

Abstract
Crimean-Congo hemorrhagic fever virus (CCHFV) is an emerging tick-borne virus of the Bunyaviridae family that is responsible for a fatal human disease for which preventative or therapeutic measures do not exist. We solved the crystal structure of the CCHFV strain Baghdad-12 nucleocapsid protein (N), a potential therapeutic target, at a resolution of 2.1 Å. N comprises a large globular domain composed of both N- and C-terminal sequences, likely involved in RNA binding, and a protruding arm domain with a conserved DEVD caspase-3 cleavage site at its apex. Alignment of our structure with that of the recently reported N protein from strain YL04057 shows a close correspondence of all folds but significant transposition of the arm through a rotation of 180 degrees and a translation of 40 Å. These observations suggest a structural flexibility that may provide the basis for switching between alternative N protein conformations during important functions such as RNA binding and oligomerization. Our structure reveals surfaces likely involved in RNA binding and oligomerization, and functionally critical residues within these domains were identified using a minigenome system able to recapitulate CCHFV-specific RNA synthesis in cells. Caspase-3 cleaves the polypeptide chain at the exposed DEVD motif; however, the cleaved N protein remains an intact unit, likely due to the intimate association of N- and C-terminal fragments in the globular domain. Structural alignment with existing N proteins reveals that the closest CCHFV relative is not another bunyavirus but the arenavirus Lassa virus instead, suggesting that current segmented negative-strand RNA virus taxonomy may need revision.
INTRODUCTION
Crimean-Congo hemorrhagic fever virus (CCHFV) is a tick-borne zoonotic virus responsible for a serious human disease characterized by hemorrhagic manifestations and multiple organ failure and is associated with a fatality rate of up to 50% (13, 46). The geographic range of CCHFV is exceptionally wide and reflects the broad distribution of the tick vector, which extends throughout 30 countries within Africa, Asia, the Middle East, and Southern Europe (25). Recent outbreaks of CCHFV infection in several Balkan states, southwestern Russia, and Turkey suggest that the activity of CCHFV is increasing, particularly in Southern Europe (24). The only treatment for CCHFV infection is postexposure administration of ribavirin, and the efficacy of this prophylaxis is in doubt (45). Development of effective treatments for prevention of CCHFV-mediated disease is now a priority for both public health and biodefense agencies.
CCHFV is a member of the Bunyaviridae family, and together with members of the Arenaviridae and Orthomyxoviridae families, these viruses are known as segmented negative-strand RNA viruses (sNSVs) by virtue of their multiple genome segments. The Bunyaviridae family contains over 350 named isolates classified within five genera, namely, Hantavirus, Nairovirus, Orthobunyavirus, Phlebovirus, and Tospovirus (41). CCHFV is a nairovirus, with a genome comprising three RNA segments: the small (S), medium (M), and large (L) segments. The S segment encodes the nucleocapsid protein (N), the M segment encodes the viral glycoproteins, and the L segment encodes an RNA-dependent RNA polymerase (RdRp; the L protein).
The genomes of sNSVs do not exist as naked RNAs but instead are encapsidated by the viral N protein to form ribonucleoprotein (RNP) complexes. RNPs associate with their cognate RdRp to form active templates for viral RNA synthesis, resulting in generation of encapsidated replication products and unencapsidated mRNAs. Genome encapsidation is also required for RNP packaging into progeny virus particles, and for bunyaviruses, virus assembly is mediated through direct association between the RNP and viral glycoproteins (18, 31, 39, 42, 44). RNP formation is thus essential for virus multiplication and therefore represents a potential therapeutic target.
In addition to RNP formation and virus assembly, the N proteins of bunyaviruses are implicated in other important functions, many of which relate to interactions with components of the host cell, including the cytoskeleton (3, 35–37, 43), cellular RNAs (26, 28), the translation machinery (8, 27), and mediators of the innate immune response (20, 30). Specifically for CCHFV, the N protein interacts with the cellular antiviral defense factor MxA (2) and recently was shown to act as a substrate for the apoptosis mediator caspase-3 (19), although the relevance of caspase-3 cleavage to the virus life cycle is unknown.
We present the 2.1-Å crystal structure of the N protein from CCHFV strain Baghdad-12. The CCHFV N structure we present displays significant differences in domain position compared to the recently reported structure of N from CCHFV strain YL04057 (15), isolated from China, and these differences may have important functional consequences, as discussed below. The structure reveals interfaces possibly involved in the critical N protein activities of N-N oligomerization and RNA binding, and these functions were tested in a minigenome assay in mammalian cells. In addition, the N structure reveals strong and unexpected structural homology with the N protein of the arenavirus Lassa virus (LASV), suggesting previously unappreciated aspects of sNSV phylogenetics, with the conclusion that current sNSV taxonomy may need revision.
MATERIALS AND METHODS
Structure determination.
The CCHFV N protein from the Baghdad-12 strain was expressed, purified, and crystallized as previously described (6). The N protein structure was solved by single isomorphous replacement with anomalous scattering (SIRAS) using a mercury derivative obtained by soaking natively grown CCHFV N crystals, suspended under the original crystallization conditions, with 5 mM thimerosal for 20 min (6). Eight prominent mercury peak sites were found, and phases from these were assigned to the native set by use of autoSHARP (50). Density modification was implemented by PARROT (9), after which the automated model building program BUCCANEER (1) was used to build 85% of the model. The model was then subjected to iterative cycles of manual building in COOT (12), and refinement in REFMAC (29) with TLS parameters applied until 98% of the protein molecules had been modeled. The TLS parameters were determined by the TLSMD server (33), which defined the globular and arm domains as separate rigid bodies. The ARP/wARP solvent program (21) was used to initially locate the bulk of water molecules, followed by manual building of the remaining solvent molecules in COOT. The fully refined model has an Rwork value of 18.4% and an Rfree value of 23.4% (Table 1) and was validated using MolProbity (7).
Table 1.
Data collection and refinement statistics
| Parameter | Valuea |
|
|---|---|---|
| Native crystal | Crystal soaked with thimerosal (8 Hg sites/AU) | |
| Wavelength (Å) | 0.9795 | 0.97950 |
| Space group | C2 | C2 |
| Cell parameters | ||
| a (Å) | 150.38 | 149.38 |
| b (Å) | 72.06 | 72.46 |
| c (Å) | 101.23 | 101.78 |
| β (°) | 110.70 | 111.05 |
| Total no. of reflections | 185,069 (27,462) | 108,583 (16,046) |
| No. of unique reflections | 56,441 (8,347) | 20,325 (2,951) |
| Resolution (Å) | 2.10 (2.21–2.10) | 3.0 (3.16–3.00) |
| Rmerge | 4.4 (46.7) | 5.6 (13.7) |
| Completeness (%) | 95.5 (97.2) | 99.0 (99.4) |
| Redundancy | 3.3 (3.3) | 5.3 (5.4) |
| I/σ(I) | 13.9 (2.5) | 19.1 (9.6) |
| VM (Å3/Da) | 2.38 | 2.39 |
| No. of molecules per AU | 2 | 2 |
| Rcrystal | 18.39 | |
| Rfree | 23.35 | |
| No. of reflections in working set | 53,584 | |
| Free R value set (% [no. of reflections]) | 5.1 (2,857) | |
| No. of nonhydrogen atoms | 7,672 | |
| No. of water molecules | 313 | |
| Mean B value (Å2) | 38.21 | |
| RMSD from ideality | ||
| Bonds (Å) | 0.022 | |
| Angles (°) | 1.559 | |
| Ramachandran statistics (%) | ||
| Preferred region | 97.02 | |
| Allowed region | 2.76 | |
| Outliers | 0.22 | |
| MolProbity score (95th percentile) | 1.65 | |
Except where noted, numbers in parentheses are for the highest resolution shell (2.21–2.10).
Plasmid construction.
The CCHFV N protein cDNA sequence in the previously described plasmid pC-N (4) was mutated using a site-directed mutagenesis approach based on structural and phylogenetic data. Selected residues were mutated to alanine (A), with the following exceptions: aspartic acid residue 272 (in the conserved DEVD caspase cleavage site) was mutated to either a glycine (G) or a glutamate (E), while isoleucine residue 304 was mutated to a glycine. The CCHFV N coding sequence was amplified from pC-N by using forward and reverse primers that introduced ClaI and XhoI sites at the 3′ and 5′ ends of the sequence, respectively. The PCR product was ligated into pCR-BLUNT II-TOPO (Invitrogen), and the coding sequence was mutated using Pfu Turbo polymerase as recommended by the manufacturer (Agilent Technologies). Where two mutations were introduced into the coding sequence, the mutagenesis was performed using a tandem approach. The mutated coding sequence was restricted out of the cloning vector by use of ClaI and XhoI and was ligated into a ClaI/XhoI-digested pC-N backbone. All clones were verified by sequencing.
Replicon assay.
Plasmid sequences and detailed cloning strategies are available upon request. Briefly, the CCHFV strain Ibar200 M segment minigenome (T7-M-Renilla) was generated by replacing the M open reading frame (ORF) with the Renilla luciferase gene (Promega), and the corresponding sequence was cloned into a vector designed to express viral sense RNAs from a T7 RNA polymerase promoter, as previously described (4). The V5-tagged L protein codons (4) were optimized for expression in human cells (Geneart), and the corresponding sequence was cloned into pCAGGS (pC-V5-L). BSR/T7 cells (Klaus Conzelmann, Max von Pettenkofer Institut, Munich, Germany) were seeded in 48-well plates 1 day prior to transfection, at a density of 3.5 × 104 cells per cm2. The cells in each well were transfected with 125 ng of pC-V5-L, 250 ng of wild-type pC-N or one of its mutants, 15 ng of T7-M-Renilla, and 15 ng of a firefly luciferase internal control plasmid (pGL3; Promega). Renilla and firefly luciferase activities were measured with a Dual-Glo luciferase assay system (Promega) on a Synergy 4 microplate reader (Biotek). CCHFV M minigenome Renilla luciferase activities were normalized over the control firefly luciferase activity and are reported as percentages of wild-type N activity.
In vitro caspase-3 cleavage and analysis.
Caspase-3 was expressed and purified according to previously published methods (10), using a plasmid encoding the full-length caspase-3 protein (53) fused to a C-terminal 6×His tag (Addgene). Expression and purification of caspase-3 were confirmed by SDS-PAGE, and its concentration was estimated by the Bradford assay (protein dye reagent; Bio-Rad) using bovine serum albumin (BSA) standards, after which the purified protein was concentrated to 10 mg ml−1. To measure caspase-3 cleavage of CCHFV N proteins with both wild-type (DEVD) and mutant (DEVE and DEVG) caspase-3 cleavage sites, a range of in vitro reactions were performed. Different molar ratios of caspase-3 protease and CCHFV N protein were added to 1.5-ml microcentrifuge tubes containing caspase-3 digestion buffer {20 mM piperazine-N,N′-bis(2-hydroxypropanesulfonic acid) (PIPES), pH 7.2, 200 mM NaCl, 20% sucrose, 0.2% (wt/vol) 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 2 mM EDTA} to reach the desired total volume of 40 μl. Reaction mixtures were incubated overnight at room temperature, and N protein digestion was analyzed using SDS-PAGE. Confirmation of caspase-3 activity was achieved by monitoring digestion of fluorogenic peptide substrate IX (Merck), derived from poly(ADP-ribose) polymerase (PARP), using a FLUOstar Optima plate reader (BMG Labtech) at excitation and absorption wavelengths of 485 nm and 520 nm, respectively.
Caspase-cleaved samples were then analyzed using an S200 10/300 GL analytical column (GE Healthcare), preequilibrated in binding buffer (100 mM NaCl, 20 mM Tris-HCl, and 1 mM EDTA, pH 7.5), at a flow rate of 0.5 ml min−1, with UV absorbance monitored at 280 nm.
Phylogenetic analysis of protein sequences.
ClustalW (47) was used to align the amino acid sequences of the N and L proteins of CCHFV with 13 other N and L protein sequences from selected negative-sense RNA viruses. The alignment was performed using default settings for multiple sequence alignment analyses. The viruses used in the comparative phylogenies were Dugbe virus, lymphocytic choriomeningitis virus, Lassa virus, Pichinde virus, Tacaribe virus, Bunyamwera virus, La Crosse virus, Rift Valley fever virus (RVFV), Hantaan virus, rice stripe virus, vesicular stomatitis virus, influenza A virus, and Thogoto virus. The accession numbers of the nucleocapsid proteins and polymerase proteins used in the analyses are quoted in the corresponding figure legends (for Fig. 2B and C). Clustal_X (22) was used to analyze the resultant alignment and to predict bootstrap confidence values of the alignment by use of the neighbor-joining algorithm. A total of 1,000 bootstrap trials were conducted on each submitted alignment file, and 111 were used as a random number generator seed. The graphical representation of the phylogeny was created using TreeView (32).
Fig 2.

Structural homology between CCHFV and LASV N proteins and phylogenetic analysis of segmented negative-strand RNA viruses by N and L protein comparison. (A) Structural alignment of CCHFV and LASV N protein structures. The CCHFV protein is shown in aquamarine. Most of the LASV N structure is shown in gray, but highlighted in yellow are the LASV N elements that move upon RNA binding (α5, loop, and α6). Note that there is no structural equivalent of the α6 RNA gate in CCHFV N. (B) The amino acid sequences of 14 viral N proteins, including those from six bunyaviruses (black text) and four arenaviruses (red text), were aligned using the ClustalW online server. Virus species abbreviations (GenBank accession numbers) are as follows: DUGV, Dugbe virus (P15190.1); CCHFV, Crimean-Congo hemorrhagic fever virus (AAB48503.1); LCMV, lymphocytic choriomeningitis virus (AAX49342.1); LASV, Lassa virus (ADY11071.1); PICV, Pichinde virus (AAC32282.1); TCRV, Tacaribe virus (AAA47903.1); BUNV, Bunyamwera virus (P16495.1); LACV, La Crosse virus (AAM94389.1); RVFV, Rift Valley fever virus (ABP88854.1); HTNV, Hantaan virus (AFA36178.1); RStV; rice stripe virus (P68559.2); VSV, vesicular stomatitis virus (AAA48470.1); IAV, influenza A virus (VHIVX1); and THOV, Thogoto virus (YP_145809.1). (C) The amino acid sequences of 14 viral polymerases, including those from six bunyaviruses (black text) and four arenaviruses (red text), were aligned using the ClustalW online server. GenBank accession numbers for the L protein sequence are as follows: DUGV, Q66431; CCHFV, AAQ98866; LCMV, AB196829; LASV, CCA30313; PICV, AAL16099.1; TCRV, AAA47901.1; BUNV, P20470; LACV, Q8JPR2; RVFV, P27316; HTNV, AAG10042; RStV, ADE60705; VSV, AAA48371.1; IAV, AAA43639; and THOV, O41353. The unrooted alignments were bootstrapped using Clustal_X, and the resultant phylograms were generated using TreeView. Bootstrap confidences are labeled at each node.
Protein structure accession number.
The atomic coordinates and structure factors for the N protein have been deposited in the Protein Data Bank (PDB) under accession number 4AKL.
RESULTS
Structure of the CCHFV N protein.
To gain mechanistic insights into its multiple functions, we determined the crystal structure of the full-length N protein (residues 1 to 482) of CCHFV strain Baghdad-12. Bacterially expressed N protein was crystallized, and the structure was solved using SIRAS. Native crystals belonging to the monoclinic space group C2 contained two N monomers in the asymmetric unit (AU). For each monomer, 474 residues were built into the model (residues 183 to 191 form a disordered loop that is missing in the electron density for both protomers), which was refined to a resolution of 2.1 Å (Table 1; Fig. 1A). N monomers consist of a globular core comprising 23 alpha helices (α1 to α11 and α18 to α29), with a prominent additional structural element we term the “arm,” comprising two long alpha helices (α15 and α17), that extends away from the core, with an exposed loop at the apex supported by a small three-helix bundle (α12 to α14) (Fig. 1A and C). The C-terminal half of N provides the helices at the core of the globular domain, which in turn are surrounded by helices from the N-terminal half. The monomers are packed in a head-to-tail arrangement, with the interface between monomers comprising helices α6 and α7 and helices α21 and α22 within the globular domain.
Fig 1.

Structure of the CCHFV N protein. (A) Structure of CCHFV N monomer in ribbon representation. Helices are shown in gray for the N-terminal portion and in dark blue for the C-terminal contribution to the globular domain and are shown in purple for the arm. The red sphere marks the N terminus, and the gold sphere marks the C terminus. Colored arrowheads show the caspase-3 DEVD cleavage motif (yellow) and the division of helices 17 and 18 (green). Panels A, B, and D were generated using PyMol. (B) Superposition (center) of the Baghdad-12 N structure (blue) and the YL04057 N structure (red) reveals very similar globular domains, whereas the arm has the same fold but is shifted relative to the globular domain. An expanded view of the region between the arm and the globular domain for each strain is shown, with relevant helices labeled. Numbering of helices for Baghdad-12 is as shown in panel C, whereas numbering of helices for YL04057 is according to the work of Guo et al. (15). (C) Primary amino acid sequences of N proteins of both strains of CCHFV, with the secondary structure shown schematically. The figure shown was made with ALINE (4a). The DEVD motif is highlighted in yellow, strain differences are highlighted in red, residues missing in the electron density are highlighted in black, and the kink between helices 17 and 18 (at Ser294) is highlighted in green. (D) The electrostatic surface potential (generated with APBS) suggests a positively charged RNA binding platform adjacent to the arm (47a), and rotation by 180 degrees suggests a positively charged cleft that we term the “pocket.” The scale bar shows the contour levels for the electrostatic potential at the solvent-exposed surface, in kT/e.
Analysis of the electrostatic surface potential of the N protein (Fig. 1D) revealed a continuous positively charged region located under the arm domain, and we refer to this region as the “platform.” An additional isolated area of positive charge is adjacent to a deep basic crevice, and this region is referred to as the “pocket.” Residues that comprise the putative RNA binding platform and pocket are conserved in all CCHFV isolates.
Structural comparison of CCHFV N proteins from strains Baghdad-12 and YL04057.
The globular domains from strains YL04057 and Baghdad-12 are very similar, with a Cα root mean square deviation (RMSD) of 0.975 Å for 357 residues with 92.16% sequence identity. In contrast, and most significantly, while the fold of the arm domain is the same (Cα RMSD of 0.642 Å, 99 residues, 77.78% identity), the position of the arm domain is radically altered; with respect to the globular domain, the arm is rotated by about 180 degrees, and the apex of the loop is shifted by a distance of 39.59 Å (at the Cα position of Asp266, at the apex of the arm, in each protein) (Fig. 1B). The main secondary structural difference that may account for this transposition is the division of a long helix in strain YL04057 into two shorter helices (α17 and α18) in strain Baghdad-12 (Fig. 1A), which may imply flexibility at the base of the arm.
Interestingly, comparison of the primary sequences of strains Baghdad-12 and YL04057 reveals 29 amino acid differences among 481 positions (Fig. 1C). Only nine of these are nonconservative, although the amino acid sequences at the division of helices 17 and 18 are identical, which could argue against strain differences being the major determining factor in arm position.
Similarity to other segmented RNA virus N proteins.
To provide insight into bunyavirus phylogenetics, we compared our CCHFV N structure to the recently reported structure of N from RVFV (14, 38), a phlebovirus. Alignment using the DALI server revealed essentially no structural homology between the CCHFV and RVFV N globular domains (Z score = 1.7), and zero similarity was detected with the arm domain. Interestingly, the DALI server identified strongest structural similarity with the N protein of the Arenaviridae family member LASV (5, 16, 17, 34), specifically within N-terminal residues 1 to 340, which comprise the RNA binding domain of LASV N (Fig. 2A) (17). Structural homology was identified for all forms (RNA-free and RNA bound) of LASV N deposited in the PDB (16, 17, 34), with Z scores falling between 12.4 and 15.3. The globular domains from the CCHFV and RNA-free LASV N proteins superposed with a Cα root mean square deviation of 3.38 Å for 237 residues with 8.01% sequence identity. Surprisingly, this suggests that CCHFV is more closely related to the arenavirus LASV than to the bunyavirus RVFV. This proposal is supported by a phylogenetic analysis of sNSV L and N sequences (Fig. 2B and C), which similarly implies that nairoviruses are more closely related to arenaviruses than to any other bunyavirus genera.
Potential for a gated RNA binding mechanism.
The electrostatic charge distribution on the CCHFV N surface revealed two possible RNA binding regions: the platform and the pocket. Structural comparison of N proteins from strains Baghdad-12 and YL04057 suggested that the arm is free to explore a range of conformations, which could significantly alter surface electrostatics and therefore RNA binding. In addition, the flexible loop between the arm and the globular domain (residues 183 to 191; not visible in either the YL04057 or Baghdad-12 N structure) could become ordered on RNA binding. Thus, the arm domain may “gate” RNA binding, similar to the LASV and RVFV gate (see Discussion).
The N-N dimer interface.
The formation of viral RNPs presumably relies on N-N interactions, and we therefore examined potential interacting surfaces that could mediate this function. The CCHFV N protein was expressed and purified as an RNA-free monomer (6). However, as described above, the AU in the C2 space group comprises two monomers (Fig. 3B), and it is plausible that the interface seen between these two non-crystallographic symmetry (NCS)-related molecules represents a biologically relevant dimer interface.
Fig 3.

In vivo effects of site-directed CCHFV N protein mutants. (A) Residues selected for mutation and testing in the minireplicon system are highlighted on the structure. Residues in the RNA binding platform are highlighted in green, residues that comprise the RNA binding pocket are shown in blue, the possible dimer interface is shown in red, and D269 of the DEVD motif is shown in orange. Alterations of residues K132, Q300, and K411 abrogated minigenome activity, and these residues are shown in magenta. (B) Ribbon representation of two protomers within the crystal lattice, suggesting the putative dimer interface investigated using the minireplicon system. (C) Histogram showing reporter gene expression for mutants, colored as described above, normalized to that of wild-type (WT) N. (D) Western blot analysis to assess relative expression levels of N protein mutants from equivalent quantities of cells expressing the minireplicon system, using a polyclonal antibody raised against the CCHFV N protein.
Analysis of the potential dimer interface by use of the PISA server reveals an average buried surface area of 1,015 Å2, a ΔG value of −9.5 kcal/mol, and a complexation significance score (CSS) of 0.0 (a score of 1 suggests a stable interface, and a score of 0 suggests a nonstable interface, i.e., crystal packing). For comparison, the oligomeric interfaces present in the RVFV N structure have an average buried surface area of 1,643 Å2, a ΔG value of −18.5 kcal/mol, and a CSS of 1.0. However, lattice contacts within the same RVFV crystal form present an average buried surface area of 437 Å2 and a ΔG value of −3.9 kcal/mol; thus, the CCHFV N interface was scored at values intermediate between the two. PISA analysis thus suggests that even the largest interface is not sufficient to generate a stable interface in solution. However, this interface may represent part of the authentic oligomerization domain, with the possibility that a conformational change on RNA binding may reveal a more extensive or rearranged interface.
Functional analysis of selected N protein residues by use of a CCHFV minigenome system.
The structure of the CCHFV N protein revealed the identity of amino acids that could potentially be involved in the critical activities of RNA binding and N-N oligomerization. To investigate the role of N protein residues in these activities, we used a CCHFV minireplicon system in which reporter gene expression was promoted by formation of RNP templates from RNA and N protein in mammalian cells (4). A panel of N proteins bearing mutations within the proposed RNA binding surfaces (Fig. 3A), the putative oligomerization interface (Fig. 3B), or the DEVD motif (Fig. 3A) was generated, with residue selection based on position within the N structure and conservation within available CCHFV N sequences. Reporter expression in the minigenome system (Fig. 3C) depends on the ability of the N protein to both bind RNA and multimerize to form an RNP template; therefore, a loss of reporter expression will reflect deficiencies in either of these activities. Of nine mutants with alterations within the RNA binding platform, six exhibited essentially unchanged replicon activity (K91A, K98A, E112A, R140A, S149A, and Y470A); however, the three remaining mutants showed replicon activity that was either significantly reduced (K90A) or effectively abrogated (K132A and Q300A). Of the selected mutants with mutations of positively charged residues within the RNA binding pocket, the K342A, K343A, and H453A mutants exhibited nearly wild-type activity; however, the K411A and H456A mutants resulted in abrogated and significantly reduced RNP activity, respectively. Additional mutants, i.e., the E387A, I304G, and W313A mutants, were also generated but were expressed poorly or undetectably in transfected cells (Fig. 3D) and were therefore not analyzed in the minigenome system.
Taken together, the results of this analysis identified three separate residues (K132, Q300, and K411) that were individually essential for replicon activity and another two residues (K90 and H456) whose mutation resulted in a significantly reduced N functionality. Because of their charge characteristics and specific location on the N protein surface, we propose that these residues likely play a role in RNA binding. Interestingly, residue K411 was recently described as contributing to DNase activity of the CCHFV N protein (15), whereas our results favor a direct role in CCHFV gene expression.
We also investigated the functional relevance of the crystallographic dimer interface (Fig. 3B) by using both single-residue mutants (E108A and K114A mutants) and double-residue mutants (K354A/E108A, K357A/E112A, K358A/R140A, and E361K/K114A mutants). None of these mutants resulted in abrogation of RNP function, suggesting that none of the altered amino acids were essential for N protein oligomerization. However, the K354A/E108A and K357/E112A double mutants showed a drop in RNP activity, suggesting that the corresponding residues may play roles in RNP assembly, possibly by promoting oligomerization.
The mutants with altered DEVD motifs (DEVG and DEVE), which are not cleavable by caspase-3 (see below), promoted levels of minigenome activity that were indistinguishable from that of wild-type N. While caspase-3 cleavage may have an effect on the life cycle of infectious virus, we do not expect to see this recapitulated in the minigenome system, due to a lack of caspase-3 induction.
Consequences of cleavage of the DEVD caspase-3 recognition site.
CCHFV replication in cell culture results in the induction of apoptosis at late time points within the infectious cycle (40), and the onset of apoptosis coincides with cleavage of the N protein by cellular caspase-3, at a conserved DEVD motif at residues 269 to 272 (19).
The structure of CCHFV N reveals that the DEVD motif is positioned at the apex of the arm domain (Fig. 1A, yellow arrowhead), in what is arguably the most exposed and accessible position on the apo molecule. The prominent position and strict conservation of the DEVD motif in all CCHFV isolates suggest that maintenance of this cleavage motif must be beneficial to the virus life cycle, and this is particularly significant due to the known plasticity of the CCHFV genome (11) and in light of the high mutation rates of RNA viruses in general. Caspase-3 cleavage resulted in the generation of N-terminal (residues 1 to 269) and C-terminal (residues 270 to 482) fragments (19), although the possible roles of these separate fragments or the cleavage event itself, within the context of the CCHFV life cycle, are unknown. To better understand the possible consequences of caspase-3 cleavage on the virus life cycle, we wanted to determine, using size exclusion chromatography (SEC), whether cleavage resulted in independent N- and C-terminal fragments or whether these N- and C-terminal fragments remained associated postcleavage. This information was important, as either of these two outcomes could have significance for N protein function downstream of caspase-3 cleavage.
Recombinant caspase-3 and purified wild-type CCHFV N protein were mixed, and cleavage at the DEVD site was monitored by analysis of digestion products separated using SDS-PAGE (Fig. 4A). Surprisingly, caspase-3 cleavage of wild-type N protein resulted in incomplete digestion even following overnight incubation at an equimolar ratio (Fig. 4A, lane 4), indicating that the CCHFV N protein was a poor substrate for cleavage despite the apparent accessibility of the DEVD motif (Fig. 1A). Caspase-3 activity was confirmed using a fluorogenic peptide derived from PARP (data not shown), and as expected, CCHFV N mutants with altered cleavage motifs (DEVE and DEVG) were uncleaved (Fig. 4B, lanes 2 and 3).
Fig 4.
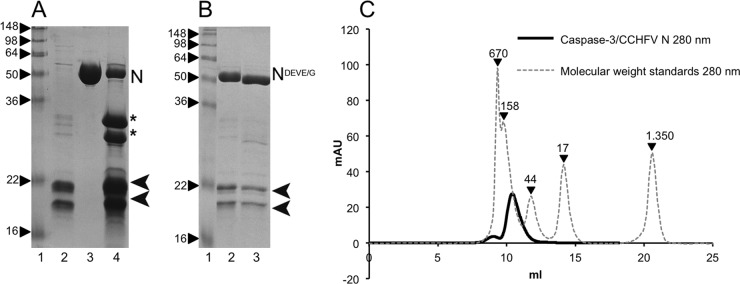
CCHFV N is cleaved by caspase-3 but remains intact. (A) SDS-PAGE gel showing cleavage of wild-type N into two fragments of the expected sizes by recombinant caspase-3. Lane 1, size markers in kDa; lane 2, purified caspase-3; lane 3, purified CCHFV N; lane 4, N and caspase-3 (1:1). N marks the full-length protein, asterisks mark the N- and C-terminal cleaved fragments, and arrowheads mark the two fragments of caspase-3. (B) DEVD mutants (DEVE or DEVG) are not cleaved by caspase-3. Lane 1, size markers in kDa; lane 2, N-DEVE plus caspase-3; lane 3, N-DEVG plus caspase-3. (C) Size exclusion chromatography of N and caspase-3 digestion products. Cleaved N protein elutes as a monomer, and cleavage products are not detected, suggesting that the N-terminal and C-terminal fragments remain in a tight complex. The following molecular mass standards (dashed line) are given in kDa: thyroglobulin, 670; β-globulin, 158; ovalbumin, 44; myoglobulin, 17; and vitamin B12, 1.35.
Analysis of the reaction products generated by caspase-3 digestion of N protein by SEC showed that all of the cleaved N protein eluted with an apparent molecular mass corresponding to that of an intact monomer (Fig. 4C). No evidence of N- or C-terminal cleavage products in solution, which would easily be resolved in later fractions due to their relatively small molecular masses (approximately 25 to 30 kDa), was detected (Fig. 4A). These findings show that following caspase cleavage, the N- and C-terminal fragments of the CCHFV N protein remain associated, likely as a consequence of close contact of helical structural elements within the globular domain, as described above (Fig. 1A).
DISCUSSION
The X-ray crystal structure of the N protein from the serious human pathogen CCHFV reveals a compact globular domain from which an extended arm domain protrudes. The globular domain contains positively charged surfaces that, because of their exposed and contiguous position on the exterior of the molecule and by the strict requirement of several residues on these surfaces for CCHFV gene expression, we propose are involved in RNA binding. In support of our identification of these putative RNA binding regions, the CCHFV N globular domain shows a high degree of structural similarity with the N-terminal RNA binding domain of the LASV N protein. The consequences of this close structural relationship are discussed further below.
In LASV, there is a proposed gating mechanism of RNA binding where helices α5 and α6 are repositioned to reveal the RNA binding surface (17). The concept of a flexible arm being involved in gating RNA binding has also been proposed for RVFV N, which has an N-terminal arm interacting with an oligomerization groove on adjacent monomers and consequently exposing an RNA binding cleft (14). CCHFV N may operate via a similar mechanism, although the structural elements involved in the gating process are likely different. The position of helix α5 in LASV corresponds to the beginning of the flexible loop leading to the CCHFV arm domain; thus, one possibility is that the arm of CCHFV N may be involved in switching between RNA-bound and unbound conformations.
Structural alignment (Fig. 2A) revealed that the CCHFV RNA binding pocket corresponds to the domain within LASV N that was initially thought to bind the cap (34) but was later suggested to represent a binding pocket involved in the interaction with a single nucleotide of a bound RNA strand (17). This raises the possibility either that CCHFV N binds RNA via two surfaces (platform and pocket) or that conformational rearrangements create a continuous RNA binding surface. It is quite possible that a combination of arm movement and structural rearrangements is required in order to reveal the appropriate RNA binding surfaces, which may not be evident in the apo crystal structures.
Because we do not know the length of RNA bound by each N monomer, or indeed the oligomeric form of N in the RNP, we cannot currently discriminate between any plausible RNA binding mechanisms or deduce what any required conformational changes might be. However, it seems reasonable, based on previous studies of RNP formation, to anticipate more details of the RNA binding mechanism to be revealed by a structure of the N protein in complex with RNA, possibly via a gating mechanism analogous to that seen for LASV.
The possibility that the arm domain can adopt different positions is supported by comparison of our N structure for strain Baghdad-12 with that for strain YL04057, reported recently (15). While the globular domains align very closely, the arm domains adopt radically different positions, being rotated by nearly 180 degrees and with the arm apex being translated by 40 Å (Fig. 1B). This rearrangement may have important consequences, as the two arm positions may represent interchangeable forms that possess different activities in critical N protein functions such as oligomerization or RNA binding. However, one possibility is that the arm positions are dictated by the different primary sequences of the respective strains.
Strain differences could account for a change in the preferred lowest energy state. However, the arm is more likely to be more mobile and free to adopt a number of conformations, taking into account the likeliness that an exposed α helix is not a stable structure in isolation. The single α helix linking the globular and arm domains is unlikely to provide a rigid link. Alternatively, these differences may have been forced by crystal packing. However, should the strain differences we observed also be reflected in solution, one consequence of the shifted arm position and sequence differences between the two N structures is an altered distribution of electrostatic surface potential, which may affect the RNA binding ability of the respective proteins. It is possible that the arm represents part of a gating mechanism allowing a switch between RNA-bound and unbound states. However, it is also plausible that the switch in arm position is responsible for conversion of monomeric N into higher-order multimers, a property that is required for RNP formation.
In addition to the radically different arm positions posing possible functional significance, the differing arm conformations of these two strains may also have a bearing on strategies for structure-based design of small-molecule inhibitors for use as antivirals.
A striking feature of the CCHFV N structure is that the arm domain displays a DEVD caspase cleavage motif at its apex, in the most accessible position on the entire molecule (Fig. 1A). This exposed position, along with its strict conservation in all CCHFV strains, suggests that the virus has evolved to present the cleavage motif to the cellular environment, which is somehow beneficial to the virus life cycle. If possession of the exposed cleavage site were not beneficial to the virus or if caspase cleavage were a host defense mechanism, a fast-mutating RNA virus such as CCHFV would be predicted to quickly eliminate the motif. The functional significance of this caspase cleavage site is therefore an intriguing feature of the CCHFV N protein. To investigate the fate of the N protein fragments following cleavage, we performed caspase cleavage of the N protein in vitro and showed that the cleavage products remained associated as a single unit. This raises the interesting possibility that N protein functions may remain unaltered following cleavage. The cleaved N protein could of course have an altered tertiary or quaternary structure, which may influence function in a variety of ways, including interaction with different protein partners, the adoption of different oligomeric states, or alteration of RNA binding affinity. In order to best understand the functional relevance of this DEVD motif in the CCHFV life cycle, we need to manipulate the CCHFV genome with a view to studying the consequence of such a change in the context of a live virus infection of intact organisms. Unfortunately, such a system currently does not exist.
Intriguingly, the nucleocapsid protein (NP) of human-infecting strains of influenza A virus also possesses caspase cleavage sites which have been shown to possess important roles in the virus life cycle (51, 52). Infectious influenza viruses bearing mutations that abrogate caspase cleavage at an N-terminal recognition site could not be rescued, indicating that such alterations are lethal to virus viability, whereas mutations within a C-terminal caspase cleavage site rapidly reverted to the wild type to restore cleavability. These findings establish an important precedent that links caspase cleavability with virus fitness, and thus pathogenesis, suggesting a second functional surface (in addition to the RNA binding site) that could be targeted by antivirals.
Structural comparisons indicate that the CCHFV N protein globular domain exhibits a high degree of homology with the N-terminal domain of the N protein of LASV, a member of the Arenaviridae family, whose members uniformly possess two RNA segments. In contrast, CCHFV N shows negligible structural similarity with the only other bunyavirus (RVFV) N protein for which high-resolution structural data are available. The finding that these bunyavirus N proteins appear essentially structurally unrelated yet the CCHFV N protein shows extremely high homology with the N protein from a virus of a different taxonomic family with a different number of genome segments is intriguing. Previous phylogenetic analyses based on L protein sequences have also described a close relationship between nairoviruses and arenaviruses (48), and this conclusion is further supported by the phylogenetic analysis of N and RdRp protein sequences presented here (Fig. 2B and C). Further supporting this close relationship, arenaviruses and nairoviruses also possess unique aspects of cellular biology that are absent from other bunyaviruses, such as the dependence of cellular SKI-1 protease processing of their respective glycoprotein precursors (23, 49). However, the high degree of structural similarity we observed between the CCHFV and LASV N proteins is the most compelling evidence yet that arenaviruses have an ancestor in common with a current or past member of the Nairovirus genus. This suggests that the current classification of all three-segment negative-strand RNA viruses as bunyaviruses may be oversimplistic and that the evident diversity within the Bunyaviridae family may warrant reevaluation of its current taxonomic status.
ACKNOWLEDGMENTS
We thank Roger Hewson (Health Protection Agency, Porton Down, United Kingdom) for the CCHFV N cDNA. We also thank Chi Trinh, Susan Matthews, and Arwen Pearson (University of Leeds, United Kingdom), as well as the beamline scientists at the Diamond synchrotron, for continued help and support with data collection.
This work was financially supported by a personal studentship to S.D.C. from The Jersey Government, by The Wellcome Trust, and by a generous donation from Jerry Knapp.
Footnotes
Published ahead of print 8 August 2012
REFERENCES
- 1. Acta Crystallographica. Section D. Biological Crystallography 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 2. Andersson I, et al. 2004. Human MxA protein inhibits the replication of Crimean-Congo hemorrhagic fever virus. J. Virol. 78:4323–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andersson I, et al. 2004. Role of actin filaments in targeting of Crimean Congo hemorrhagic fever virus nucleocapsid protein to perinuclear regions of mammalian cells. J. Med. Virol. 72:83–93 [DOI] [PubMed] [Google Scholar]
- 4. Bergeron E, Albarino CG, Khristova ML, Nichol ST. 2010. Crimean-Congo hemorrhagic fever virus-encoded ovarian tumor protease activity is dispensable for virus RNA polymerase function. J. Virol. 84:216–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a. Bond CS, Schuttelkopf AW. 2009. ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr. D Biol. Crystallogr. 65:510–512 [DOI] [PubMed] [Google Scholar]
- 5. Brunotte L, et al. 2011. Structure of the Lassa virus nucleoprotein revealed by X-ray crystallography, small-angle X-ray scattering, and electron microscopy. J. Biol. Chem. 286:38748–38756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carter SD, Barr JN, Edwards TA. 2012. Expression, purification and crystallization of the Crimean-Congo hemorrhagic fever virus nucleocapsid protein. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68:569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen VB, et al. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng E, et al. 2011. Characterization of the interaction between hantavirus nucleocapsid protein (N) and ribosomal protein S19 (RPS19). J. Biol. Chem. 286:11814–11824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cowtan K. 2010. Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr. 66:470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Denault JB, Salvesen GS. 2003. Expression, purification, and characterization of caspases. Curr. Protoc. Protein Sci. Chapter 21:Unit 21.13 [DOI] [PubMed] [Google Scholar]
- 11. Deyde VM, Khristova ML, Rollin PE, Ksiazek TG, Nichol ST. 2006. Crimean-Congo hemorrhagic fever virus genomics and global diversity. J. Virol. 80:8834–8842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 13. Ergonul O, Tuncbilek S, Baykam N, Celikbas A, Dokuzoguz B. 2006. Evaluation of serum levels of interleukin (IL)-6, IL-10, and tumor necrosis factor-alpha in patients with Crimean-Congo hemorrhagic fever. J. Infect. Dis. 193:941–944 [DOI] [PubMed] [Google Scholar]
- 14. Ferron F, et al. 2011. The hexamer structure of the Rift Valley fever virus nucleoprotein suggests a mechanism for its assembly into ribonucleoprotein complexes. PLoS Pathog. 7:e1002030 doi:10.1371/journal.ppat.1002030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo Y, et al. 2012. Crimean-Congo hemorrhagic fever virus nucleoprotein reveals endonuclease activity in bunyaviruses. Proc. Natl. Acad. Sci. U. S. A. 109:5046–5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO. 2011. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. U. S. A. 108:2396–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hastie KM, et al. 2011. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc. Natl. Acad. Sci. U. S. A. 108:19365–19370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hepojoki J, et al. 2010. Cytoplasmic tails of hantavirus glycoproteins interact with the nucleocapsid protein. J. Gen. Virol. 91:2341–2350 [DOI] [PubMed] [Google Scholar]
- 19. Karlberg H, Tan YJ, Mirazimi A. 2011. Induction of caspase activation and cleavage of the viral nucleocapsid protein in different cell types during Crimean-Congo hemorrhagic fever virus infection. J. Biol. Chem. 286:3227–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kochs G, Janzen C, Hohenberg H, Haller O. 2002. Antivirally active MxA protein sequesters La Crosse virus nucleocapsid protein into perinuclear complexes. Proc. Natl. Acad. Sci. U. S. A. 99:3153–3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lamzin VS, Wilson KS. 1997. Automated refinement for protein crystallography. Methods Enzymol. 277:269–305 [DOI] [PubMed] [Google Scholar]
- 22. Larkin MA, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 23. Lenz O, ter Meulen J, Klenk HD, Seidah NG, Garten W. 2001. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. U. S. A. 98:12701–12705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maltezou HC, Papa A. 2010. Crimean-Congo hemorrhagic fever: risk for emergence of new endemic foci in Europe? Travel Med. Infect. Dis. 8:139–143 [DOI] [PubMed] [Google Scholar]
- 25. Mild M, Simon M, Albert J, Mirazimi A. 2010. Towards an understanding of the migration of Crimean-Congo hemorrhagic fever virus. J. Gen. Virol. 91:199–207 [DOI] [PubMed] [Google Scholar]
- 26. Mir MA, Duran WA, Hjelle BL, Ye C, Panganiban AT. 2008. Storage of cellular 5′ mRNA caps in P bodies for viral cap-snatching. Proc. Natl. Acad. Sci. U. S. A. 105:19294–19299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mir MA, Panganiban AT. 2008. A protein that replaces the entire cellular eIF4F complex. EMBO J. 27:3129–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mir MA, Sheema S, Haseeb A, Haque A. 2010. Hantavirus nucleocapsid protein has distinct m7G cap- and RNA-binding sites. J. Biol. Chem. 285:11357–11368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255 [DOI] [PubMed] [Google Scholar]
- 30. Ontiveros SJ, Li Q, Jonsson CB. 2010. Modulation of apoptosis and immune signaling pathways by the Hantaan virus nucleocapsid protein. Virology 401:165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Overby AK, Pettersson RF, Neve EP. 2007. The glycoprotein cytoplasmic tail of Uukuniemi virus (Bunyaviridae) interacts with ribonucleoproteins and is critical for genome packaging. J. Virol. 81:3198–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Page RD. 1996. TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12:357–358 [DOI] [PubMed] [Google Scholar]
- 33. Painter J, Merritt EA. 2006. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62:439–450 [DOI] [PubMed] [Google Scholar]
- 34. Qi X, et al. 2010. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 468:779–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramanathan HN, et al. 2007. Dynein-dependent transport of the Hantaan virus nucleocapsid protein to the endoplasmic reticulum-Golgi intermediate compartment. J. Virol. 81:8634–8647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramanathan HN, Jonsson CB. 2008. New and Old World hantaviruses differentially utilize host cytoskeletal components during their life cycles. Virology 374:138–150 [DOI] [PubMed] [Google Scholar]
- 37. Ravkov EV, Nichol ST, Peters CJ, Compans RW. 1998. Role of actin microfilaments in Black Creek Canal virus morphogenesis. J. Virol. 72:2865–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raymond DD, Piper ME, Gerrard SR, Smith JL. 2010. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc. Natl. Acad. Sci. U. S. A. 107:11769–11774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ribeiro D, Borst JW, Goldbach R, Kormelink R. 2009. Tomato spotted wilt virus nucleocapsid protein interacts with both viral glycoproteins Gn and Gc in planta. Virology 383:121–130 [DOI] [PubMed] [Google Scholar]
- 40. Rodrigues R, Paranhos-Baccala G, Vernet G, Peyrefitte CN. 2012. Crimean-Congo hemorrhagic fever virus-infected hepatocytes induce ER-stress and apoptosis crosstalk. PLoS One 7:e29712 doi:10.1371/journal.pone.0029712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schmaljohn CS, Nichol ST. 2007. Bunyaviridae, p 1741–1789 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 42. Shi X, Kohl A, Li P, Elliott RM. 2007. Role of the cytoplasmic tail domains of Bunyamwera orthobunyavirus glycoproteins Gn and Gc in virus assembly and morphogenesis. J. Virol. 81:10151–10160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simon M, Johansson C, Lundkvist A, Mirazimi A. 2009. Microtubule-dependent and microtubule-independent steps in Crimean-Congo hemorrhagic fever virus replication cycle. Virology 385:313–322 [DOI] [PubMed] [Google Scholar]
- 44. Snippe M, Willem Borst J, Goldbach R, Kormelink R. 2007. Tomato spotted wilt virus Gc and N proteins interact in vivo. Virology 357:115–123 [DOI] [PubMed] [Google Scholar]
- 45. Soares-Weiser K, Thomas S, Thomson G, Garner P. 2010. Ribavirin for Crimean-Congo hemorrhagic fever: systematic review and meta-analysis. BMC Infect. Dis. 10:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Swanepoel R, et al. 1987. Epidemiologic and clinical features of Crimean-Congo hemorrhagic fever in southern Africa. Am. J. Trop. Med. Hyg. 36:120–132 [DOI] [PubMed] [Google Scholar]
- 47. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47a. Unni S, et al. 2011. Web servers and services for electrostatics calculations with APBS and PDB2PQR. J. Comput. Chem. 32:1488–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vieth S, Torda AE, Asper M, Schmitz H, Gunther S. 2004. Sequence analysis of L RNA of Lassa virus. Virology 318:153–168 [DOI] [PubMed] [Google Scholar]
- 49. Vincent MJ, et al. 2003. Crimean-Congo hemorrhagic fever virus glycoprotein proteolytic processing by subtilase SKI-1. J. Virol. 77:8640–8649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vonrhein C, Blanc E, Roversi P, Bricogne G. 2007. Automated structure solution with autoSHARP. Methods Mol. Biol. 364:215–230 [DOI] [PubMed] [Google Scholar]
- 51. Zhirnov OP, Konakova TE, Garten W, Klenk H. 1999. Caspase-dependent N-terminal cleavage of influenza virus nucleocapsid protein in infected cells. J. Virol. 73:10158–10163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhirnov OP, Syrtzev VV. 2009. Influenza virus pathogenicity is determined by caspase cleavage motifs located in the viral proteins. J. Mol. Genet. Med. 3:124–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou Q, et al. 1997. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J. Biol. Chem. 272:7797–7800 [DOI] [PubMed] [Google Scholar]