCCTAATCCTCTAC; region 3 (164771 to 165045) forward, AGAGGATTAGGGTTGGTAAAGGTAG; and region 3 reverse, ACCATTACTCTAAACTCTCCTCACC. The PCR product was sent for direct sequencing to BaseClear (Leiden, The Netherlands).
Plasmids.
Plasmid DNA was purified on maxiprep columns according to the manufacturer's protocol (Qiagen, Venlo, The Netherlands). pSG5 and pcDNA3.1 were obtained from Stratagene (La Jolla, CA) and Invitrogen, respectively. The SG5-R and SG5-Z expression vectors (kindly provided by S. D. Hayward) containing the B95.8 BRLF1 and BZLF1 open reading frames, respectively, and SG5-R (aa 1 to 550) expressing R deleted for the transcriptional activation domain were previously described (23, 54). The BARF1 promoter region from −678 to the ATG start site (164367 to 165045) (GenBank accession no. NC007605) was cloned into pCpG.LUC, a CpG-free luciferase reporter vector kindly provided by M. Rehli (34), using a forward primer with a SpeI site (CTGACTAGTCTCATCACGCAACACCCACTGTTT) and a reverse primer with a BglII site (AATAGATCTGCTCTGGACTCTCCTCACCCAG). To construct deletion mutants, the following forward primers with a SpeI site were used: ATG-633 (CTGACTAGTAAGTCAGTCAGGCTGGCCAGG), ATG-582 (CTGACTAGTGATCTTGGCATGCCGCCCAGC), ATG-468 (CTGACTAGTACCGCAAACACCACTGTGTAGC), ATG-410 (CTGACTAGTGGTCGTTGTACACTGCGCGCAG), ATG-350 (CTGACTAGTCGATGTCGGCTGTCCTGCAGG), ATG-327 (CTGACTAGTAGCTCCGCGTACAGCTTCCTATCC), ATG-261 (CTGACTAGTGGCAAAGGCAGGTCTTTCTCATCC), ATG-220 (CTGACTAGTCATGGCCCTGAACATGAGGTAGC), ATG-156 (CTGACTAGTCACGCCTCGACCGGGGTC), and ATG-63 (CTGACTAGTTGATAAAATGGGCGTGGCAG). The plasmid was propagated in PIR-expressing bacteria (Invitrogen). RRE mutants of the BARF1 promoter reporter construct were created using QuikChange Lightning multisite-directed mutagenesis according to the manufacturer's instructions (Stratagene) to incorporate specific mutations in pCpG.BARF1p(ATG-582).LUC.
In vitro DNA methylation.
The use of a CpG-free reporter construct enables study of the effect of promoter methylation without silencing due to backbone methylation. In vitro DNA methylation of the luciferase constructs was accomplished with CpG methylase (SssI methyltransferase; New England BioLabs, Ipswich, MA), by following the procedure recommended by the manufacturer. Completion of DNA methylation was confirmed by digestion with the restriction enzyme HpaII (New England BioLabs), which cleaves its recognition sequence only if the DNA is not methylated at the cytosine residue within the CpG motif.
Transfections.
Cells were seeded the day prior to transfection. Transfections of HeLa cells for reporter assays were performed by use of Fugene 6 (Roche). Other cell lines were transfected by use of Lipofectamine 2000 (Invitrogen). Transfections were performed according to the manufacturer's instructions, except that for reporter assays the reagent/DNA ratio was 1.5 μl:0.5 μg in 100 μl Opti-MEM for 2 × 105 cells plated in 1 ml medium in a 12-well plate.
Luciferase assays.
Luciferase assays were performed 48 h after transfection by using extracts prepared by freeze-thawing the cell pellet in reporter lysis buffer according to the instructions of the manufacturer (Promega, Madison, WI). Luciferase activity was assayed using the luciferase reporter assay system (Promega) as suggested by the manufacturer.
ChIP assay.
Chromatin immunoprecipitation (ChIP) was performed as described previously (54). Briefly, 293 BRLF1-stop cells were transfected with pSG5 or pSG5-R and cross-linked after 24 h in EGS (ethylene glycolbis [succinimidyl succinate]) followed by fresh 1% paraformaldehyde. Following cell lysis and DNA fragmentation by sonication, DNA-protein complexes were immunoprecipitated with anti-BRLF1 (Argene, Shirley, NY) and control anti-IgG (Santa Cruz, Santa Cruz, CA) antibodies. Protein-DNA cross-linking was reversed at 65°C overnight, and DNA was purified using a Qiagen gel extraction kit. The presence of BARF1 promoter DNA fragments in each precipitate was detected using PCR with forward primer GGCCCTGAACATGAGGTAGC and reverse primer TCTGGACTCTCCTCACCCAG (164829 to 165042), and primers for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were forward primer TCACCACCATGGAGAAGGCT and reverse primer GCCATCCACAGTCTTCTGGG.
Electrophoretic mobility shift assays (EMSAs).
R550 protein extract and control extract were created as previously described by Chen et al. (11) by lysis of SG5-R550- and SG5-transfected HeLa cells. Cells were harvested, centrifuged, and snap-frozen at −80°C. Frozen cell pellets were suspended in lysis buffer containing 0.42 M NaCl, 20 mM HEPES (pH 7.5), 25% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail (Roche), followed by 15 min of maximum-speed centrifugation at 4°C. Supernatants were stored at −80°C, and protein concentrations were determined by the Bradford method (Bio-Rad, Hercules, CA). Annealed double-stranded oligonucleotides (see Fig. 3C) were end labeled with 32P using T4 polynucleotide kinase (New England BioLabs) and desalted with G-25 Sephadex columns (Roche). Binding reactions were performed in buffer containing 10 mM HEPES (pH 7.5), 50 mM NaCl, 2 mM MgCl2, 2.5 μM ZnSO4, 0.5 M EDTA, 1 mM DTT, 15% glycerol, and 0.5 μg poly(dI-dC), using 15 μg total cell lysate followed, after 5 min at room temperature, by 11,000 cpm of labeled nucleotide in a total volume of 50 μl. For supershift reactions, anti-BRLF1 (Argene) was added 20 min following addition of the probe. The reaction mixtures were incubated for 40 min at room temperature before being loaded onto a 4% polyacrylamide gel in 0.5× Tris-borate-EDTA buffer at 35 mA. Gels were dried on Whatman paper under vacuum conditions and exposed to autoradiography film for 12 to 40 h at −80°C.
Fig 3.

R binds to RREs on the BARF1 promoter. (A) Potential R-responsive elements (R1 to R7) are depicted on the BARF1 promoter region. Black vertical lines represent methylation sites. Rounded gray indicators point to the deletion mutants made from the BARF1 reporter construct, shortening the BARF1 promoter sequence from the original −679 to −63 relative to the ATG start site. The asterisk indicates the R ChIP sequence peak as determined by Heilman et al. (25). (B) ChIP assays were performed using extracts from HEK293 BRLF1-stop cells transfected with an R expression vector (R) or a control vector (−). R was immunoprecipitated by a control antibody (Ab) or anti-R antibody, and coimmunoprecipitated DNA was PCR amplified. The band in the last lane indicates that the BARF1 promoter region DNA specifically precipitated with R. (C) The RRE optimal sequence and the consensus sequence according to Chen et al. (11) are shown. Oligonucleotides of the potential RREs in the BARF1 promoter with surrounding nucleotides were used in EMSA. To create double-stranded probes, oligonucleotides were hybridized with their respective opposite strands. The oligonucleotide of the BMLF1 promoter RRE served as a positive control. CpG sites are indicated by stars. (D) The ability of in vitro-translated C-terminal-truncated R (R550) to bind to 23P-end-labeled probes of the potential RREs in the BARF1 promoter was examined by EMSA. Extracts were made of HeLa cells transfected with a control (−) or with R550 (+). A probe containing the BMLF1 promoter RRE served as a positive control. Four of the seven RREs (RRE 1 to 4) on the BARF1 promoter showed binding (R). S, supershift; F, unbound oligonucleotide. (E) Deletion mutants of the BARF1 promoter reporter construct were made as indicated in panel B. AGS cells were transfected with the deletion mutant luciferase constructs and with or without R expression vector. The R-induced luciferase activity (fold) was measured 48 h after transfection. Data represent the results of a representative experiment.
Quantitative RT-PCR.
Cells were plated in 6-well plates and transfected with either SG5-R or SG5 as a control. At designated time points, cells were harvested in 1 ml TRIzol (Invitrogen) and stored at −80°C until further processing. Guanidinium isothiocyanate-phenol-chloroform extraction was performed to isolate total cellular RNA, followed by DNase (Promega) treatment and ethanol precipitation. cDNA was synthesized using avian myeloblastosis virus (AMV) reverse transcriptase (RT; Promega) and sequence-specific primers BARF1 forward (GCCTCTAACGCTGTCTGTCC) and reverse (GAGAGGCTCCCATCCTTTTC) (165414 to 165433) (GenBank accession no. NC007605), R forward (TGATGATTCCTGCCACCATA) and reverse (GAGGACGGGATAGGTGAACA) (92289 to 92507), and U1A forward (CAGTATGCCAAGACCGACTCAGA) and reverse (GGCCCGGCATGTGGTGCATAA). RT-PCR was performed with SybrGreen (Roche) and the aforementioned primers using a LightCycler 480 system (Roche) After quantification to known concentrations of the corresponding gene constructs, values were normalized to U1A.
SDS-PAGE and Western blot analysis.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitor cocktail (Roche) and sonicated, after which cell debris was removed by centrifugation. Supernatants were diluted in 2× loading buffer (Bio-Rad) with β-mercaptoethanol, denatured for 5 min at 95°C, and separated on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. After transfer to Hybond ECL nitrocellulose membrane (GE Healthcare, Little Chalfont, United Kingdom), the membrane was blocked in phosphate-buffered saline (PBS) with 0.05% Tween 20 (PBST) containing 3% nonfat dried milk for 1 h at room temperature, after which the following antibodies were incubated for 2 h at room temperature in PBST containing 5% bovine serum albumin (BSA): anti-BRLF1 (Argene) (1:250), anti-BZLF1 (catalog no. sc-53904; Santa Cruz Biotechnology) (1:250), and anti-β-actin peroxidase (Sigma-Aldrich) (1:10,000). After incubation with peroxidase-labeled secondary antibody rabbit anti-mouse (Dako, Glostrup, Denmark), bands were visualized with ECL Plus (GE Healthcare).
RESULTS
The BARF1 promoter is highly methylated in cell lines and carcinomas.
The methylation status of EBV promoters influences regulation by various transcription factors. Analysis of the number of CpGs in the BARF1 promoter by using Methprimer (36) showed two large CpG islands spanning the whole putative promoter region up to −607 relative to the BARF1 ATG start site (Fig. 1A). Bisulfite sequencing PCR (BSP) primers were developed to completely analyze the region from ATG-632 to the ATG start site comprising the CpG regulatory elements. Two cell lines, HH514 Burkitt's lymphoma cells and C666.1 cells consistently harboring EBV, were studied next to C15 and C17 mouse xenograft NPC tumor material (7). BARF1 mRNA was expressed in the C666.1 NPC cell line and in C15 and C17 NPC mouse tumor (Fig. 1B). After bisulfite treatment, the CpG-rich regions were amplified for sequence analysis. Almost all CpGs in the investigated samples were methylated (Fig. 1C, black dots), in both the carcinoma and B cell lines, indicating that BARF1 was expressed, despite methylation of its promoter. CpG site I was (partly) unmethylated (white dot) in C666.1 cells and C17 tumor material. CpG site II was unmethylated in HH514 cells. Treatment of HH514 cells with the histone deacetylase inhibitor sodium butyrate (NaB) combined with TPA for 24 h induced the early lytic cycle, which is associated with hypomethylation of the viral DNA (44). BSP methylation analysis of NaB-TPA-treated HH514 cells demonstrated that the vast majority of the BARF1 promoter was demethylated (Fig. 1D). Quantitative analysis confirmed that expression of the lytic cycle immediate-early gene R and the early gene BARF1 mRNA was induced by NaB-TPA treatment (Fig. 1B).
Fig 1.

Methylation of the BARF1 promoter region. (A) Methprimer analysis of CpG islands in the BARF1 promoter showed two large CpG islands spanning the whole promoter up to −607 nucleotides relative to the BARF1 ATG start site. Obs, observed; Exp, expected. (B) Quantitative PCR of BARF1 and R mRNA normalized to U1A. BARF1 mRNA was detected both in C15 and C17 NPC mouse tumor material and in C666.1 NPC cells. R levels in C15 mouse tumor material were below the detection limit. BARF1 mRNA was present at low levels in HH514 cells but could be expressed upon treatment with NaB and TPA. (C) Bisulfite sequencing PCR (BSP) using primers as indicated (arrows) was performed to analyze the CpG islands on the BARF1 promoter from ATG-632 to the ATG start site. Almost all CpGs in the investigated samples were methylated (black dots), both in HH514 Burkitt's lymphoma cells and in C666.1 cells consistently harboring EBV and C15 and C17 mouse xenograft NPC tumor material. CpG site I was unmethylated (white dots) in C666.1 cells and C17 tumor material. CpG site II was unmethylated in HH514 cells. (D) BSP analysis of 24-h NaB-TPA-treated HH514 cells shows that the BARF1 promoter was partially (gray dots) or completely (white dots) demethylated.
R, but not Z, activates the BARF1 promoter, independently of methylation status.
To examine if and which of the immediate early proteins were responsible for the induction of the BARF1 gene in the lytic cycle, the BARF1 promoter sequence (up to −679 nucleotides from the ATG start site) was inserted upstream of the luciferase gene in a pCpG-free reporter construct. EBV-positive cells have always a certain amount of baseline lytic activity; 1% to 5% of the cells express Z and/or R. The constitutive activity of the BARF1 promoter was higher in EBV-positive cells (Fig. 2A), indicating that an EBV gene was responsible for the activity. The reporter construct was cotransfected with an expression vector containing the R gene in multiple EBV-positive and -negative cell lines. Induction of luciferase activity was evaluated 48 h after transfection. Equal R expression levels were confirmed by Western blot analysis (data not shown). R induced 50- to 250-fold upregulation of luciferase activity in both EBV-positive and -negative cell lines (Fig. 2B). The BARF1 promoter region was found to be highly methylated. To examine how methylation affects its ability to be activated, the pCpG luciferase construct containing the BARF1 promoter was ex vitro methylated using methyltransferase or mock treated. Both the constitutive activity and the R-induced activity of the BARF1 promoter-driven luciferase construct were weaker in the methylated construct, leaving the fold induction mostly unaffected by methylation (Fig. 2C and D). With some promoters, Z preferentially binds to the methylated versus the unmethylated Z response elements (4, 15). A minor induction of expression of the methylated construct was seen upon cotransfection with the Z expression vector (Fig. 2D), and, depending on the cell line, Z mediated an average of a 45-fold induction of luciferase activity of the methylated construct (data not shown). Cotransfection with both Z and R resulted in an induction of luciferase activity lower than that seen when R alone was expressed (data not shown).
Fig 2.

R, but not Z, activates the BARF1 promoter, independent of methylation status and in the context of the viral genome. A BARF1 promoter reporter construct was created by inserting the promoter sequence up to −679 nucleotides from the ATG start site upstream of the luciferase gene in a CpG-free reporter construct. (A) Luciferase assays showed that the constitutive activity of the BARF1 promoter in pCpG.LUC versus pCpG.LUC empty was highest in EBV-positive C666.1 and SNU-719 cells. (B) Cotransfection of the reporter construct with an R expression vector induced a 50- to 250-fold upregulation of luciferase activity in most cell lines and up to 3,000-fold upregulation in HeLa cells. For HeLa, AGS, and SNU-719 cells, n = 2; for C666.1 and Hone1 cells, n = 3. Standard errors of the means (SEM) are shown. (C) An ex vitro-methylated reporter construct maintained its activation when cotransfected with an R expression vector. (D) Representative example of unmethylated or methylated promoter construct cotransfected either with an R or Z expression vector in AGS cells. Both empty vector (−) and R vector relative light unit (RLU) values were lower when the promoter luciferase construct was methylated, leaving the fold activation mostly unaffected. SDS-PAGE Western blot analysis confirmed equal expression levels of Z and R protein. (E) EBV-positive C666.1 and AGS B95-8 cells expressed more BARF1 mRNA 48 h after transfection with R, indicating that R upregulates BARF1 in the context of EBV infection. con, control. (F) Parallel experiments using Z-defective HEK293 cells (Z-stop) and Z knockout (ZKO) AGS cells demonstrated autonomous R-transactivating activity of BARF1 that was independent of the lytic cycle. When only 1% of the transfected DNA consisted of R expression vector, induction of BARF1 was reduced, indicating that BARF1 induction was R dosage dependent.
R alone can induce BARF1 expression in EBV-infected epithelial cells.
To obtain conclusive evidence that R activates BARF1 RNA expression in the context of EBV infection, EBV-positive cells were transfected with R expression vector and quantitative RT-PCR was performed. CNE Akata (not shown) and AGS B95.8 cells, both carrying recombinant EBV, and C666.1 cells, a NPC cell line consistently harboring EBV, showed induction of BARF1 mRNA above basal expression levels when R was transfected (Fig. 2E). To rule out the possibility that this induction was independent of viral replication, cells with a Z-defective EBV, 293 Zstop and AGS ZKO, incapable of lytic induction were transfected with the R expression vector. The strong induction of BARF1 mRNA confirms that R is capable of transactivating the BARF1 promoter in the context of the viral genome, independently of Z or viral replication (Fig. 2F). The increase of BARF1 RNA expression was R dosage dependent.
Multiple RREs are mapped between −544 and −327 nucleotides relative to the ATG start site.
The BARF1 promoter region harbors multiple potential R-responsive elements (RREs) based on consensus sequence gNcc-N9-ggNg (11) as depicted in Fig. 3A. To determine if R directly binds to the BARF1 promoter region in EBV-harboring cells, chromatin immunoprecipitation (ChIP) assays were performed using extracts from BRLF1-stop cells transfected with an R expression vector or a control vector. R was precipitated by anti-R antibody, and cross-linked DNA was PCR amplified. As shown in Fig. 3B, the ChIP assays demonstrated that R binds to the BARF1 promoter region or in its immediate proximity.
To map the precise locations of the RREs in the BARF1 promoter, a series of probes was designed spanning each of the various RREs and surrounding nucleotides in the BARF1 promoter (Fig. 3C). The ability of R to bind to these RRE probes was evaluated in an electromobility shift assay (EMSA), using lysate of cells transfected with an R550 (aa 1 to 550) expression vector, which lacks the activation domain and was previously shown to bind with higher affinity than full-length R in EMSA (11). A probe containing the EBV BMLF1 (SM) promoter RRE, previously found to be a strong RRE (11), served as a positive control. The probes that showed detectable R binding contained BARF1 promoter sequences from nucleotides −544 to −327 relative to the BARF1 transcriptional ATG start site (Fig. 3D). Specific interaction between the oligonucleotides and R550 was confirmed by supershift (S) with antibody to R. Remarkably, the probe containing a predicted RRE sequence with the best homology to the optimal R binding element (R7) (11) did not show detectable binding in EMSA (not shown). The BARF1 RRE probes also demonstrated binding to unknown cellular factors, some of which have the same migration properties as R. Therefore, the supershift bands with antibody against R are more visible. These unknown factors are not further described in this report. The BMLF1 oligonucleotide forms, apart from R, a complex with YY1 (11).
To further identify the RRE responsible for BARF1 promoter activation by R, deletion mutants of the reporter construct were made, shortening the BARF1 promoter sequence from the original −679 to −63 nucleic acids relative to the ATG start site in small, 23- to 114-nucleotide steps (Fig. 3B). After removal of the region between ATG-679 and -582, an initial 3-fold increase of luciferase activity to 900-fold was seen (Fig. 3E), which can be explained by loss of interfering proteins or suppressors. When R1 and R2 (between 5′ deletion ATG-582 and -468) were deleted, luciferase activity showed a 14-fold decrease and continued to slowly decrease down to ATG-327 (Fig. 3E), indicating that multiple RREs might work in synergism.
RREs are required for R-dependent BARF1 promoter activation.
To illustrate the importance of the RREs for BARF1 promoter activation by R, mutants of the reporter construct were made in which single RREs or the combination of all four were mutated in the CG-rich motif (Fig. 4A). These constructs were transfected into AGS cells together with the expression vector for R, and luciferase activity was measured. Mutation of RRE 2 and RRE 3 showed a decrease in luciferase activity compared to the control (Fig. 4B). Mutation of RRE 1 and 4 showed an increase in luciferase activity. It is possible that these single mutations allowed more R binding space for RRE 2 and 3 or that new cellular transcription factor binding sites were created. When all four RREs were mutated, luciferase activity was reduced by 84%.
Fig 4.
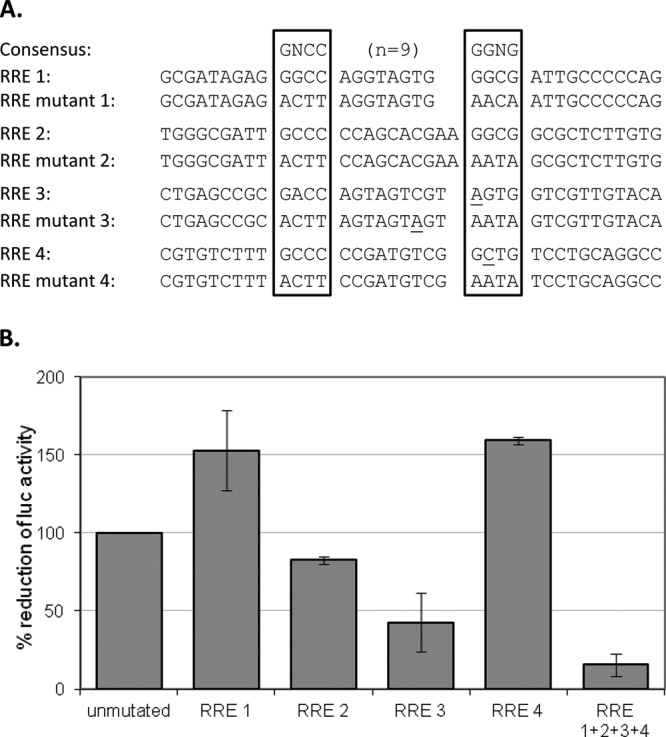
RREs are required for R-dependent BARF1 promoter activation. (A) Site-directed mutations of the RREs in a BARF1 promoter ATG-582 luciferase reporter construct were made. Seven nucleotides, predominantly in the core sequences, were selected for mutation. The original sequences and their respective mutations are shown. (B) AGS cells were transfected with the mutated reporter constructs in combination with either the empty vector or the R expression vector. The amount of luciferase activity was determined 48 h after transfection. The induction of luciferase activity in 3 experiments is shown.
BARF1 expression kinetics in response to R.
The kinetics of R-responsive genes differs among cell types and depends on the type of lytic stimuli used (11, 31, 39). The abundance of BARF1 RNA was examined at various time points after transfection with an R expression vector in AGS B95.8 wild-type (wt) cells and compared to that seen with cells that were transfected with control vector. Expression of BARF1 mRNA in these cells was induced 15 h after transfection with R expression vector, and fold induction was low (∼35) compared to empty vector control induction (Fig. 5A). AGS B95.8 wt cells have some lytic cells in the pool (37), expressing R protein (Fig. 5B) and BARF1, which could shield new induction of BARF1 RNA by R. To obtain a more accurate and sensitive readout of BARF1 expression kinetics by R, AGS Z knockout cells, which do not express Z and have no background expression of R protein, were used (Fig. 5D). Baseline levels of R mRNA were 630-fold lower in AGS ZKO cells than in AGS B95.8 wt cells (Fig. 5E), and baseline levels of BARF1 RNA were 38-fold lower in AGS ZKO cells than in AGS B95.8 wt cells (Fig. 5F). In AGS ZKO cells, induction of BARF1 RNA can be detected as early as 6 h after transfection with R expression vector (Fig. 5C), confirming that expression of BARF1 as an early gene during lytic reactivation is activated by immediate-early R-transactivating protein.
Fig 5.

R transactivates BARF1 mRNA with early kinetics. (A) AGS B95.8 cells were transfected with R expression vector or empty vector. Cells were collected at different time points after transfection, and RNA was isolated and converted to cDNA using gene-specific primers. Quantitative RT-PCR was performed, and BARF1 values were normalized to U1A. The graph represents the fold induction of BARF1 mRNA by R (data represent averages of the results of two experiments). (B) SDS-PAGE Western blot analysis of R expression levels in AGS B95.8 cells after transfection with empty vector (−) and at various time points after transfection with R expression vector. (C) AGS ZKO cells were transfected with R expression vector or empty vector, and RT-PCR was performed on samples harvested at various time points as described for panel A. Values of fold induction of BARF1 mRNA by R, representing averages of the results of three experiments, are shown. (D) SDS-PAGE Western blot analysis of R expression levels in AGS ZKO cells after transfection with empty vector (−) and at various time points after transfection with R expression vector. R protein was detectable 6 h after transfection. (E) R mRNA levels indicated a 630-fold difference in R baseline expression between AGS ZKO and AGS B95.8 cells. (F) BARF1 mRNA levels indicated a 38-fold difference in BARF1 baseline expression between AGS ZKO and AGS B95.8 cells.
DISCUSSION
Secreted BARF1 protein is a potent immune modulator, and, intracellularly, BARF1 may drive carcinogenesis. BARF1 is considered an early lytic gene, but detailed information about its epigenetic regulation and its transcriptional activation in the lytic cycle is absent (39). Latency of EBV is enabled by dense methylation of the viral episome. The methylation status of an EBV promoter influences its regulation by various transcription factors, and it generally induces transcriptional repression. In latency, BARF1 is expressed; however, this is observed only in EBV-related carcinomas and not in lymphomas (67). In carcinomas, BARF1 expression is not related to lytic cycle activation (55). The transcription of BARF1 in latent carcinomas might be explained by different levels of epigenetic regulation and/or involvement of different host cell transcription factors. The methylation status of the BARF1 gene control region in various cell lines was investigated, and almost all CpGs were methylated, in both carcinoma and B cell lines, indicating that a BARF1 transcription factor(s) must be able to overcome methylation-induced repression. When NaB and TPA were used to induce the lytic cycle, virtually all CpGs were demethylated. However, this can be a combined effect of the histone deacetylase-inhibiting function of NaB in parallel with lytic cycle-induced demethylation.
The switch from latent to lytic EBV infection is mediated by the viral immediate-early proteins Z and R inducing a series of nonstructural viral genes preparing the cell for survival and viral DNA replication, finally resulting in the production and release of infectious viral particles (28, 32). BARF1 belongs to the group of nonstructural EBV genes, functioning in apoptosis resistance and immune modulation (26a, 56, 59, 63). This study reveals that R, and not Z, is the BARF1 trancriptional activator. R transactivates BARF1 by direct binding to its promoter, and methylation does not influence R-transactivating activity of the BARF1 promoter. EBV viral genome methylation differentially affects BZLF1 (Z) versus BRLF1 (R) activation of lytic viral promoters. Z has an enhanced ability to bind to methylated versus unmethylated ZREs of the Na and R promoters but not other EBV promoters (4, 15), while R activation of lytic promoters can be inhibited by methylation (BMRF1) or even put to a stop (BALF2, BLRF2) (Wille, Herpes Meeting, Poland). Assays using a BARF1 promoter reporter construct, either methylated or unmethylated, showed R, and not Z, to be responsible for the induction of BARF1 transcription. Z was only moderately capable of inducing the methylated reporter construct, and the Z-responsive elements found in the BARF1 promoter area are reversed oriented (3). Methylation reduced both the constitutive activity (approximately 4-fold lower; data not shown) and the R-induced activity of the BARF1 promoter reporter construct, leaving the fold induction by R mostly unaffected by methylation and indicating that other cellular transcription factors are affected by the methylation.
In induced HH514 BL cells, C666.1 NPC cells, and C17 xenografts, BARF1 as well as R mRNA was present (Fig. 1B). However, in the C15 xenografts, only BARF1 mRNA could be detected. BARF1 is highly expressed in NPC and GC, in which EBV displays a latency phenotype with no expression of lytic cycle genes (13, 55, 58, 64, 67). In carcinomas, BARF1 expression is likely regulated by a cell type-specific transcription factor(s) other than the R early gene, explaining the presence of R-independent BARF1 mRNA in the C15 xenografts.
Many RREs in the BARF1 promoter as well as other gene promoters contain CpG motifs in both the core and linker regions, and R binding to these RREs is not affected by methylation. Differences in levels of epigenetic modification in gene control regions, ranging from the RRE to the TATA box and polymerase binding site, might explain why methylation is not inhibitory for BARF1 transcriptional activation whereas it is for other R-responsive genes. The relative distance to the TATA box and the quality and quantity of RREs combined with epigenetic modifications all together influence the interaction with other factors of the transcriptional apparatus.
Expression of R in cells harboring full or Z knockout EBV episomes demonstrated that R is capable of transactivating the BARF1 promoter in the context of the viral genome and independently of Z or viral replication. Chromatin immunoprecipitation further demonstrated that R binds to the BARF1 promoter region. The R transcriptional activator binds to a specific DNA sequence. Chen et al. previously published the consensus and optimal RRE sequences, based on mutagenesis of the core and 9-nucleotide linker (N9) sequences in the RRE in the BMLF1 promoter (11). Based on this consensus sequence, leaving room for error, we found that the BARF1 promoter region harbors seven potential RREs (Fig. 3B). EMSA showed R binding to four probes containing BARF1 promoter sequences from −544 to −327 relative to the BARF1 transcriptional ATG start site. Experiments with 5′ deletion mutants of the reporter construct showed a 14-fold decrease of activity when RRE 1 and RRE 2 were lost and an additional 4-fold decrease after loss of RRE 3 and RRE 4, indicating that the RREs work in synergism. Results seen with mutations of the RREs in the BARF1 promoter reporter construct illustrate the importance of the RREs in promoter activation by R. Single mutation of RRE 2 or RRE 3 results in loss of transactivating activity, and mutation of all four RREs reduces luciferase activity to 16% of the activity of the unmutated control. We found that the major RREs responsible for R activation of the BARF1 promoter were RRE 2, located between −516 and −498 relative to the BARF1 transcriptional ATG start site, and RRE 3, located between −426 and −409. Independently, Heilmann et al. (26) indicated that R binds to the bidirectional BALF2/BARF1 promoter. The BALF2 primer set used to determine R binding by ChIP analysis flanks RRE 2 and overlaps RRE 3, with the high-confidence ChIP sequence peak in the middle, as indicated by the asterisk in Fig. 3A. Although cell type differences and limitations of ChIP have to be considered, our findings largely agree with their data.
Since RRE 7 overlaps best with the optimal RRE, it is remarkable that it does not show binding in EMSA. Also, the 5′ deletion mutants of the BARF1 promoter reporter construct indicate RRE 7 to be nonfunctional as an R binding site. On the other hand, our group identified in the BARF1 promoter four RREs which all differ from the consensus RRE but did show binding in EMSA and were together capable of a strong promoter induction. RRE 1 and RRE 2, although having consensus core sequences, enclose internal spaces which deviate from the consensus in length. The second core sequence of RRE 3 is agtg instead of the consensus ggNg, and also the second core sequence of RRE 4 (gctg) differs from the consensus. Our four RRE oligonucleotides showed a low binding affinity in EMSA compared to the BMLF1 positive-control oligonucleotide. Nevertheless, R was found to induce a 50- to 250-fold upregulation of luciferase activity. Most likely, the four RREs in the BARF1 promoter region work in synergism, with RRE 2 and RRE 3 the dominant responsible sites. Furthermore, the EMSA radiographs of all four BARF1 RRE oligonucleotides showed additional complexes of various sizes with other cellular factors (Fig. 3D). Future studies utilizing DNA pulldown assays combined with mass spectrometry proteomics analysis might elucidate the identity and importance of the unknown factors in BARF1 gene regulation.
Previously, Chen et al. indicated that the kinetics of expression of R-transactivated genes did not correlate with RRE affinity and that other components might interfere with transcriptional activation (11). Although the affinities of R to the RREs in the BARF1 promoter region were not strong, BARF1 mRNA expression was detected rapidly at 6 h after transfection with an R expression vector in the AGS ZKO cell line. The maximum levels of BARF1 RNA were observed at 15 h. The BARF1 expression kinetics induced by R were similar, as observed by a microarray analysis of EBV lytic gene transcription induced by IgG cross-linking of Akata cells which resulted in BARF1 mRNA expression starting from 6 h after induction and reaching maximum levels after 12 h (39). The classification of BARF1 as an early gene indicates that the action of BARF1 protein in a biological context, inhibiting apoptosis and acting as an immune modulator, is necessary in the early hours of lytic replication.
In conclusion, expression of the BARF1 protein during lytic replication is directly regulated by R, and not by the major IE protein Z, independently of BARF1 promoter methylation. The R-responsive elements were mapped between −544 and −327 relative to the BARF1 transcriptional ATG start site. The fast kinetics of BARF1 expression induced by R indicated that BARF1 protein expression is necessary early during the viral replication cycle. Future experiments are necessary to unravel the role of cellular factors in BARF1 transcriptional regulation, both during lytic reactivation by R and in latent EBV-positive carcinoma.
ACKNOWLEDGMENTS
This study was supported by the Dutch Cancer Society (grant VU2007-3776).
We thank Sandra Verkuijlen and Elisabeth Barlow for their excellent technical support.
Footnotes
Published ahead of print 15 August 2012
REFERENCES
- 1. Adamson AL, et al. 2000. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J. Virol. 74:1224–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ambinder RF, Robertson KD, Tao Q. 1999. DNA methylation and the Epstein-Barr virus. Semin. Cancer Biol. 9:369–375 [DOI] [PubMed] [Google Scholar]
- 3. Bergbauer M, et al. 2010. CpG-methylation regulates a class of Epstein-Barr virus promoters. PLoS Pathog. 6:e1001114 doi:10.1371/journal.ppat.1001114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. 2004. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat. Genet. 36:1099–1104 [DOI] [PubMed] [Google Scholar]
- 5. Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. 2005. BZLF1 activation of the methylated form of the BRLF1 immediate-early promoter is regulated by BZLF1 residue 186. J. Virol. 79:7338–7348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bird AP, Wolffe AP. 1999. Methylation-induced repression—belts, braces, and chromatin. Cell 99:451–454 [DOI] [PubMed] [Google Scholar]
- 7. Busson P, et al. 1988. Establishment and characterization of three transplantable EBV-containing nasopharyngeal carcinomas. Int. J. Cancer 42:599–606 [DOI] [PubMed] [Google Scholar]
- 8. Chang LK, et al. 2008. Enhancement of transactivation activity of Rta of Epstein-Barr virus by RanBPM. J. Mol. Biol. 379:231–242 [DOI] [PubMed] [Google Scholar]
- 9. Chang Y, et al. 2004. Induction of Epstein-Barr virus latent membrane protein 1 by a lytic transactivator Rta. J. Virol. 78:13028–13036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang YN, Dong DL, Hayward GS, Hayward SD. 1990. The Epstein-Barr virus Zta transactivator: a member of the bZIP family with unique DNA-binding specificity and a dimerization domain that lacks the characteristic heptad leucine zipper motif. J. Virol. 64:3358–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen LW, Chang PJ, Delecluse HJ, Miller G. 2005. Marked variation in response of consensus binding elements for the Rta protein of Epstein-Barr virus. J. Virol. 79:9635–9650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cohen JI, Lekstrom K. 1999. Epstein-Barr virus BARF1 protein is dispensable for B-cell transformation and inhibits alpha interferon secretion from mononuclear cells. J. Virol. 73:7627–7632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Decaussin G, Sbih-Lammali F, de Turenne-Tessier M, Bouguermouh A, Ooka T. 2000. Expression of BARF1 gene encoded by Epstein-Barr virus in nasopharyngeal carcinoma biopsies. Cancer Res. 60:5584–5588 [PubMed] [Google Scholar]
- 14. DeWire SM, McVoy MA, Damania B. 2002. Kinetics of expression of rhesus monkey rhadinovirus (RRV) and identification and characterization of a polycistronic transcript encoding the RRV Orf50/Rta, RRV R8, and R8.1 genes. J. Virol. 76:9819–9831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dickerson SJ, et al. 2009. Methylation-dependent binding of the epstein-barr virus BZLF1 protein to viral promoters. PLoS Pathog. 5:e1000356 doi:10.1371/journal.ppat.1000356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ernberg I, et al. 1989. The role of methylation in the phenotype-dependent modulation of Epstein-Barr nuclear antigen 2 and latent membrane protein genes in cells latently infected with Epstein-Barr virus. J. Gen. Virol. 70(Pt 11):2989–3002 [DOI] [PubMed] [Google Scholar]
- 17. Farrell PJ, Rowe DT, Rooney CM, Kouzarides T. 1989. Epstein-Barr virus BZLF1 trans-activator specifically binds to a consensus AP-1 site and is related to c-fos. EMBO J. 8:127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feederle R, et al. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 19:3080–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flemington E, Speck SH. 1990. Autoregulation of Epstein-Barr virus putative lytic switch gene BZLF1. J. Virol. 64:1227–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Flemington E, Speck SH. 1990. Evidence for coiled-coil dimer formation by an Epstein-Barr virus transactivator that lacks a heptad repeat of leucine residues. Proc. Natl. Acad. Sci. U. S. A. 87:9459–9463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gruffat H, Manet E, Rigolet A, Sergeant A. 1990. The enhancer factor R of Epstein-Barr virus (EBV) is a sequence-specific DNA binding protein. Nucleic Acids Res. 18:6835–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gruffat H, Sergeant A. 1994. Characterization of the DNA-binding site repertoire for the Epstein-Barr virus transcription factor R. Nucleic Acids Res. 22:1172–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hardwick JM, Lieberman PM, Hayward SD. 1988. A new Epstein-Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J. Virol. 62:2274–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hatfull G, Bankier AT, Barrell BG, Farrell PJ. 1988. Sequence analysis of Raji Epstein-Barr virus DNA. Virology 164:334–340 [DOI] [PubMed] [Google Scholar]
- 25. Heilmann AM, Calderwood MA, Johannsen E. 2010. Epstein-Barr virus LF2 protein regulates viral replication by altering Rta subcellular localization. J. Virol. 84:9920–9931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heilmann AM, Calderwood MA, Portal D, Lu Y, Johannsen E. 2012. Genome-wide analysis of Epstein-Barr virus Rta DNA binding. J. Virol. 86:5151–5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26a. Hoebe EK, et al. Epstein-Barr virus encoded BARF1 protein is a decoy receptor for macrophage colony stimulating factor and interferes with macrophage differentiation and activation. Viral Immunol., in press [DOI] [PubMed] [Google Scholar]
- 27. Hung CH, Liu ST. 1999. Characterization of the Epstein-Barr virus BALF2 promoter. J. Gen. Virol. 80(Pt 10):2747–2750 [DOI] [PubMed] [Google Scholar]
- 28. Israel BF, Kenney SC. 2011. EBV lytic infection, p 571–611 In Robertson ES. (ed), Epstein-Barr virus. Caister Academic Press, Philadelphia, PA [Google Scholar]
- 29. Kalla M, Gobel C, Hammerschmidt W. 2012. The lytic phase of epstein-barr virus requires a viral genome with 5-methylcytosine residues in CpG sites. J. Virol. 86:447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kalla M, Schmeinck A, Bergbauer M, Pich D, Hammerschmidt W. 2010. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc. Natl. Acad. Sci. U. S. A. 107:850–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kenney S, Holley-Guthrie E, Mar EC, Smith M. 1989. The Epstein-Barr virus BMLF1 promoter contains an enhancer element that is responsive to the BZLF1 and BRLF1 transactivators. J. Virol. 63:3878–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2654 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott, Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 33. Klein G, et al. 1974. Direct evidence for the presence of Epstein-Barr virus DNA and nuclear antigen in malignant epithelial cells from patients with poorly differentiated carcinoma of the nasopharynx. Proc. Natl. Acad. Sci. U. S. A. 71:4737–4741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klug M, Rehli M. 2006. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 1:127–130 [DOI] [PubMed] [Google Scholar]
- 35. Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79:1296–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li LC, Dahiya R. 2002. MethPrimer: designing primers for methylation PCRs. Bioinformatics 18:1427–1431 [DOI] [PubMed] [Google Scholar]
- 37. Li Y, Webster-Cyriaque J, Tomlinson CC, Yohe M, Kenney S. 2004. Fatty acid synthase expression is induced by the Epstein-Barr virus immediate-early protein BRLF1 and is required for lytic viral gene expression. J. Virol. 78:4197–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu C, Sista ND, Pagano JS. 1996. Activation of the Epstein-Barr virus DNA polymerase promoter by the BRLF1 immediate-early protein is mediated through USF and E2F. J. Virol. 70:2545–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu CC, et al. 2006. Genome-wide transcription program and expression of the Rta responsive gene of Epstein-Barr virus. Virology 345:358–372 [DOI] [PubMed] [Google Scholar]
- 40. Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312 [DOI] [PubMed] [Google Scholar]
- 41. Manet E, Rigolet A, Gruffat H, Giot JF, Sergeant A. 1991. Domains of the Epstein-Barr virus (EBV) transcription factor R required for dimerization, DNA binding and activation. Nucleic Acids Res. 19:2661–2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Middeldorp JM, Brink AA, van den Brule AJ, Meijer CJ. 2003. Pathogenic roles for Epstein-Barr virus (EBV) gene products in EBV-associated proliferative disorders. Crit. Rev. Oncol. Hematol. 45:1–36 [DOI] [PubMed] [Google Scholar]
- 43. Middeldorp JM, Herbrink P. 1988. Epstein-Barr virus specific marker molecules for early diagnosis of infectious mononucleosis. J. Virol. Methods 21:133–146 [DOI] [PubMed] [Google Scholar]
- 44. Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. 2007. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv. Cancer Res. 97:81–109 [DOI] [PubMed] [Google Scholar]
- 45. Minarovits J, et al. 1992. RNA polymerase III-transcribed EBER 1 and 2 transcription units are expressed and hypomethylated in the major Epstein-Barr virus-carrying cell types. J. Gen. Virol. 73(Pt 7):1687–1692 [DOI] [PubMed] [Google Scholar]
- 46. Minarovits J, et al. 1991. Host cell phenotype-dependent methylation patterns of Epstein-Barr virus DNA. J. Gen. Virol. 72(Pt 7):1591–1599 [DOI] [PubMed] [Google Scholar]
- 47. Ooka T. 2005. Biological role of the BARF1 gene encoded by Epstein-Barr virus, p 613 In Robertson ES. (ed), Epstein-Barr virus. Caister Academic Press, Philadelphia, PA [Google Scholar]
- 48. Park JG, et al. 1997. Establishment and characterization of human gastric carcinoma cell lines. Int. J. Cancer 70:443–449 [DOI] [PubMed] [Google Scholar]
- 49. Petosa C, et al. 2006. Structural basis of lytic cycle activation by the Epstein-Barr virus ZEBRA protein. Mol. Cell 21:565–572 [DOI] [PubMed] [Google Scholar]
- 50. Raab-Traub N. 2002. Epstein-Barr virus in the pathogenesis of NPC. Semin. Cancer Biol. 12:431–441 [DOI] [PubMed] [Google Scholar]
- 51. Ragoczy T, Miller G. 2001. Autostimulation of the Epstein-Barr virus BRLF1 promoter is mediated through consensus Sp1 and Sp3 binding sites. J. Virol. 75:5240–5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott, Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 53. Robertson KD, Ambinder RF. 1997. Methylation of the Epstein-Barr virus genome in normal lymphocytes. Blood 90:4480–4484 [PubMed] [Google Scholar]
- 54. Robinson AR, Kwek SS, Hagemeier SR, Wille CK, Kenney SC. 2011. Cellular transcription factor Oct-1 interacts with the Epstein-Barr virus BRLF1 protein to promote disruption of viral latency. J. Virol. 85:8940–8953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Seto E, et al. 2005. Epstein-Barr virus (EBV)-encoded BARF1 gene is expressed in nasopharyngeal carcinoma and EBV-associated gastric carcinoma tissues in the absence of lytic gene expression. J. Med. Virol. 76:82–88 [DOI] [PubMed] [Google Scholar]
- 56. Sheng W, Decaussin G, Sumner S, Ooka T. 2001. N-terminal domain of BARF1 gene encoded by Epstein-Barr virus is essential for malignant transformation of rodent fibroblasts and activation of BCL-2. Oncogene 20:1176–1185 [DOI] [PubMed] [Google Scholar]
- 57. Sixbey JW, Nedrud JG, Raab-Traub N, Hanes RA, Pagano JS. 1984. Epstein-Barr virus replication in oropharyngeal epithelial cells. N. Engl. J. Med. 310:1225–1230 [DOI] [PubMed] [Google Scholar]
- 58. Stevens SJ, et al. 2006. Noninvasive diagnosis of nasopharyngeal carcinoma: nasopharyngeal brushings reveal high Epstein-Barr virus DNA load and carcinoma-specific viral BARF1 mRNA. Int. J. Cancer 119:608–614 [DOI] [PubMed] [Google Scholar]
- 59. Strockbine LD, et al. 1998. The Epstein-Barr virus BARF1 gene encodes a novel, soluble colony-stimulating factor-1 receptor. J. Virol. 72:4015–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Swenson JJ, Holley-Guthrie E, Kenney SC. 2001. Epstein-Barr virus immediate-early protein BRLF1 interacts with CBP, promoting enhanced BRLF1 transactivation. J. Virol. 75:6228–6234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takacs M, et al. 2010. Epigenetic regulation of latent Epstein-Barr virus promoters. Biochim. Biophys. Acta 1799:228–235 [DOI] [PubMed] [Google Scholar]
- 62. Tao Q, Robertson KD, Manns A, Hildesheim A, Ambinder RF. 1998. The Epstein-Barr virus major latent promoter Qp is constitutively active, hypomethylated, and methylation sensitive. J. Virol. 72:7075–7083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang Q, et al. 2006. Anti-apoptotic role of BARF1 in gastric cancer cells. Cancer Lett. 238:90–103 [DOI] [PubMed] [Google Scholar]
- 64. Wang Y, Luo B, Zhao P, Huang BH. 2004. Expression of Epstein-Barr virus genes in EBV-associated gastric carcinoma. Ai Zheng 23:782–787 (In Chinese.) [PubMed] [Google Scholar]
- 65. Whitehouse A, Carr IM, Griffiths JC, Meredith DM. 1997. The herpesvirus saimiri ORF50 gene, encoding a transcriptional activator homologous to the Epstein-Barr virus R protein, is transcribed from two distinct promoters of different temporal phases. J. Virol. 71:2550–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4:757–768 [DOI] [PubMed] [Google Scholar]
- 67. zur Hausen A, Brink AA, Craanen ME, Middeldorp JM, Meijer CJ, van den Brule AJ. 2000. Unique transcription pattern of Epstein-Barr virus (EBV) in EBV-carrying gastric adenocarcinomas: expression of the transforming BARF1 gene. Cancer Res. 60:2745–2748 [PubMed] [Google Scholar]