

Abstract
The double-stranded RNA virus mammalian reovirus displays broad cell, tissue, and host tropism. A critical checkpoint in the reovirus replication cycle resides within viral cytoplasmic inclusions, which are biosynthetic centers of genome multiplication and new-particle assembly. Replication of strain type 3 Dearing (T3) is arrested in Madin-Darby canine kidney (MDCK) cells at a step subsequent to inclusion development and prior to formation of genomic double-stranded RNA. This phenotype is primarily regulated by viral replication protein μ2. To understand how reovirus inclusions differ in productively and abortively infected MDCK cells, we used confocal immunofluorescence and thin-section transmission electron microscopy (TEM) to probe inclusion organization and particle morphogenesis. Although no abnormalities in inclusion morphology or viral protein localization were observed in T3-infected MDCK cells using confocal microscopy, TEM revealed markedly diminished production of mature progeny virions. T3 inclusions were less frequent and smaller than those formed by T3-T1M1, a productively replicating reovirus strain, and contained decreased numbers of complete particles. T3 replication was enhanced when cells were cultivated at 31°C, and inclusion ultrastructure at low-temperature infection more closely resembled that of a productive infection. These results indicate that particle assembly in T3-infected MDCK cells is defective, possibly due to a temperature-sensitive structural or functional property of μ2. Thus, reovirus cell tropism can be governed by interactions between viral replication proteins and the unique cell environment that modulate efficiency of particle assembly.
INTRODUCTION
A common strategy shared by numerous viruses is the formation of specialized sites within a host cell to complete viral replication. Animal double-stranded RNA (dsRNA) viruses generate nonmembranous intracytoplasmic structures—termed inclusions or factories—that have a characteristic morphology, contain viral proteins and RNA, and constitute the presumed site of negative-strand RNA synthesis and particle assembly (11, 35, 48). We are studying replication mechanisms of the dsRNA virus mammalian orthoreovirus (reovirus) to better understand the function of viral inclusions and the influence of these novel organelles on viral tropism.
Reoviruses are nonenveloped, double-shelled, icosahedral particles containing a genome of 10 dsRNA segments (40). The viral replication cycle is entirely cytoplasmic. Following internalization of virions, the viral outer capsid disassembles to generate transcriptionally active core particles (3, 15, 49), which are released into the cytoplasm and synthesize full-length, message-sense, capped and nonpolyadenylated single-stranded RNAs (ssRNAs) (32, 42, 45). Viral inclusions are detectable as early as 4 h postinfection by confocal immunofluorescence microscopy, lack a delimiting membrane, and contain viral proteins and dsRNA, virion particles at various stages of morphogenesis, and paracrystalline arrays of virion particles at late times of infection (12, 13, 37). Studies of viral inclusions in infected cells and viral inclusion-like structures formed by ectopic protein expression indicate that higher-order multimers of the viral nonstructural protein μNS establish inclusions by forming an essential matrix to which μ2, nonstructural protein σNS, and other viral structural proteins are recruited (1, 4, 5, 7–9, 13, 16, 17, 22, 23, 26, 27, 34, 37, 41, 43, 44, 46). The μNS, μ2, and σNS proteins participate in inclusion formation and maturation as indispensable components of viral replication (23). However, the nature of ribonucleoprotein (RNP) intermediates linking the initial round of viral gene expression to the emergence of mature progeny virions has been only partially defined.
Although viral RNA assortment, genome replication, secondary transcription, and particle formation are presumed to occur within reovirus inclusions (2, 13, 14, 29, 30, 37, 58, 59), individual steps of particle assembly remain largely unknown. However, this process likely begins with assortment of positive-sense viral RNAs by a specific mechanism. Available data are congruous with an assembly model wherein equimolar amounts of the 10 viral mRNAs associate with nonstructural and core proteins and condense to form “replicase particles” (2, 14) capable of synthesizing complementary negative-sense RNA to generate the dsRNA genome. Populations of particles with distinctive complements of viral RNA and protein, as well as specific transcriptional activities (i.e., positive- or negative-strand synthesis), can be separated physiochemically and are thought to represent intermediates on a pathway toward virion assembly (29, 30, 58, 59). Current concepts of sequential steps in virion assembly are rooted mainly in biochemical analyses of these subvirion forms. However, the proposed assembly intermediates have not been correlated with specific morphogenic events in viral inclusions.
Reovirus inclusions are implicated functionally as determinants of viral cell tropism (33), where tropism is defined as the range of hosts, tissues, and cells productively infected. Reovirus strains type 1 Lang (T1) and type 3 Dearing (T3) display differential capacities to replicate in Madin-Darby canine kidney (MDCK) cells (33, 39). Strain T1 replicates efficiently, while T3 replicates poorly. Analysis of recombinant reovirus strains derived from reverse genetics revealed that the M1-encoded μ2 protein of strain T1 in a T3 genetic background independently promotes reovirus replication in MDCK cells (33). The L1-encoded λ3 protein is a modulator of μ2 effects on replication efficiency, although λ3 does not display an independent association with replication capacity in MDCK cells. Furthermore, the influence of λ3 on μ2-mediated replication efficiency is manifest only in the T1 genetic background. These findings indicate that μ2 is the primary determinant of reovirus replication capacity in MDCK cells and that λ3 is a conditional coregulator of infectivity dependent on the viral genetic context. In the same study, we found that the μ2-sensitive step in replication occurs at a point in the reovirus life cycle subsequent to primary rounds of viral transcription and translation but prior to dsRNA synthesis, which suggests that μ2 and λ3 mediate differences in strain-specific replication efficiency at a life cycle stage occurring within viral inclusions.
The reovirus RNA-dependent RNA polymerase (RdRp) is thought to be composed of two subunits, λ3 and μ2. The λ3 protein is the RdRp catalytic subunit (47, 50, 56), and numerous biochemical, structural, and genetic studies indicate that μ2 lies proximal to λ3 and forms a subunit of the fully assembled polymerase complex (6, 10, 21, 31, 36, 54, 56). The μ2 protein also represents an essential component of viral inclusions required for productive viral replication (24, 34). It binds inclusion-forming protein μNS (9) and determines strain-specific differences in inclusion morphology—filamentous or globular—based on the relative capacity to bind microtubules and tether inclusions to the cytoskeleton (34). Although the μ2 protein coordinates diverse events during the reovirus replication cycle, a mechanistic understanding of the role of μ2 in replication remains unknown, primarily because its functions have not been linked to specific steps in the viral life cycle.
In the work described here, we used confocal immunofluorescence and transmission electron microscopy (TEM) to more precisely define the role of μ2 protein in productive reovirus replication and virion particle production. The results indicate that particle assembly in T3-infected MDCK cells is defective and dependent on μ2 protein in a cell-type-specific and temperature-dependent manner. Thus, permissivity of some cell types to reovirus infection is governed by interactions between viral replication proteins and the unique cell environment that promote genesis of functional viral inclusions.
MATERIALS AND METHODS
Cells and viruses.
L929 cells were grown in Joklik's modified Eagle's minimal essential medium (Lonza, Walkersville, MD) supplemented to contain 5% fetal calf serum (Cellgro, Manassas, VA), 2 mM l-glutamine (Gibco, Grand Island, NY), 100 U/ml penicillin (Gibco), 100 μg/ml streptomycin (Gibco), and 0.25 μg/ml amphotericin B (Sigma, St. Louis, MO). MDCK cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented to contain 4.5 μg/ml sodium pyruvate (Cellgro), 10% fetal calf serum, 2 mM l-glutamine, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B. Strains T3 and T3-T1M1 were recovered by reverse genetics as described previously (22, 25). Viral titers were determined by plaque assay using L929 cells (51).
Quantification of virus infectivity.
Monolayers of L929 cells or MDCK cells (approximately 5 × 105 cells) seeded in 24-well plates (Corning, Tewksbury, MA) were adsorbed with virus at a multiplicity of infection (MOI) of 2 PFU/cell at room temperature (RT) for 1 h. The viral inoculum was removed, cells were washed with phosphate-buffered saline (PBS), fresh medium was added, and cells were incubated at 31°C or 37°C for various intervals, followed by removal to −80°C. Viral titers in cell lysates were determined by plaque assay. Viral yield was calculated as the difference between log10 titer at various time points of infection and log10 titer at 0 h.
Immunofluorescence detection of reovirus infection.
L929 cells plated on untreated glass coverslips and MDCK cells plated on poly-l-lysine (Sigma)-treated glass coverslips in 24-well plates were adsorbed with virus at an MOI of 20 PFU/cell. Following incubation at 37°C for 24 h, cells were fixed with 10% formalin and stained with guinea pig σNS-specific antibody (4) and either rabbit μNS-specific (23), rabbit μ2-specific (57), mouse σ3-specific (4F2) (52), mouse μ1-specific (8H6) (52), or rabbit λ2-specific (7F4) (52) antibody, followed by Alexa Fluor 488-conjugated goat anti-guinea pig secondary antibody (Invitrogen, Eugene, OR), Alexa Fluor 546-conjugated goat anti-rabbit secondary antibody (Invitrogen), or Alexa Fluor 546-conjugated goat anti-mouse antibody (Invitrogen). ToPro3 (Invitrogen) was used to stain the nucleus. Images were acquired using a Zeiss LSM 510 Meta inverted confocal microscope.
Electron microscopy.
L929 cells or MDCK cells grown in T75 flasks (Corning) were adsorbed at RT for 1 h with virus at an MOI of 100 PFU/cell. Following adsorption, cells were incubated at 31°C or 37°C for 24 or 72 h. Cells were trypsinized, pelleted, washed with PBS, fixed at room temperature for at least 1 h with 2.5% glutaraldehyde buffered with 0.1 M sodium cacodylate, and stored at 4°C. Following fixation, cells were washed with 0.1 M sodium cacodylate, postfixed in 1% osmium tetroxide–0.1 M sodium cacodylate for 1 h, washed in 0.1 M sodium cacodylate, and dehydrated using a graded series of ethanols. After dehydration, cells were subjected to two changes of propylene oxide and embedded in epoxy resin. Ultrathin (65 to 70 nm) serial sections were obtained using a Leica Ultracut microtome and collected on copper grids. Samples were contrast stained with 2% uranyl acetate followed by Reynold's lead citrate and examined using a Phillips CM10 transmission electron microscope or FEI T-12 transmission electron microscope operated at 80 KeV.
Quantification of TEM images.
The fraction of cytoplasm containing virion particles or inclusion matrix in electron micrographs was quantified using point counting stereology (53). Images were collected in a systematic, unbiased fashion using a FEI T-12 transmission electron microscope at a magnification of ×30,000. Thirty-six images were collected for each sample. A grid of points was superimposed over the images (grid size, 43 by 43 nm), and points overlying cytoplasm or virus-specific material were counted. The volume of virus-occupied cytoplasm was calculated by dividing the number of points that superimposed on virus-specific material by the total number points on cytoplasm. Particle diameters were measured using ImageJ (http://rsb.info.nih.gov/ij/index.html). Student's t test was used to determine statistical significance of differences among groups. The fraction of the viral inclusion matrix occupied by particles was calculated from the images collected for point counting. Particle area was estimated by assuming that particles are spherical and calculating area using πr2, where r is half the mean diameter of mature and empty particles measured using ImageJ. Areas of mature and empty particles were calculated independently using their respective diameters and combined to yield total particle area.
RESULTS
Reovirus replication in MDCK cells.
Our previous studies of T3-infected MDCK cells revealed that inclusions produced by strain T3 display normal morphological and kinetic development despite an apparent functional abnormality leading to arrest of replication (33). To more precisely define viral inclusion composition and ultrastructure in T3-infected MDCK cells, we used both confocal immunofluorescence microscopy and thin-section TEM. Strain T3-T1M1, containing the T1 M1 gene in an otherwise T3 genetic background, served as a reference for efficient μ2-regulated reovirus replication. Consistent with our previous results (33), T3-T1M1 replicated to high titers, whereas T3 exhibited minimal titer increase over a 48-h incubation interval (Fig. 1A).
Fig 1.

Reovirus replication and inclusion formation. (A) MDCK cells were adsorbed with T3 or T3-T1M1 at an MOI of 2 PFU/cell, and viral titers in cell lysates were determined at the times shown by plaque assay using L929 cells. Results represent the mean of triplicate experiments. Error bars indicate standard deviations. (B and C) MDCK cells were adsorbed with T3 (B) or T3-T1M1 (C) at an MOI of 100 PFU/cell, fixed at 24 h postinfection, stained with anti-σNS (green) and anti-μNS (red) antibodies and ToPro3 (blue; nuclear) and imaged using confocal microscopy. Scale bar, 50 μm.
Viral protein recruitment in MDCK cells.
To determine whether protein recruitment to inclusions differs in productively and nonproductively infected cells, we visualized individual viral proteins using confocal microscopy. Nonstructural proteins σNS and μNS physically associate in the formation of an inclusion scaffold (27). In cells infected with T3 or T3-T1M1, σNS and μNS displayed colocalization, forming large inclusions with usual strain-dependent morphology (globular for T3 and filamentous for T3-T1M1) (34]) at 24 h postinfection (Fig. 1B and C). No alterations in σNS or μNS localization were apparent in T3-infected cells. These data indicate that the viral proteins required to form inclusions are properly recruited and assembled in T3-infected MDCK cells.
We next examined the localization of viral core proteins μ2 and λ2 and outer capsid proteins μ1 and σ3 in T3- and T3-T1M1-infected MDCK cells (Fig. 2A) and L929 cells (Fig. 2B). In MDCK cells, the μ2 and λ2 proteins of both T3 and T3-T1M1 were found throughout the cytoplasm and concentrated in viral inclusions (Fig. 2A). A similar distribution of these proteins was observed in L929 cells. Outer capsid protein σ3 was highly abundant in both T3- and T3-T1M1-infected MDCK cells. It was found distributed throughout the cytoplasm, colocalized with σNS in inclusion puncta, and frequently in the nucleus (Fig. 2A). The distribution of σ3 in L929 cells was similar except for reduced distribution to the nucleus compared with MDCK cells. Outer capsid protein μ1 was observed diffusely throughout the cytoplasm and in punctate structures associated with inclusions (Fig. 2). Fewer μ1-containing punctate structures were observed in T3-infected MDCK cells than in T3-T1M1-infected MDCK cells (Fig. 2A). However, this quantitative difference correlated with diminished levels of μ1 expression overall. Viral proteins also were visualized at earlier times postinfection. Inclusions were less developed, but patterns of protein distribution were similar to those observed at 24 h (data not shown). In summary, no qualitative differences in the distribution of nonstructural, core, and outer capsid viral proteins were discernible in comparison of T3- and T3-T1M1-infected MDCK cells, indicating that viral protein recruitment to inclusions occurs normally for both strains.
Fig 2.
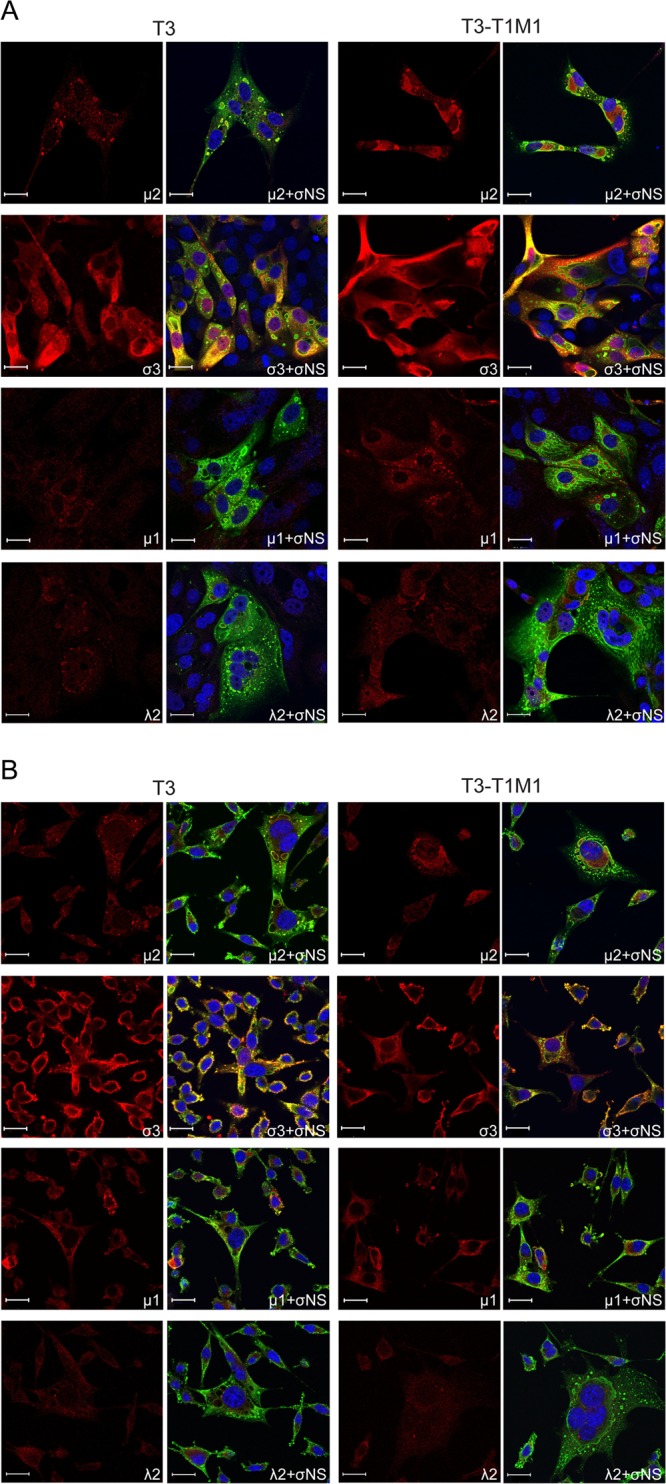
Distribution of reovirus proteins μ2, σ3, μ1, and λ2 in infected cells. MDCK cells (A) and L929 cells (B) were absorbed with either T3 or T3-T1M1 at an MOI of 20 PFU/cell, fixed at 24 h postinfection, stained with anti-σNS (green) and anti-μ2, anti-σ3, anti-μ1, or anti-λ2 (red) antibodies. Nuclei were visualized using ToPro3 (blue). Images were captured using confocal microscopy. Scale bar, 20 μm.
Ultrastructural analysis of viral inclusions in MDCK cells.
As confocal microscopy did not reveal overt abnormalities in the morphology or viral protein composition of T3 inclusions in MDCK cells, we used thin-section TEM to visualize viral particles within inclusions. In both T3- and T3-T1M1-infected cells, mature virion particles (i.e., containing electron-dense centers representing genomic viral RNA) (13, 38) and empty virion particles were observed in a variety of arrangements. Some sections contained only a few scattered mature virions, whereas others contained large groups of viral particles surrounded by material having a consistency clearly distinct from cytoplasm. The latter structures presumably correspond to the filamentous or globular inclusions observed using confocal microscopy; we refer to these as particle-containing inclusions. Representative images of particle-containing inclusions in T3-T1M1-infected MDCK cells are shown in Fig. 3A. These large aggregates of virions contain a significant fraction of complete particles in paracrystalline array. In marked contrast, T3 particle-containing inclusions were less frequent, smaller, displayed decreased organization, and harbored fewer mature particles. Inclusions containing T3 particles like those shown in Fig. 3A were rare and required extensive searching to locate. The sizes, frequencies, and particle contents of T3 and T3-T1M1 inclusions in L929 cells were similar to each other and to those in T3-T1M1-infected MDCK cells (Fig. 3B).
Fig 3.
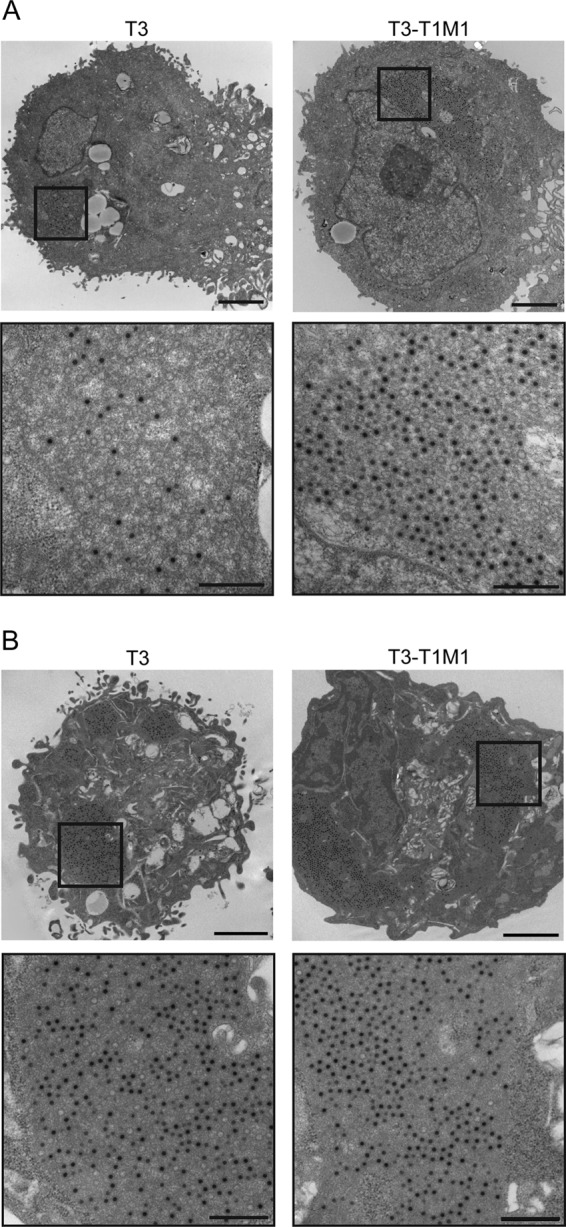
Ultrastructural analysis of viral inclusions. MDCK cells (A) and L929 cells (B) were adsorbed with either T3 or T3-T1M1 at an MOI of 100 PFU/cell and fixed at 24 h postinfection. Ultrathin sections (65 to 70 nm) were examined using transmission electron microscopy. Boxed regions correspond to high-magnification images shown below. Scale bars, 2 μm (low-magnification images) and 500 nm (high-magnification images).
To systematically confirm our impressions of T3 and T3-T1M1 inclusion ultrastructural morphology, we quantified certain features of inclusions captured in electron micrographic images. The fraction of cytoplasm containing virion particles or inclusion matrix was measured using the point counting stereology method (53). We defined the viral fraction of cytoplasm to be viral particles and surrounding milieu (i.e., inclusion matrix) that was clearly distinct from cytoplasm. Structures resembling viral inclusions but devoid of viral particles were counted as cytoplasm since we could not be certain that these were of viral origin. However, such “empty inclusions” were infrequent. In T3-T1M1-infected MDCK cells, 19.2% of the cytoplasm was occupied by inclusion matrix, whereas this fraction was only 2.9% in T3-infected cells (Table 1). The corresponding values in L929 cells were 27.8% and 14.9%, respectively. The proportion of inclusion matrix occupied by particles was 11.2% and 17.8% in T3- and T3-T1M1-infected MDCK cells, respectively, and 23.4% and 32.1%, respectively, in L929 cells. These data are consistent with our observation that particles within inclusions of T3-infected MDCK cells are less densely packed. Finally, we determined the ratios of complete and empty particles in inclusions formed in both cell types. The fraction of complete particles in T3- and T3-T1M1-infected MDCK cells was 36.3% and 76.7%, respectively. The corresponding values in L929 cells were 79.7% and 94.9%, respectively. The smaller fraction of inclusion matrix observed in the cytoplasm of T3-infected MDCK cells suggests that particle assembly is defective and dependent on μ2 protein. While T3 infection of L929 cells resulted in lower values for the inclusion matrix fraction, particle density, and particle maturity than T3-T1M1 infection, differences in the fraction of inclusion matrix and mature particles were clearly more pronounced in MDCK cells infected with these strains. Furthermore, T3 particles assumed definite order in inclusions observed in L929 cells, unlike the irregular arrangements typical of T3-infected MDCK cells. Taken together, qualitative and quantitative TEM analysis of reovirus inclusions under conditions of productive and abortive infection of MDCK cells points to viral assembly as a μ2-sensitive step in viral replication.
Table 1.
Quantitative analysis of reovirus replication using thin-section electron micrographs of reovirus-infected MDCK and L929 cellsa
| Cell type and virus strain | Fraction of cytoplasm containing virion particles or inclusion matrix (%) | Fraction of viral inclusion matrix occupied by particles (%) | Mature virion particle fraction (%) |
|---|---|---|---|
| MDCK | |||
| T3 | 2.9 | 11.2 | 36.3 |
| T3-T1M1 | 19.2 | 17.8 | 76.7 |
| L929 | |||
| T3 | 14.9 | 23.4 | 79.7 |
| T3-T1M1 | 27.8 | 32.1 | 94.9 |
A total of 36 images was examined for each sample condition.
While examining the ultrastructure of reovirus inclusions in MDCK cells, we observed that empty T3 particles appeared smaller than empty T3-T1M1 particles. Therefore, we quantified and compared the diameters of complete and empty particles. A high-magnification image of virion particles in T3-T1M1-infected MDCK cells (Fig. 4) illustrates the specific diameter measurements recorded: (i) electron-dense centers of mature particles, (ii) outer rim of mature particles, and (iii) outer rim of empty particles. Large numbers of observations (110 to 417) for each category were made to statistically validate the size differences. The mean inner diameter of mature T3 and T3-T1M1 particles in both MDCK and L929 cells was ∼38 nm, whereas the mean outer diameter of mature particles was ∼72 nm (MDCK cells) or ∼71 nm (L929 cells) (Table 2). In contrast, the mean diameter of empty particles in T3-infected MDCK cells was significantly less than the empty-particle diameters in T3-T1M1-infected MDCK cells (67.3 ± 0.27 nm versus 73.5 ± 0.34 nm, respectively). Diameters of empty particles of T3 and T3-T1M1 in L929 cells, i.e., 71.2 ± 0.40 nm and 72.0 ± 0.42 nm, respectively, were not significantly different. These results reveal a difference in the size of T3 and T3-T1M1 empty particles produced in MDCK cells but not L929 cells, indicating that some aspect of T3 particle assembly in MDCK cells follows an anomalous path. Furthermore, these findings are consistent with a μ2-associated block to morphogenesis of mature T3 virions in MDCK cells.
Fig 4.
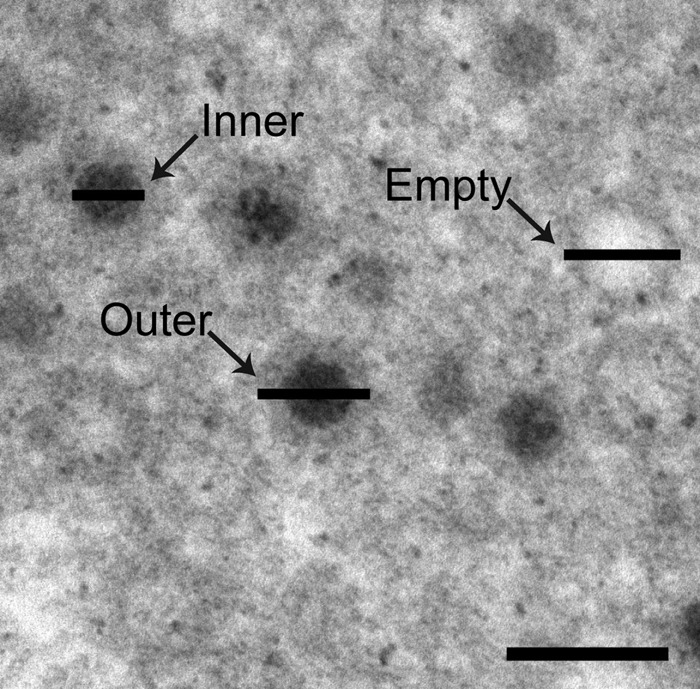
High-magnification image of T3-T1M1-infected MDCK cells. The diameter of the inner electron-dense center of complete particles (Inner), the outer diameter of complete particles (Outer), and the outer diameter of empty particles (Empty) are demarcated by horizontal bars. Scale bar, 100 nm.
Table 2.
Quantification of viral particle diameters in reovirus-infected MDCK and L929 cells
| Cell type and virus strain | Particle size (nm)a |
||
|---|---|---|---|
| Mature particle |
Empty particle outer diam (n) | ||
| Outer diam (n) | Inner diam (n) | ||
| MDCK | |||
| T3 | 72.3 ± 0.44 (110) | 38.3 ± 0.22 (116) | 67.3 ± 0.27 (306) |
| T3-T1M1 | 72.0 ± 0.21b (311) | 37.9 ± 0.12b (323) | 73.5 ± 0.34c (177) |
| L929 | |||
| T3 | 71.3 ± 0.24 (380) | 38.5 ± 0.11 (417) | 71.2 ± 0.40 (119) |
| T3-T1M1 | 71.0 ± 0.22b (385) | 38.3 ± 0.11b (413) | 72.0 ± 0.42b (138) |
Values represent means ± standard error of the means. n, number of particles measured.
Not significant versus T3 (t test).
P < 0.0001 versus T3 (t test).
Replication of reovirus at low temperature.
A previous study suggested that the T3 μ2 protein is subject to temperature-dependent misfolding (28), which led us to consider whether strain-dependent differences in viral replication efficiency in MDCK cells might be influenced by the temperature of infection. To test this hypothesis, viral titers were monitored in MDCK cells (Fig. 5A and B) and L929 cells (Fig. 5C and D) infected with either T3 or T3-T1M1 and incubated at either 31°C (Fig. 5A and C) or 37°C (Fig. 5B and D) for 96 h postinfection. Replication of T3 in MDCK cells at 31°C greatly exceeded that at 37°C, closely approaching the yield of T3-T1M1 at 31°C. In L929 cells infected at 31°C, T3 displayed significantly lower yield at 24 h postinfection than T3-T1M1 but attained a similarly high level of replication at later times postinfection. T3-T1M1 replicated efficiently in both cell types at 31°C. However, the rise in titer was delayed relative to 37°C. Taken together, these results indicate that replication of T3 in MDCK cells is temperature dependent.
Fig 5.

Temperature dependence of reovirus replication in MDCK cells. MDCK cells (A and B) or L929 cells (C and D) were adsorbed at an MOI of 2 PFU/cell and incubated at 31°C (A and C) or 37°C (B and D) for the times shown. Viral titers in cell lysates were determined by plaque assay using L929 cells. Viral yields were calculated as the difference between the log10 titers at the indicated times and log10 titer at 0 h. Results represent the mean of triplicate experiments. Error bars indicate standard deviations. *, P < 0.05 in comparison to T3-T1M1 (Student's t test).
We used thin-section TEM of MDCK cells infected at the lower temperature to determine whether enhanced replication kinetics of T3 in MDCK cells at 31°C correlates with restoration of normal inclusion ultrastructure. At 72 h postinfection, large particle-containing inclusions enclosing mature and empty particles were observed in both T3- and T3-T1M1-infected cells (Fig. 6). The fraction of cytoplasm occupied by the viral inclusion matrix was 17% and 28% in T3- and T3-T1M1-infected cells, respectively, while the proportion of inclusion matrix occupied by particles was 12.7% in T3-infected cells and 16.8% in T3-T1M1-infected cells (Table 3). Complete particles comprised 29.7% and 36.3% of total virions in T3- and T3-T1M1-infected cells, respectively. The substantial increase in the fraction of inclusion matrix from 2.9% to 17% in T3-infected MDCK cells incubated at 31°C versus 37°C provides further evidence that the restriction to T3 replication in MDCK cells relaxes at low temperature. Additionally, the mean diameters of empty particles in MDCK cells infected with T3 (72.6 ± 0.29 nm) and T3-T1M1 (71.9 ± 0.20 nm) were comparable at reduced incubation temperature (Table 4). Thus, lowering the temperature of infection partially relieved μ2-mediated restriction of T3 replication in MDCK cells and reversed the block to mature particle assembly, consistent with the hypothesis that the conformational state of μ2 influences efficiency of the reovirus life cycle through a mechanism involving virion morphogenesis.
Fig 6.
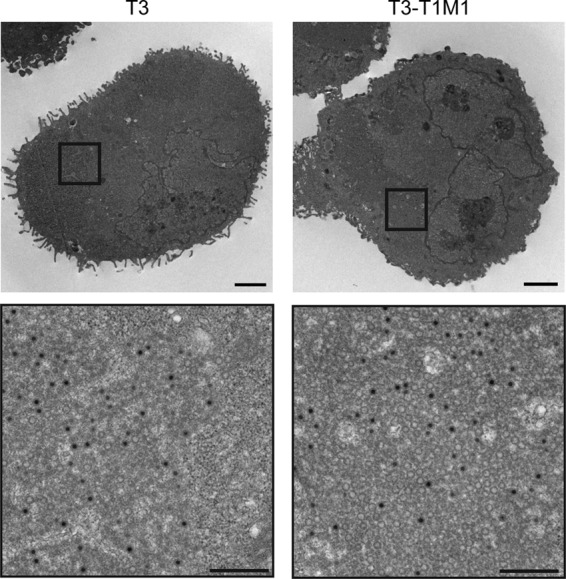
Ultrastructural analysis of viral inclusions in MDCK cells infected at low temperature. MDCK cells were adsorbed with either T3 or T3-T1M1 at an MOI of 100 PFU/cell, incubated at 31°C, and fixed at 72 h postinfection. Ultrathin sections (65 to 70 nm) were examined using transmission electron microscopy. Boxed regions correspond to high-magnification images shown below. Scale bars, 2 μm (low-magnification images) and 500 nm (high-magnification images).
Table 3.
Quantitative analysis of reovirus replication at 31°C using thin-section electron micrographs of reovirus-infected MDCK cellsa
| Virus strain | Fraction of cytoplasm containing virion particles or inclusion matrix (%) | Fraction of viral inclusion matrix occupied by particles (%) | Mature virion particle fraction (%) |
|---|---|---|---|
| T3 | 17.2 | 12.7 | 29.7 |
| T3-T1M1 | 28.0 | 16.8 | 36.3 |
A total of 36 images was examined for each sample condition.
Table 4.
Quantification of viral particle diameters following infection of MDCK cells at 31°C
| Virus strain | Particle size (nm)a |
||
|---|---|---|---|
| Mature particle |
Empty particle outer diam (n) | ||
| Outer diam (n) | Inner diam (n) | ||
| T3 | 71.9 ± 0.35 (203) | 38.0 ± 0.16 (225) | 72.6 ± 0.29 (284) |
| T3-T1M1 | 71.5 ± 0.23b (306) | 37.9 ± 0.13b (312) | 71.9 ± 0.20b (472) |
Values represent means ± standard errors of the means. n, number of particles measured.
Not significant versus T3 (t test).
DISCUSSION
A common strategy of virus replication is formation of neo-organelles—i.e., inclusions—that concentrate and protect viral components required for genome multiplication and particle assembly. An understanding of viral inclusion composition and organization is essential to illuminate strategies by which viruses direct a successful replication program. In this study, we characterized a postentry-determined, strain-specific replication defect in MDCK cells to enhance knowledge of critical activities within reovirus inclusions that depend on the particular cell environment. Our findings suggest that reovirus tropism for some types of cells is governed by interactions between viral replication proteins and cell-type-specific factors that favor development of inclusions capable of generating fully assembled infectious particles.
Original studies of reovirus replication in MDCK cells revealed a genetic association of polymerase proteins μ2 and λ3 with viral replication efficiency (39). We subsequently showed that the μ2-sensitive step of replication in these cells occurs following the formation of viral inclusions but prior to dsRNA synthesis, leading us to propose that reovirus strain-specific replication efficiency in MDCK cells is determined at a postentry replication checkpoint responsive to the composition of the viral polymerase complex (33). Here, we report findings of a more comprehensive analysis of viral protein trafficking to inclusions conducted to determine whether one or more inclusion components displays abnormal localization in nonproductively infected MDCK cells. Using confocal immunofluorescence microscopy, no discernible cell-type-specific differences in the distribution of μ2, the other major inclusion proteins μNS and σNS, core protein λ2, or outer capsid proteins μ1 and σ3 were observed in T3-infected MDCK cells (Fig. 1 and 2A). Although a relative reduction (compared with T3-T1M1) in the expression of μ1 was observed, the significance of this reduction to T3 replication in MDCK cells is unclear as expression of all viral proteins markedly declines at late times postinfection (33). Thus, results of our confocal microscopy studies confirm the expected pattern of viral protein localization to T3 inclusions in MDCK cells, indicating structurally or functionally abnormal interactions within the inclusion that prevent production of infectious progeny.
TEM analysis of MDCK cells infected with T3 revealed a much smaller fraction of cytoplasm containing particles and inclusion matrix than cells infected with strain T3-T1M1 (2.9% versus 19.2%) (Fig. 3 and Table 1). Mature particles with electron-dense (i.e., genome-containing) centers and empty particles were observed in MDCK cells infected with either T3 or T3-T1M1. However, the mature fraction of T3 particles was less than that of T3-T1M1 (36% versus 77%) (Table 1). Assembly intermediates that escape the restrictive step appear competent to reach full structural maturity, consistent with the finding that conditions can be modified to permit partial relief of the restriction to T3 replication in MDCK cells. Collectively, the substantial decrease in particle-containing inclusion matrix and mature virion particles suggests that assembly of T3 particles is restricted in MDCK cells, leading to a significant reduction in viral yield. It is possible that decreased protein synthesis leads to diminished mature particle yields in T3-infected cells. However, we think our results are more consistent with a model wherein replication fails due to inefficient production of replicase particles. We previously observed that new T3 virion proteins are expressed through 12 h postinfection in MDCK cells (33). Additionally, results of the current study confirm that no overt impediments exist to the morphogenesis of T3 inclusions in these cells, and we found no obvious abnormalities in the levels or localization of T3 proteins relative to T3-T1M1 (Fig. 1, 2). Biochemical analysis of reovirus RNA synthesis has demonstrated that 80 to 95% of viral mRNA is transcribed by progeny particles made in the infected cells (55), and these transcripts serve as templates for protein translation during later portions of the viral life cycle (40). Taken together, results from our work and from other published studies best support the conclusion that diminished viral RNA and protein production in T3-infected MDCK cells is secondary to a defect in particle assembly.
Although results of confocal microscopy studies indicate that a majority of cells are infected (Fig. 1 and 2), not all TEM sections contained identifiable viral structures. We defined inclusions in electron micrographs by the presence of virion particles. Structures resembling particle-free inclusions were rarely observed, and inclusion matrix could not be determined with complete confidence in the absence of particles. It is possible that our TEM method lacks the sensitivity necessary to identify a particle-free inclusion matrix. Alternatively, the large difference in section thickness between the two methods (900-nm confocal z-slice versus 70-nm electron microscopy thin section) may explain preferential detection of infected cells using confocal microscopy.
The observation that empty particles are smaller in T3-infected MDCK cells than in T3-T1M1-infected MDCK cells offers corroborating evidence of aberrant assembly of T3 virion particles. It is not known whether empty particles represent precursors to mature virions or replication by-products, but the latter possibility is generally favored (18, 40). In either case, these structures inevitably share some assembly steps with complete virion particles and highlight the potential for a global μ2-dependent defect in particle assembly. Possible explanations for the smaller size of empty T3 particles include missing or stoichiometrically incorrect protein subassemblies or misfolding of key protein constituents. The cell-type-specific nature of this phenotype further suggests that discrete cellular factors are essential to coordinate successful particle maturation.
TEM ultrastructural analysis revealed a 6.6-fold decrease in the fraction of cytoplasm containing virion particles or an inclusion matrix in T3-infecected MDCK cells in comparison to T3-T1M1-infected cells (Table 1). This difference correlates with an approximate 100-fold decrease in T3 viral yield compared with T3-T1M1 (Fig. 5). Numerous factors may contribute to disproportionate changes in virion production as measured by TEM and viral plaque assay. First, the mature, and presumably infectious, fraction of virion particles is significantly less in T3-infected (36%) than in T3-T1M1-infected (77%) MDCK cells. Also, TEM almost exclusively identifies intracellular virus, whereas replication experiments measure both intracellular and released virus. Finally, the two analytical methods are subject to nonequivalent sampling bias. For example, in the replication experiments, virus particles moving randomly in a fluid matrix are equally likely to undergo sampling in a plaque titration assay, in contrast to TEM-based virus detection, where virion particles are immobilized within structures that have nonuniform boundaries and represent fewer members of the total virus population available for quantification. These technical differences could account in part for the greater discriminatory power of a viral replication assay than that of TEM for enumeration of viable virion particles.
The capacity of T3 to replicate in MDCK cells at 31°C provides insight into a possible mechanism of the μ2-regulated replication phenotype. The T3 μ2 protein is highly ubiquitylated in a temperature-dependent manner, and ubiquitylation is reduced when the temperature of viral infection is lowered (28). These findings suggest that T3 μ2 misfolds at higher temperature and raise the possibility that temperature-sensitive conformations of μ2 influence a critical early step in viral assembly. The μ2 ubiquitylation phenotype segregates with a polymorphism at amino acid position 208 (28), while temperature-dependent replication efficiency in MDCK cells is regulated by amino acid position 347, without detectable contribution by the residue at position 208 (33). The effect of amino acid variability at position 347 on μ2 conformational states has not been examined. However, reversal of the block to T3 replication at decreased temperature suggests that residue 347 modulates a temperature-sensitive property of μ2, possibly protein-folding, which in turn governs reovirus strain-specific interactions with the host cell environment during viral assembly. These interactions could involve stimulation of replication complexes containing T1 μ2 or inhibition of complexes containing T3 μ2. Alternatively, the replication block could be passive in nature, caused by a failure of T3 μ2 to successfully interact with cellular factors. In either model, conformational variation in μ2 could conceivably influence a critical association of viral and host cell factors required for virion assembly.
The T3 replication phenotype in MDCK cells is similar to that of reovirus strain tsH11.2, which contains lesions in μ2 protein that restrict viral replication to low temperature. Like T3 infection of MDCK cells, tsH11.2 synthesizes normal amounts of ssRNA but displays significantly diminished dsRNA production and secondary translation (10). However, unlike T3 infection of MDCK cells, tsH11.2 μ2 does not concentrate in viral inclusions at restrictive temperature (26). Thus, the replication block to tsH11.2 might operate through mechanisms affecting μ2 localization. Although the precise step in viral replication blocked in tsH11.2 infection may differ from or complement that of T3 in MDCK cells at nonpermissive temperature, both of these models of conditional reovirus replication confirm the importance of μ2 protein for progression of the viral life cycle beyond primary rounds of RNA and protein synthesis.
Reovirus inclusions are generally thought to contain cellular proteins repurposed for viral use. Identities of these molecules remain mostly unknown. Early studies using electron microscopy identified inclusion-associated cytoskeletal elements, including coated microtubules and thick, kinked fibrils interspersed among viral particles (13, 16). Known cellular proteins recruited to reovirus inclusions include chaperone Hsc70 (20) and clathrin (19). The roles played by these proteins in inclusion formation and function await further investigation. A comprehensive study of cellular factors required for reovirus replication, in particular those required for inclusion development and maturation, will further define postentry mechanisms of viral tropism and the precise activities of μ2 and other viral replication proteins in virion morphogenesis.
In summary, we used microscopic techniques to characterize functionally abnormal reovirus inclusions that form in nonpermissive cells to more fully understand the role played by μ2 in promoting productive viral replication and tropism for host cells. Findings made in this study establish virion particle assembly as a μ2-regulated step in the reovirus replication program and suggest that a temperature-sensitive property of μ2, perhaps protein conformation, controls key events within inclusions that culminate in assembly of mature particles. Additional characterization of this system may identify novel cellular regulators of reovirus infection that participate in virion morphogenesis and serve as determinants of viral cell tropism.
ACKNOWLEDGMENTS
Acquisition and analysis of confocal and transmission electron microscopic images were made possible in part through use of the Vanderbilt University Medical Center Cell Imaging Shared Resource that is supported in part by Public Health Service awards DK58404, HD15052, DK59637, and EY08126. Funding for this work was provided by the Department of Pathology, Microbiology, and Immunology, Elizabeth B. Lamb Center for Pediatric Research, and Public Health Service awards R01 AI32539 (T.S.D.) and K08 AI062862 (J.D.C.). Additional support was provided by Public Health Service awards CA68485 for the Vanderbilt-Ingram Cancer Center and DK20593 for the Vanderbilt Diabetes Research and Training Center.
Footnotes
Published ahead of print 25 July 2012
REFERENCES
- 1. Anderson N, Doane FW. 1966. An electron-microscope study of reovirus type 2 in L cells. J. Pathol. Bacteriol. 92:433–439 [DOI] [PubMed] [Google Scholar]
- 2. Antczak JB, Joklik WK. 1992. Reovirus genome segment assortment into progeny genomes studied by the use of monoclonal antibodies directed against reovirus proteins. Virology 187:760–776 [DOI] [PubMed] [Google Scholar]
- 3. Baer GS, Ebert DH, Chung CJ, Erickson AH, Dermody TS. 1999. Mutant cells selected during persistent reovirus infection do not express mature cathepsin L and do not support reovirus disassembly. J. Virol. 73:9532–9543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Becker MM, et al. 2001. Reovirus σNS protein is required for nucleation of viral assembly complexes and formation of viral inclusions. J. Virol. 75:1459–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Becker MM, Peters TR, Dermody TS. 2003. Reovirus σNS and μNS proteins form cytoplasmic inclusion structures in the absence of viral infection. J. Virol. 77:5948–5963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brentano L, Noah DL, Brown EG, Sherry B. 1998. The reovirus μ2, encoded by the M1 gene, is an RNA-binding protein. J. Virol. 72:8354–8357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Broering TJ, et al. 2005. Carboxyl-proximal regions of reovirus nonstructural protein μNS necessary and sufficient for forming factory-like inclusions. J. Virol. 79:6194–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Broering TJ, et al. 2004. Reovirus nonstructural protein μNS recruits viral core surface proteins and entering core particles to factory-like inclusions. J. Virol. 78:1882–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Broering TJ, Parker JS, Joyce PL, Kim J, Nibert ML. 2002. Mammalian reovirus nonstructural protein μNS forms large inclusions and colocalizes with reovirus microtubule-associated protein μ2 in transfected cells. J. Virol. 76:8285–8297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coombs KM. 1996. Identification and characterization of a double-stranded RNA-reovirus temperature-sensitive mutant defective in minor core protein μ2. J. Virol. 70:4237–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coombs KM. 2006. Reovirus structure and morphogenesis. Curr. Top. Microbiol. Immunol. 309:117–167 [DOI] [PubMed] [Google Scholar]
- 12. Dales S. 1963. Association between the spindle apparatus and reovirus. Proc. Natl. Acad. Sci. U. S. A. 50:268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dales S, Gomatos P, Hsu KC. 1965. The uptake and development of reovirus in strain L cells followed with labelled viral ribonucleic acid and ferritin-antibody conjugates. Virology 25:193–211 [DOI] [PubMed] [Google Scholar]
- 14. Dermody TS, Parker J, Sherry B. Orthoreovirus. In Knipe DM, et al. (ed), Fields virology, 6th ed, in press Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 15. Ebert DH, Deussing J, Peters C, Dermody TS. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 277:24609–24617 [DOI] [PubMed] [Google Scholar]
- 16. Fields BN. 1971. Temperature-sensitive mutants of reovirus type 3 features of genetic recombination. Virology 46:142–148 [DOI] [PubMed] [Google Scholar]
- 17. Gomatos PJ, Tamm I, Dales S, Franklin RM. 1962. Reovirus type 3: physical characteristics and interactions with L cells. Virology 17:441–454 [DOI] [PubMed] [Google Scholar]
- 18. Hazelton PR, Coombs KM. 1995. The reovirus mutant tsA279 has temperature-sensitive lesions in the M2 and L2 genes: the M2 gene is associated with decreased viral protein production and blockade in transmembrane transport. Virology 207:46–58 [DOI] [PubMed] [Google Scholar]
- 19. Ivanovic T, et al. 2011. Recruitment of cellular clathrin to viral factories and disruption of clathrin-dependent trafficking. Traffic 12:1179–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaufer S, Coffey CM, Parker JS. 2012. The cellular chaperone Hsc70 is specifically recruited to reovirus viral factories independently of its chaperone function. J. Virol. 86:1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim J, Parker JS, Murray KE, Nibert ML. 2004. Nucleoside and RNA triphosphatase activities of orthoreovirus transcriptase cofactor μ2. J. Biol. Chem. 279:4394–4403 [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi T, et al. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kobayashi T, Chappell JD, Danthi P, Dermody TS. 2006. Gene-specific inhibition of reovirus replication by RNA interference. J. Virol. 80:9053–9063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kobayashi T, Ooms LS, Chappell JD, Dermody TS. 2009. Identification of functional domains in reovirus replication proteins μNS and μ2. J. Virol. 83:2892–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mbisa JL, Becker MM, Zou S, Dermody TS, Brown EG. 2000. Reovirus μ2 protein determines strain-specific differences in the rate of viral inclusion formation in L929 cells. Virology 272:16–26 [DOI] [PubMed] [Google Scholar]
- 27. Miller CL, Broering TJ, Parker JS, Arnold MM, Nibert ML. 2003. Reovirus σNS protein localizes to inclusions through an association requiring the μNS amino terminus. J. Virol. 77:4566–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller CL, et al. 2004. Increased ubiquitination and other covariant phenotypes attributed to a strain- and temperature-dependent defect of reovirus core protein μ2. J. Virol. 78:10291–10302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morgan EM, Zweerink HJ. 1975. Characterization of transcriptase and replicase particles isolated from reovirus infected cells. Virology 68:455–466 [DOI] [PubMed] [Google Scholar]
- 30. Morgan EM, Zweerink HJ. 1974. Reovirus morphogenesis: core-like particles in cells infected at 39° with wild-type reovirus and temperature-sensitive mutants of groups B and G. Virology 59:556–565 [DOI] [PubMed] [Google Scholar]
- 31. Noble S, Nibert ML. 1997. Core protein μ2 is a second determinant of nucleoside triphosphatase activities by reovirus cores. J. Virol. 71:7728–7735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nonoyama M, Millward S, Graham AF. 1974. Control of transcription of the reovirus genome. Nucleic Acids Res. 1:373–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ooms LS, Kobayashi T, Dermody TS, Chappell JD. 2010. A post-entry step in the mammalian orthoreovirus replication cycle is a determinant of cell tropism. J. Biol. Chem. 285:41604–41613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parker JS, Broering TJ, Kim J, Higgins DE, Nibert ML. 2002. Reovirus core protein m2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J. Virol. 76:4483–4496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Patton JT, Silvestri LS, Tortorici MA, Vasquez-Del Carpio R, Taraporewala ZF. 2006. Rotavirus genome replication and morphogenesis: role of the viroplasm. Curr. Top. Microbiol. Immunol. 309:169–187 [DOI] [PubMed] [Google Scholar]
- 36. Reinisch KM, Nibert ML, Harrison SC. 2000. Structure of the reovirus core at 3.6 Å resolution. Nature 404:960–967 [DOI] [PubMed] [Google Scholar]
- 37. Rhim JS, Jordan LE, Mayor HD. 1962. Cytochemical, fluorescent-antibody and electron microscopic studies on the growth of reovirus (ECHO 10) in tissue culture. Virology 17:342–355 [DOI] [PubMed] [Google Scholar]
- 38. Rhim JS, Smith KO, Melnick JL. 1961. Complete and coreless forms of reovirus (ECHO 10): ratio of number of virus particles to infective units in the one-step growth cycle. Virology 15:428–435 [DOI] [PubMed] [Google Scholar]
- 39. Rodgers SE, et al. 1997. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J. Virol. 71:2540–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schiff LA, Nibert ML, Tyler KL. 2007. Orthoreoviruses and their replication, p 1853–1915 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 41. Sharpe AH, Chen LB, Fields BN. 1982. The interaction of mammalian reoviruses with the cytoskeleton of monkey kidney CV-1 cells. Virology 120:399–411 [DOI] [PubMed] [Google Scholar]
- 42. Shatkin AJ, LaFiandra AJ. 1972. Transcription by infectious subviral particles of reovirus. J. Virol. 10:698–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silverstein SC, Dales S. 1968. The penetration of reovirus RNA and initiation of its genetic function in L-strain fibroblasts. J. Cell Biol. 36:197–230 [PMC free article] [PubMed] [Google Scholar]
- 44. Silverstein SC, Schur PH. 1970. Immunofluorescent localization of double-stranded RNA in reovirus-infected cells. Virology 41:564–566 [DOI] [PubMed] [Google Scholar]
- 45. Skehel JJ, Joklik WK. 1969. Studies on the in vitro transcription of reovirus RNA catalyzed by reovirus cores. Virology 39:822–831 [DOI] [PubMed] [Google Scholar]
- 46. Spendlove RS, Lennette EH, Knight CO, Chin JN. 1963. Development of viral antigens and infectious virus on HeLa cells infected with reovirus. J. Immunol. 90:548–553 [PubMed] [Google Scholar]
- 47. Starnes MC, Joklik WK. 1993. Reovirus protein λ3 is a poly(C)-dependent poly(G) polymerase. Virology 193:356–366 [DOI] [PubMed] [Google Scholar]
- 48. Stuart DI, Grimes JM. 2006. Structural studies on orbivirus proteins and particles. Curr. Top. Microbiol. Immunol. 309:221–244 [DOI] [PubMed] [Google Scholar]
- 49. Sturzenbecker LJ, Nibert ML, Furlong DB, Fields BN. 1987. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J. Virol. 61:2351–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tao Y, Farsetta DL, Nibert ML, Harrison SC. 2002. RNA synthesis in a cage—structural studies of reovirus polymerase λ3. Cell 111:733–745 [DOI] [PubMed] [Google Scholar]
- 51. Virgin HW, Bassel-Duby IVR, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J. Virol. 62:4594–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Virgin HW, Tyler KL. 1991. Role of immune cells in protection against and control of reovirus infection in neonatal mice. J. Virol. 65:5157–5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weibel ER. 1979. Stereological methods: practical methods for biological morphometry. Academic Press, London, United Kingdom [Google Scholar]
- 54. Yin P, Cheang M, Coombs KM. 1996. The M1 gene is associated with differences in the temperature optimum of the transcriptase activity in reovirus core particles. J. Virol. 70:1223–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zarbl H, Millward S. 1983. The reovirus multiplication cycle, p 107–196 In Joklik WK. (ed), The Reoviridae. Plenum Press, New York, NY [Google Scholar]
- 56. Zhang X, Walker SB, Chipman PR, Nibert ML, Baker TS. 2003. Reovirus polymerase λ3 localized by cryo-electron microscopy of virions at a resolution of 7.6 Å. Nat. Struct. Biol. 10:1011–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zou S, Brown EG. 1996. Stable expression of the reovirus μ2 protein in mouse L cells complements the growth of a reovirus ts mutant with a defect in its M1 gene. Virology 217:42–48 [DOI] [PubMed] [Google Scholar]
- 58. Zweerink HJ, Ito Y, Matsuhisa T. 1972. Synthesis of reovirus double-stranded RNA within virion-like particles. Virology 50:349–358 [DOI] [PubMed] [Google Scholar]
- 59. Zweerink HJ, Morgan EM, Skyler JS. 1976. Reovirus morphogenesis: characterization of subviral particles in infected cells. Virology 73:442–453 [DOI] [PubMed] [Google Scholar]