

Abstract
We previously reported that the macrolide antibiotic clarithromycin (CAM) enhanced the mucosal immune response in pediatric influenza, particularly in children treated with the antiviral neuraminidase inhibitor oseltamivir (OSV) with low production of mucosal antiviral secretory IgA (S-IgA). The aims of the present study were to confirm the effects of CAM on S-IgA immune responses, by using influenza A virus (IAV) H1N1-infected mice treated with or without OSV, and to determine the molecular mechanisms responsible for the induction of mucosal IgA class switching recombination in IAV-infected CAM-treated mice. The anti-IAV S-IgA responses and expression levels of IgA class switching recombination-associated molecules were examined in bronchus-lymphoid tissues and spleens of infected mice. We also assessed neutralization activities of S-IgA against IAV. Data show that CAM enhanced anti-IAV S-IgA induction in the airway of infected mice and restored the attenuated antiviral S-IgA levels in OSV-treated mice to the levels in the vehicle-treated mice. The expression levels of B-cell-activating factor of the tumor necrosis factor family (BAFF) molecule on mucosal dendritic cells as well as those of activation-induced cytidine deaminase and Iμ-Cα transcripts on B cells were enhanced by CAM, compared with the levels without CAM treatment, but CAM had no effect on the expression of the BAFF receptor on B cells. Enhancement by CAM of neutralization activities of airway S-IgA against IAV in vitro and reinfected mice was observed. This study identifies that CAM enhances S-IgA production and neutralizing activities through the induction of IgA class switching recombination and upregulation of BAFF molecules in mucosal dendritic cells in IAV-infected mice.
INTRODUCTION
Influenza brings repeating global threats to humans through annual epidemics, and there have been several pandemics, with considerable morbidity and mortality. In order to prevent complications and aggravation of the flu symptoms (25, 36), it is not uncommon, in Japan, to prescribe clarithromycin (CAM), a macrolide antibiotic developed by modification of erythromycin (11), combined with oseltamivir (OSV) as an antiviral neuraminidase inhibitor. In this regard, we have previously reported that administration of CAM in influenza A virus (IAV)-infected mice resulted in suppression of tumor necrosis factor alpha and augmentation of interleukin-12 production in the blood, resulting in alleviation of the flu symptoms (18), while oral treatment with OSV attenuated the induction of respiratory anti-IAV specific secretory IgA (S-IgA) immune responses (39). Furthermore, we have recently verified in IAV-infected children that oral CAM augmented the nasopharyngeal mucosal immune responses, while OSV suppressed the production of mucosal anti-IAV S-IgA (37). Of interest, we have also reported that 75% of patients treated with the combination of CAM and OSV showed increases in S-IgA production to levels similar to those seen in patients treated with CAM alone (37). Others have also reported that CAM acted on the viral replication cycles, resulting in inhibition of progeny virus production in vitro (25, 26), and modulated airway inflammation in IAV infection by reduction of the viral receptor, sialic acid with an α2,6 linkage on the airway epithelial cells, through inhibition of nuclear factor kappa B (NF-κB) expression and increase in intraendosomal pH (45). However, there is little information on the mechanisms of CAM-boosted induction of mucosal anti-IAV S-IgA.
Nasopharyngeal-associated lymphoreticular tissue and Peyer's patches are known as mucosal inductive sites where IgA-committed B cells undergo μ- to α-isotype class switching recombination (CSR). The IgA-committed B cells subsequently migrate to diffuse mucosal effector tissues, including the nasal passages (NPs) and intestinal lamina propria (iLP) (3, 22). In addition to these mucosal inductive tissues, T-cell-independent IgA CSR occurs in the iLP (8, 9, 12). Similarly, B cells of the isolated lymphoid follicles, scattered throughout the intestine, can undergo IgA CSR either from actual bacterial infection or from constant surveillance of commensals (10, 40). In this regard, both in vitro and in vivo studies have shown that B-cell-activating factor of the tumor necrosis factor family (BAFF) and the proliferation-inducing ligand (APRIL), members of the tumor necrosis factor ligand superfamily, promote T-cell-independent CSR of IgA via engagement of BAFF receptor (BAFF-R), transmembrane activator and calcium modulator cyclophilin ligand interactor (TACI), and B-cell maturation antigen (Ag) (BCMA) (4, 5, 24). In addition, BAFF and APRIL on dendritic cells (DCs) can induce the expression of activation-induced cytidine deaminase (AID) expression in murine B cells (24, 44). Recent studies have also reported that retinoic acid-producing DCs from mucosa-associated lymphoreticular tissue induce surface IgA and gut homing receptor expression on B cells in a T-cell-independent manner (17, 29). BAFF and APRIL on DCs interact with BAFF-R, BCMA, and TACI on B cells and induce IgA CSR (2).
The aims of the present study were to confirm the effects of CAM on S-IgA immune responses, by using IAV (H1N1)-infected weanling mice, and to determine the cellular and molecular mechanisms responsible for the induction of IgA CSR in IAV-infected mice treated with CAM.
MATERIALS AND METHODS
Animals and viral infection.
All experiments were conducted in accordance with the animal care committee guidelines of Tokushima University. Specific-pathogen-free 4-week-old weanling BALB/c female mice were obtained from Japan SLC. The mice were nasally inoculated with 25 PFU of mouse-adapted IAV/PR8/34(H1N1) in 15 μl of saline under ketamine anesthesia at day 0. At 20 h after infection, the mice were divided into the following four groups and treated orally daily for 5 days (Fig. 1A): the CAM group (n = 10; 150 μg/head, every 12 h), the OSV group (n = 10, 50 μg/head, every 12 h), the CAM-OSV group (n = 10, CAM, 150 μg/head/12 h; OSV, 50 μg/head/12 h), and the vehicle–0.5% methylcellulose 400 (MC) group (n = 10, 100 μl of MC/head). OSV and CAM were dissolved in sterilized MC solution. The body weight was measured on the morning of each day after viral infection (Fig. 1B) (mean ± standard deviation [SD] at day 0, 14.9 ± 0.6 g).
Fig 1.
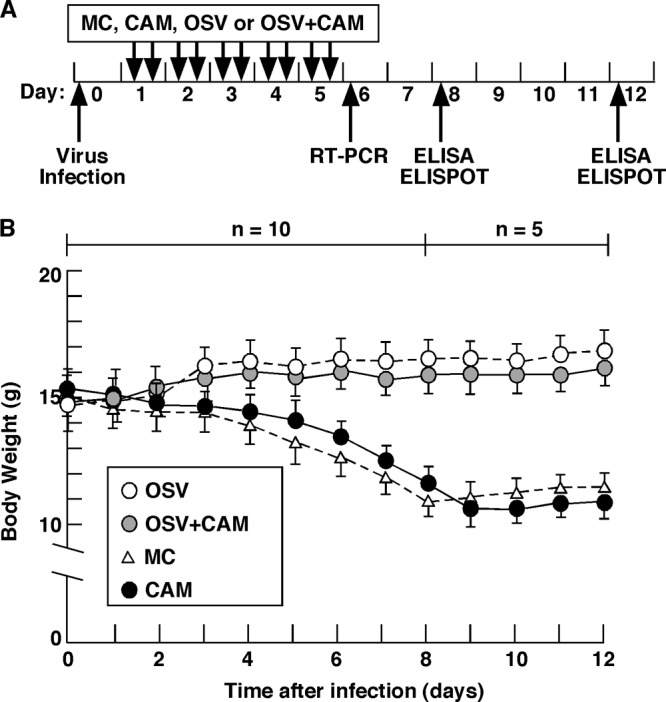
Experimental protocol for infection with IAV/PR8/34 virus. (A) At 20 h after infection with 25 PFU of IAV, mice were treated orally with 150 μg of CAM, 50 μg of OSV, OSV (50 μg) plus CAM (150 μg) in 100 μl of sterilized MC (n = 10), or 100 μl of MC as a control (n = 10). The dose was given twice daily for 5 days. (B) Changes in body weight in the four treatment groups. Data are means ± SEM (from day 0 to 7, n = 10; from day 8 to 12, n = 5).
For viral reinfection experiments, the mice were initially infected intranasally with 5 PFU of IAV/PR8/34(H1N1) in 15 μl of saline and then orally treated with OSV, CAM, OSV-CAM, and MC as described above for 5 days at 20 h after infection. Two weeks after the initial infection, the mice were infected again with 50× the 50% lethal dose (LD50; 500 PFU) of IAV/PR8/34(H1N1) in saline instilled into the nostril. After infection, loss of body weight and survival of animals were monitored. The effects of the initial treatment of infected mice with OSV, CAM, OSV-CAM, and MC were analyzed at day 14 after reinfection. The mice were euthanized, and isolated lungs were fixed with 4% paraformaldehyde for histopathological evaluation as described previously (35).
Cell isolation from mucosal lymphoid tissues.
At days 6, 8, and 12 after infection, mononuclear cells were isolated from mucosal lymphoid tissues (Fig. 1A). Mononuclear cells from the mediastinal lymph nodes (MeLNs) and spleen were isolated under aseptic conditions by the mechanical dissociation method as described previously (14, 17). Nasal passages (NPs) were isolated using a modification of the dissociation method described previously (14, 15, 17). Mononuclear cells were isolated from the lungs by a combination of an enzymatic dissociation procedure using collagenase type IV (0.5 mg/ml; Sigma-Aldrich, St. Louis, MO) followed by discontinuous Percoll (Amersham Biosciences, Piscataway, NJ) gradient centrifugation. To obtain B cells or DCs from the mucosal lymph nodes, mononuclear cells from the MeLNs, lungs, NPs, and spleen were incubated with anti-B220-conjugated or anti-CD11c-labeled microbeads (Miltenyi Biotec, Auburn, CA), based on the protocol supplied by the manufacturer (16) and were then positively selected with AutoMACS. The purified cell fraction consisted of >97% B220+ or CD11c+ cells, with >99% cell viability.
Analysis of mucosal immune responses to IAV by ELISA or ELISPOT assay.
At days 8 and 12 after infection, nasal washes (NWs), bronchoalveolar fluids (BALF), and plasma were collected as described previously (27, 45) and subjected to enzyme-linked immunosorbent assay (ELISA) to determine anti IAV-specific S-IgA and IgG levels (27, 33, 39). Mononuclear cells isolated from the MeLNs, lungs, NPs, and spleen were subjected to an enzyme-linked immunosorbent spot (ELISPOT) assay to determine the numbers of IgA and IgG antibody (Ab)-forming cells (AFCs), as described previously (15, 17, 39).
Measurement of BAFF expression in BMDCs treated with or without CAM and poly(I·C).
Bone-marrow-derived dendritic cells (BMDCs) from naïve mice were prepared as described previously (28). BMDCs were stimulated with CAM (15 μM) or poly(I·C) (Amersham Bioscience, Piscataway, NJ) (20 μg/ml) for 6 h at 37°C. A concentration of 15 μM CAM is the maximum serum concentration of CAM in clinical use (500 mg of oral administration) (13). The expression levels of BAFF were measured by quantitative reverse transcription (RT)-PCR (43).
RT-PCR and quantitative RT-PCR assays.
Total RNAs were extracted from mononuclear cells isolated from the MeLNs, lung, NPs, spleen, and BMDCs. The proliferation-inducing ligand (AID; 349 bp), Iμ-Cα (267 bp), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 355 bp) transcripts in mucosal B cells were amplified using specific primers as described previously (9, 20, 30, 34, 38). The following primers were used: AID, sense, 5′-GGCTGAGGTTAGGGTTCCATCTCAG-3′, and antisense, 5′-GAGGGAGTCAAGAAAGTCACGCTGGA-3′; Iμ-Cα, sense, 5′-CTCTGGCCTGCTTATTGTTG-3′, and antisense, 5′-GAGCTGGTGGGAGTGTCAGTG-3′; and GAPDH, sense 5′-CATCACCATCTTCCAGGA-3′, and antisense, 5′-GAGGGGCCATCCACAGTCTTC-3′. In order to examine the expression of BAFF and APRIL on mucosal DCs or their receptors on B cells, the cDNAs purified from DCs and B cells were amplified with a pair of specific primers for BAFF (285 bp), APRIL (597 bp), BAFF-R (521 bp), TACI (172 bp), and BCMA (167 bp) and by an RT-PCR method (16). The following primers were used: BAFF, sense, 5′-GAGAACAAAATAGTGGTGAGGCA-3′, and antisense, 5′-GTCGTCTCCGTTGCGTGAA-3′; APRIL, sense, 5′-ACCCTCCCTGCTACCTCTTGG-3′, and antisense, 5′-TCCTTCCCGAGATACCACCTG-3′; BAFF-R, sense, 5′-GACATGGGCGCCAGGAGACTCCGGGTCCGA-3′, and antisense, 5′-TGGGCCAGCTGTCTTGGTGGTCACCACCAGCTC-3′; TACI, sense, 5′-CCAGGATTGAGGCTAAGTAGCG-3′, and antisense, GGGGAGTTTGCTTGTGACC-3′; and BCMA, sense, 5′-CAAGCGTGACCAGTTCAGTGA-3′, and antisense, 5′-CGATCCGTCAAGCTGACCTG-3′.
Neutralizing activities for IAV.
To assess IAV neutralization by mucosal S-IgA, Madin-Darby canine kidney (MDCK) cells were incubated with mixtures of IAV and S-IgA purified from NWs and BALF, as described previously (1). The S-IgA from NWs and BALF was purified with the Kaptiv-AE IgA affinity column according to the protocol provided by the manufacturer (Tecnogen S.p.A., Piacenza, Italy). Anti-influenza virus nucleoprotein monoclonal antibody (MAb) (QED Bioscience, San Diego, CA) and horseradish peroxidase-labeled anti-mouse IgG were used to detect the infected cells. The viral protein was visualized using the TrueBlue peroxidase substrate (KPL, Inc., Gaithersburg, MD), and the numbers of cell foci/well were counted under a stereomicroscope.
Statistical analysis.
The results are expressed as means ± 1 standard error of the mean (SEM). Data from the treatment groups were compared with those from the control, using an unpaired Mann-Whitney U test with Statview software (StatView 5.0 software (SAS Institute, Cary, NC). A P value of <0.05 was considered significant.
RESULTS
CAM significantly enhances airway S-IgA immune responses in the early stage of IAV infection, but not systemic IgG immune responses.
After IAV infection, mice given OSV with or without CAM showed a slight but continuous increase in body weight during the 12-day experimental period (Fig. 1B). On the other hand, mice treated with CAM alone or MC as a control exhibited continuous loss of body weight from day 0 up to 26.7 to 27.4% loss at day 9 or 10 postinfection, followed by slow recovery (Fig. 1B). None of the mice died during the experimental period. These results suggest that OSV treatment for 5 days in the early stage of infection lessens the rate of complications and aggravation of flu symptoms in mice by suppression of viral replication.
To investigate the effect of the 5-day CAM treatment on antigen (Ag)-specific mucosal and systemic immune responses, we measured Ag-specific S-IgA levels in NWs and BALF as well as IgA and IgG levels in serum of mice at days 8 and 12 postinfection. CAM significantly increased anti-IAV-specific S-IgA levels in NWs and BALF of OSV-treated mice at days 8 and 12, compared with the levels in mice treated only with OSV (Fig. 2A). CAM also significantly increased S-IgA levels in NWs and BALF compared with the levels in MC-treated mice at day 12, the maximal phase of mucosal S-IgA induction (19, 37), although the effect was not significant at day 8, the intermediate phase (Fig. 2A). Furthermore, these findings were confirmed by Ag-specific ELISPOT assays. CAM significantly increased the numbers of Ag-specific IgA AFCs in respiratory effector tissues compared with MC and OSV treatment at day 8 and/or 12. On the other hand, there were no significant differences in the total numbers of S-IgA AFCs in these lymphoid tissues among the groups (data not shown). These results indicate that CAM upregulates Ag-specific S-IgA responses in mucosal effector tissues. However, CAM did not have a significant effect on the elevated levels of systemic Ag-specific IgA and IgG responses in serum of infected animals, compared with the control, such as MC or OSV treatment, although OSV treatment tended to suppress slightly the production of Ag-specific IgG and AFCs (Fig. 3A and B). Ag-specific Abs in airway secretions and serum at day 0 were below the detection levels. These experiments confirmed our previous findings of attenuated induction of antiviral S-IgA at days 8 and 12 in OSV-treated mice, compared with the vehicle-treated mice (39). Taken together, these findings indicate that CAM predominantly enhances mucosal immunity of both the attenuated antiviral S-IgA production in the OSV-treated mice and the S-IgA production in the MC-treated mice.
Fig 2.

Anti-IAV-specific S-IgA immune response in external secretions and mucosal lymphoid tissues. Mice infected with IAV at day 0 were treated on five (from day 1 to 5) consecutive days with CAM, OSV, OSV-CAM, or MC. (A) At days 8 and 12, IAV-specific S-IgA levels were determined in nasal washings (NWs) and bronchoalveolar lavage fluid (BALF) by ELISA. Data are means ± SEM (n = 5). *, P < 0.05, versus MC; # and ##, P < 0.05 and P < 0.01, respectively, versus OSV. (B) At days 8 and 12, mononuclear cells isolated from the nasal passages (NPs), mediastinal lymph nodes (MeLNs), and lung were subjected to IAV-specific ELISPOT assay to determine the numbers of IgA AFCs. Data are means ± SEM (n = 5). *, P < 0.05 versus MC; # and ##, P < 0.05 and P < 0.01, respectively, versus OSV.
Fig 3.

Systemic IAV-specific immune responses in mice after infection. Mice infected at day 0 were treated for five consecutive days (from days 1 to 5) with CAM, OSV, OSV-CAM, or MC. (A) IAV-specific IgA and IgG levels in plasma determined by ELISA. Data are means ± SEM (n = 5). There were no significant differences between CAM and MC and between OSV and OSV-CAM. (B) Mononuclear cells isolated from the spleen were subjected to the ELISPOT assay to determine the numbers of IgA and IgG AFCs. Data are means ± SEM (n = 5). There were no significant differences between CAM and MC and between OSV and OSV-CAM.
CAM induces IgA CSR in airway-associated lymphoid tissues.
We next examined CSR from the μ- to α-chain in B cells from MeLNs, lung, NPs, and spleen (i.e., effector lymphoid tissues) of IAV-infected mice treated with CAM. The mRNA levels of AID and IμCα transcripts in mucosal B cells were analyzed by semiquantitative RT-PCR. Importantly, CAM significantly increased the levels of CSR-associated molecule-specific mRNAs in B cells from MeLNs, lung, and NPs, compared with the levels from MC- and OSV-treated mice, respectively (Fig. 4A), but had no such effect on in B cells from the spleen (Fig. 4B).
Fig 4.

Preferential presence of AID, Iμ-Cα transcripts in B cells from the nasopharyngeal-tracheal effector sites in IAV-infected-mice treated with CAM, OSV, OSV-CAM, or MC. Total RNA extracted from the mediastinal lymph nodes (MeLNs), lung, nasal passages (NPs), and spleen of IAV-infected mice at day 6 was subjected to RT-PCR analysis. The left panels show representative results, and the graphs on the right show the expression level of the respective CSR-associated molecules relative to the density of GAPDH expression, calculated as 100 with ChemiDoc XRS Quantity One Analysis software (Bio-Rad). Data are means ± SEM from 5 mice for each group and represent a total of five separate experiments. *, P < 0.05 versus MC; # and ##, P < 0.05 and P < 0.01, respectively, versus OSV.
OSV significantly suppressed the expression levels of CSR-associated molecule-specific mRNAs in B cells from MeLNs, lung, and NPs, compared with the MC group (Fig. 4AA. In addition, OSV-CAM significantly increased the expression of AID- and IμCα-specific mRNAs in B cells from MeLNs, lung, and NPs compared with the OSV group (Fig. 4A). These results indicate that CAM predominantly enhances the recovery of reduced IgA CSR by OSV in mucosal effector tissues of IAV-infected mice.
CAM increases BAFF molecule expression on DCs from MeLNs, lung, and NPs, but not that of BAFF receptors on B cells.
It is known that IgA CSR occurs through both a T-cell-dependent engagement with CD40-CD40 ligand and a T-cell-independent pathway (6, 12, 24). We have also shown that the APRIL molecule on oral-nasopharyngeal DCs mediates the T-cell-independent IgA CSR for innate immunity by interaction with the TACI molecule on B cells of mucosal effector tissues (16). In this regard, we next examined whether CAM induces the expression of BAFF and APRIL molecules on DCs from MeLNs, lung, and NPs at the early phase of IAV infection. Although the expression levels of BAFF and APRIL on DCs from MeLNs, lung, and NPs of IAV-infected OSV-treated mice were similar to those of the MC-treated mice, a significantly higher expression level of BAFF, but not APRIL, was noted on DCs from MeLNs, lung, and NPs of IAV-infected OSV-CAM-treated mice and CAM-treated mice compared with OSV- and MC-treated mice, respectively (Fig. 5).
Fig 5.

Expression of B-cell-activating factor (BAFF) and proliferation-inducing ligand (APRIL) in CD11c+ DCs on mucosal effector sites. The day after the last dose of CAM, OSV, OSV-CAM, and MC (i.e., at day 6), mononuclear cells were isolated from the mediastinal lymph nodes (MeLNs), lung, and nasal passages (NPs) of IAV-infected mice, and mucosal DCs were purified with AutoMacs using anti-CD11c MAb-labeled microbeads, as described in Materials and Methods. Total RNA extracted from CD11C+ DCs was subjected to RT-PCR analysis. The left panels show representative results, and the graphs on the right show the results of quantitative analysis. Data are means ± SEM from 5 mice for each group and represent a total of five separate experiments. *, P < 0.05 versus MC; #, P < 0.05 versus OSV.
We next analyzed by quantitative RT-PCR whether the effect of CAM on upregulation of BAFF on DCs is direct or indirect (Table 1). Although treatment of BMDCs with 20 μg/ml of poly(I·C) as a positive control (43) significantly increased BAFF expression on the cells, treatment with 15 μM CAM, the maximum serum concentration in clinical use (13), did not increase the expression. These results suggest that CAM indirectly enhances BAFF expression on DCs in the airway-associated lymphoid tissues and that BAFF-expressing mucosal DCs induce IgA CSR in the airway effector tissues.
Table 1.
Quantitative RT-PCR analyses of BAFF expression in BMDCs
| Treatment | Relative mRNA expression rate ata: |
|
|---|---|---|
| 0 h | 6 h | |
| Vehicle (0.05% DMSO)b | 0.30 ± 0.04 | 0.40 ± 0.16 |
| CAM (15 μM) | 0.25 ± 0.02 | 0.24 ± 0.07 |
| Poly(I·C) (20 μg/ml) | 0.30 ± 0.10 | 1.47 ± 0.37* |
After the cells had been treated with each reagent for 0 and 6 h, total RNA was isolated, and the expression levels of BAFF mRNA normalized to GAPDH mRNA were detected. Data are means ± SEM from four independent experiments. *, P < 0.05 versus vehicle at 6 h.
DMSO, dimethyl sulfoxide.
We also examined B cells from the MeLNs, lung, and NPs for their expression of BAFF-R, TACI, and BCMA, representing receptors for BAFF molecules on mucosal B cells. The results showed no significant change in the expression levels irrespective of the treatment (Fig. 6).
Fig 6.

Expression of B-cell-activating factor (BAFF) and proliferation-inducing ligand (APRIL) receptor molecules (BAFF-R, transmembrane activator and calcium modulator cyclophilin ligand interactor [TACI], and B-cell maturation antigen [BCMA]) in mucosal B220+ B cells. The day after the last dose of CAM, OSV, OSV-CAM, or MC, mononuclear cells were isolated from the mediastinal lymph nodes (MeLNs), lung, and nasal passages (NPs) of IAV-infected mice. The mucosal B cells were purified with AutoMacs using anti-B220 MAb-conjugated microbeads. Total RNA extracted from B220+ B cells was subjected to RT-PCR analysis. The left panels show representative results, and the graphs on the right show the results of quantitative analysis. Data are means ± SEM of 5 mice for each group and represent a total of five separate experiments.
Reinforcement of anti-virus S-IgA by CAM exhibits neutralizing activities.
Since mucosal S-IgA acts as a neutralizing Ab to pathogens and exotoxin, we determined in vitro the effects of treatment on the neutralizing activities of S-IgA. Incubation of MDCK cells with mixtures of IAV and S-IgA purified from NWs of the MC- and CAM-treated groups resulted in significant reductions in the numbers of infected-cell foci, compared with that from uninfected mice (Fig. 7, top). In contrast, similar in vitro experiments using S-IgA from the OSV-treated group showed significantly lower neutralizing activities than the MC- and CAM-treated groups. Even under the suppressed neutralizing activities in the OSV-treated mice, it is noted that additional combination of CAM with OSV significantly enhanced the neutralization activities of S-IgA fractions purified from BALF (Fig. 7, bottom). The combined treatment with CAM and OSV also tended to increase the neutralization activities of S-IgA fractions from NWs, but this was not significant (Fig. 7, top). These results indicate that CAM predominantly boosts mucosal S-IgA neutralization activities in BALF of IAV-infected OSV-treated mice.
Fig 7.

Neutralization activities of S-IgA in nasal washings (NWs) and bronchoalveolar lavage fluid (BALF) against IAV/PR8/34(H1N1) infection in MDCK cells. At day 12 postinfection, S-IgA was purified from NWs and BALF of mice treated with CAM, OSV, OSV-CAM, or MC or from naïve mice (uninfected), and 2 μg of purified S-IgA from each group was then preincubated with IAV (1,450 PFU, 100 μl) for 1 h at 37°C. Each mixture was subsequently incubated with confluent monolayers of MDCK cells at 37°C for 1 h for infection. Sixteen hours after infection, the cells were fixed with 4% paraformaldehyde–phosphate-buffered saline (PBS), permeabilized with 0.3% Triton X-100–PBS, and then visualized by TrueBlue peroxidase substrate. NWs and BALF from uninfected mice were used as the control. Original magnification, ×40. The values in the graph are mean numbers of infected cells ± SEM (n = 4). *, P < 0.01 versus uninfected; # and ##, P < 0.05 and P < 0.01, respectively, versus MC or CAM; †, P < 0.05 versus OSV-CAM.
We next confirmed reinforcement of protective immunity by CAM in reinfection experiments in vivo (Fig. 8). After initial IAV infection of mice followed by treatment with OSV, CAM, OSV-CAM, and MC for 5 days, the mice were reinfected with a lethal dose (50× LD50; 500 PFU) of IAV at day 14 after the initial infection. All preinfected animals in the four treatment groups, such as the OSV group, the CAM group, the OSV-CAM group, and the MC group, survived with continuous increases in body weight (Fig. 8A). Although all mice (n = 10) in the four treatment groups showed resistance against a lethal dose of virus challenge, inflammatory cell migration in the lungs was most severe in the OSV-treated group at day 14 after reinfection (Fig. 8B). Lung inflammation was mild in the CAM-OSV-treated group, the CAM-treated group, and the MC-treated group compared with the lungs of noninfected mice.
Fig 8.
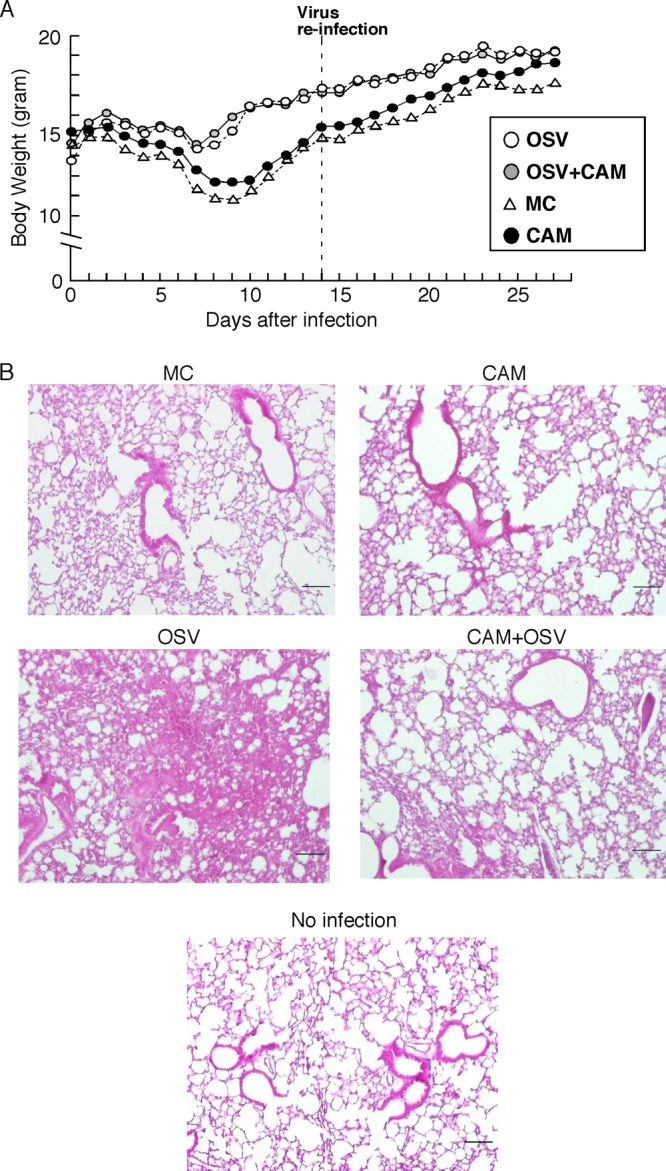
Reinforcement of protective immunity by CAM in reinfected mice. (A) At 20 h after infection with 5 PFU of IAV, mice were treated orally with CAM, OSV, OSV-CAM, or MC as described in the legend to Fig. 1. (A) Changes in body weight in the four treatment groups (n = 10). Data are means ± SEM. (B) Hematoxylin-and-eosin-staining sections in the lungs of four groups of mice at day 14 after reinfection and noninfection control mice. Each result is representative of 10 animals in each group. Bar, 100 μm.
DISCUSSION
Mucosal immunity works as the body's frontline of defense against invasion of pathogens. Against variant virus infections, mucosal S-IgA confers cross-protection, and thus may provide an overall higher protective activity than systemic IgG (23, 33). Furthermore, mucosal S-IgA is known as a noninflammatory Ab (21). Although heterosubtypic protection from influenza is not only mediated by cytotoxic T cells (31), mucosal S-IgA and respiratory B cells induced by nasal vaccination and natural infection (7, 32) are crucial in protecting against IAV infection. In addition, humoral and protective immunity associated with antihemagglutinin specific IgA elicited by primary infection or vaccination is important for the prevention of secondary infection with different virus subtypes (41). Therefore, we consider that induction of mucosal S-IgA is critical for immune protection against IAV infection.
In the present study, using the IAV-infected mice model, we demonstrated that CAM boosted the induction of sufficient mucosal S-IgA for protection against IAV infection in the respiratory tract (Fig. 2 and 7). As shown in the results in Fig. 2 and our recent animal (45) and human (3) studies, OSV treatment attenuated S-IgA production in the airway. In contrast, CAM enhanced respiratory S-IgA responses in OSV-treated mice and naïve mice. However, no significant differences were seen between the OSV- and OSV-CAM-treated mice with regard to the percentages of CD11c+ DCs on NPs and MeLNs at day 6 (data not shown). The findings of reinforcement of neutralizing activities of S-IgA by CAM (Fig. 7) were supported by the results of reinfection experiments. The lung inflammation of IAV-infected OSV-CAM-treated mice was definitely mild in reinfection compared with that of IAV-infected OSV-treated mice (Fig. 8). These results support boosting of protective immunity by CAM in IAV-infected mice.
Previous studies indicated that AID is essential for CSR, which specifically occurs in activated B cells (4, 12, 24), and that Iμ-Cα transcript is a hallmark for active IgA CSR in vivo (20). To study the mechanisms of CAM-enhanced S-IgA production, we investigated the expression of AID and Iμ-Cα molecules involved in the induction of mucosal IgA CSR by B cells of the MeLNs, lung, and NPs. The results showed higher expression of AID and Iμ-Cα molecules in B cells from the mucosal lymph nodes in the CAM- and OSV-CAM-treated groups, but surprisingly low expression in the OSV-treated group, compared with the MC vehicle-treated group. These findings indicate that CAM induces T-cell-independent IgA CSR at the respiratory effector sites. In this regard, previous studies described a T-cell-independent IgA CSR pathway, in addition to the classical T-cell-dependent CSR in the intestinal mucosa (6), and we have also recently reported the upregulation of T-cell-independent μ- to α-isotype CSR on B-1 B cells of oral-nasopharyngeal effector tissues following the use of naïve cholera toxin as a nasal adjuvant, with T-cell-independent Ag (16). Furthermore, the present study showed that OSV inhibited the induction of T-cell-independent IgA CSR in respiratory effector sites, whereas OSV-CAM restored the suppressed induction. These findings suggest that the combination of CAM and OSV enhances anti-IAV S-IgA production, whereas OSV suppresses such processes.
It has been reported that both intestinal DCs and epithelial cells induce CD40-independent IgA CSR through BAFF and APRIL molecules (12, 24). In addition, we have also reported that APRIL-expressing DCs in oral-nasopharyngeal mucosal effector tissues induced IgA CSR on mucosal B-1 B cells following the use of naïve cholera toxin with T-cell-independent Ag as a nasal adjuvant (16). We also studied the cellular and molecular mechanisms of T-cell-independent IgA CSR in the MeLNs, lung, and NPs in CAM-treated mice by focusing on DCs from these mucosal lymph nodes of mice treated with CAM and their potential roles in the induction of T-cell-independent μ- to α-isotype CSR. The results showed that CAM increased the expression of BAFF molecules on DCs of the mucosal lymphoid tissues, compared with the other treatment groups (Fig. 5). However, CAM did not increase the expression of APRIL on DCs of the mucosal lymph nodes. These results suggest that BAFF-expressing DCs play a key role in IgA CSR by B cells in respiratory effector tissues. In this regard, a recent study reported that intestinal DCs activated through Toll-like receptor 5 signaling produce retinoic acid and induce IgA production by peritoneal B cells (42). Thus, it is possible that CAM stimulates virus-specific S-IgA responses through the production of retinoic acid by activated DCs in the MeLNs, lung, and NPs.
Although it is important to identify the receptors for the BAFF molecule on B cells that are responsible for the induction of IgA CSR, stable expression of BAFF-R, TACI, and BMCA molecules was noted on B cells from the MeLNs, lung, and NPs in IAV-infected mice treated with CAM, OSV, OSV-CAM, or MC, and no significant differences in BAFF-R, TACI, and BCMA expression were evident among these groups. Taken together, the results indicate that CAM stimulates DCs in the mucosal lymphoid tissues and plays an important role in IgA CSR by upregulation of expression of BAFF molecule. However, data in the experiments to clarify whether the effect of CAM on upregulation of BAFF on DCs is a direct or indirect effect suggest that CAM indirectly enhances BAFF expression on mucosal DCs (Table 1). Further screening studies for the target molecule(s) of CAM are currently in progress.
In summary, using IAV-infected weanling mice, the present study demonstrated that orally administered CAM enhances mucosal IAV-specific S-IgA immune responses and has neutralization activities. Thus, based on the production of mucosal S-IgA Ab, we consider that the combination of CAM-OSV treatment for IAV could potentially prevent complications and aggravation of the flu symptoms at an early stage of infection.
ACKNOWLEDGMENTS
We thank Rebekah S. Gilbert for editorial assistance, scientific discussion, and critiques during the preparation of the manuscript.
The authors declare they have no potential conflicts of interest.
Financial support for this work was provided by Grant-in-Aid 24249059 and Special Coordination Funds for the Promotion of Science and Technology from the Ministry of Education, Culture, Sports, and Science and Technology of Japan.
Footnotes
Published ahead of print 15 August 2012
REFERENCES
- 1. Al-Garawi AA, et al. 2009. Acute, but not resolved, influenza A infection enhances susceptibility to house dust mite-induced allergic disease. J. Immunol. 182:3095–3104 [DOI] [PubMed] [Google Scholar]
- 2. Barone F, Patel P, Sanderson JD, Spencer J. 2009. Gut-associated lymphoid tissue contains the molecular machinery to support T-cell-dependent and T-cell-independent class switch recombination. Mucosal Immunol. 2:495–503 [DOI] [PubMed] [Google Scholar]
- 3. Brandtzaeg P, Farstad IN, Haraldsen G. 1999. Regional specialization in the mucosal immune system: primed cells do not always home along the same track. Immunol. Today 20:267–277 [DOI] [PubMed] [Google Scholar]
- 4. Castigli E, et al. 2004. Impaired IgA class switching in APRIL-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 101:3903–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Castigli E, et al. 2005. TACI and BAFF-R mediate isotype switching in B cells. J. Exp. Med. 201:35–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cerutti A. 2008. The regulation of IgA class switching. Nat. Rev. Immunol. 8:421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Choi YS, Baumgarth N. 2008. Dual role for B-1a cells in immunity to influenza virus infection. J. Exp. Med. 205:3053–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crouch EE, et al. 2007. Regulation of AID expression in the immune response. J. Exp. Med. 04:1145–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fagarasan S, Kinoshita K, Muramatsu M, Ikuta K, Honjo T. 2001. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature 413:639–643 [DOI] [PubMed] [Google Scholar]
- 10. Hamada H, et al. 2002. Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J. Immunol. 168:57–64 [DOI] [PubMed] [Google Scholar]
- 11. Hashisaki GT. 1995. Update on macrolide antibiotics. Am. J. Otolaryngol. 16:153–157 [DOI] [PubMed] [Google Scholar]
- 12. He B, et al. 2007. Intestinal bacteria trigger T cell-independent immunoglobulin A2 class switching by inducing epithelial cell secretion of the cytokine APRIL. Immunity 26:812–826 [DOI] [PubMed] [Google Scholar]
- 13. Honeybourne D, Kees F, Andrews JM, Baldwin D, Wise R. 1994. The levels of clarithromycin and its 14-hydroxy metabolite in the lung. Eur. Respir. J. 7:1275–1280 [DOI] [PubMed] [Google Scholar]
- 14. Kataoka K, et al. 2011. Nasal dendritic cell targeting flt3 ligand as a safe adjuvant elicits effective protection against fatal pneumococcal pneumoniae. Infect. Immun. 79:2819–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kataoka K, et al. 2007. Nasal cholera toxin elicits IL-5 and IL-5 receptor alpha-chain expressing B-1a B cells for innate mucosal antibody responses. J. Immunol. 178:6058–6065 [DOI] [PubMed] [Google Scholar]
- 16. Kataoka K, et al. 2011. Oral-nasopharyngeal dendritic cells mediate T cell-independent IgA class switching on B-1 B cells. PLoS One 6:e25396 doi:10.1371/journal.pone.0025396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kataoka K, et al. 2004. Nasal Flt3 ligand cDNA elicits CD11c+CD8+ dendritic cells for enhanced mucosal immunity. J. Immunol. 172:3612–3619 [DOI] [PubMed] [Google Scholar]
- 18. Kido H, Okumura Y, Yamada H, Le TQ, Yano M. 2007. Protease essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr. Pharm. Des. 13:405–414 [DOI] [PubMed] [Google Scholar]
- 19. Kido H, et al. 2011. Attenuation of respiratory immune responses by antiviral neuraminidase inhibitor treatment and boost of mucosal immunoglobulin A response by co-administration of immuno-modulator clarithromycin in pediatric influenza. Influenza Other Respir. Viruses 5:240–243 [Google Scholar]
- 20. Kinoshita K, Harigai M, Fagarasan S, Muramatsu M, Honjyo T. 2001. A hallmark of active class switch recombination: transcripts directed by I promoters on looped-out circular DNAs. Proc. Natl. Acad. Sci. U. S. A. 98:12620–12633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kiyono H, Kunisawa J, McGhee JR, Mestecky J. 2008. The mucosal immune system, p 983–1030 In Paul WE. (ed), Fundamental immunology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 22. Kunkel EJ, Butcher EC. 2003. Plasma-cell homing. Nat. Rev. Immunol. 3:822–829 [DOI] [PubMed] [Google Scholar]
- 23. Liew FY, Russel SM, Appleyard G, Brand CM, Beale J. 1984. Cross-protection in mice infected with influenza A virus by the respiratory route is correlated with local IgA antibody rather than serum antibody or cytotoxic T cell reactivity. Eur. J. Immunol. 14:350–356 [DOI] [PubMed] [Google Scholar]
- 24. Litinskiy MB, et al. 2002. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 3:822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maeda S, Yamada Y, Nakamura H, Maeda T. 1999. Efficacy of antibiotics against influenza-like illness in an influenza epidemic. Pediatr. Int. 41:274–276 [DOI] [PubMed] [Google Scholar]
- 26. Miyamoto D, et al. 2008. Clarithromicin inhibits progeny virus production from human influenza virus-infected host cells. Biol. Pharm. Bull. 31:217–222 [DOI] [PubMed] [Google Scholar]
- 27. Mizuno D, Ide-Kurihara M, Ichinomiya T, Kubo I, Kido H. 2006. Modified pulmonary surfactant is a potent adjuvant that stimulates the mucosal IgA production in response to the influenza virus antigen. J. Immunol. 176:1122–1130 [DOI] [PubMed] [Google Scholar]
- 28. Mizuno D, et al. 2011. Surfactant protein C is an essential constituent for mucosal adjuvanticity of Surfacten, acting as an antigen delivery vehicle and inducing both local and systemic immunity. Vaccine 29:5368–5378 [DOI] [PubMed] [Google Scholar]
- 29. Mora JR, et al. 2006. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314:1157–1160 [DOI] [PubMed] [Google Scholar]
- 30. Muramatsu M, et al. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNS-editing deaminase family in germinal center B cells. J. Biol. Chem. 274:18470–18476 [DOI] [PubMed] [Google Scholar]
- 31. Nguyen HH, et al. 1999. Heterosubtypic immunity to lethal influenza A virus infection is associated with virus-specific CD8+ cytotoxic T lymphocyte responses induced in mucosa-associated tissues. Virology 254:50–60 [DOI] [PubMed] [Google Scholar]
- 32. Nguyen HH, van Ginkel FW, Vu HL, McGhee JR, Mestecky J. 2001. Heterosubtypic immunity to influenza A virus infection requires B cells but not CD8+ cytotoxic T lymphocytes. J. Infect. Dis. 183:368–376 [DOI] [PubMed] [Google Scholar]
- 33. Nishino M, et al. 2009. Influenza vaccine with Surfacten, a modified pulmonary surfactant, induces systemic and mucosal immune responses without side effects in minipigs. Vaccine 27:5620–5627 [DOI] [PubMed] [Google Scholar]
- 34. Okumura Y, et al. 2006. Serase-1B, a new splice variant of polyserase-1/TMPRSS9, activates urokinase-type plasminogen activator and proteolytic activation is negatively regulated by glycosaminoglycans. Biochem. J. 400:551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pan H-Y, et al. 2011. Up-regulation of ectopic trypsins in the myocardium by influenza A virus infection triggers acute myocarditis. Cardiovasc. Res. 89:595–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sato K, et al. 1998. Therapeutic effect of erythromycin of influenza virus-induced lung injury in mice. Am. J. Respir. Crit. Care Med. 157:853–857 [DOI] [PubMed] [Google Scholar]
- 37. Sawabuchi T, et al. 2009. Boost of mucosal secretory immunoglobulin A response by clarithromycin in pediatric influenza. Respirology 14:1173–1179 [DOI] [PubMed] [Google Scholar]
- 38. Shikina T, et al. 2004. IgA class switch occurs in the organized nasopharynx- and gut-associated lymphoid tissue, but not in the diffuse lamina propria of airways and gut. J. Immunol. 172:6259–6264 [DOI] [PubMed] [Google Scholar]
- 39. Takahashi E, et al. 2010. Attenuation of inducible respiratory immune responses by oseltamivir treatment in mice infected with influenza A virus. Microbes Infect. 12:778–783 [DOI] [PubMed] [Google Scholar]
- 40. Tsuji M, et al. 2008. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29:261–271 [DOI] [PubMed] [Google Scholar]
- 41. Tumpey TM, Renshaw M, Clements JD, Katz JM. 2001. Mucosal delivery of inactivated influenza vaccine induces B-cell-dependent heterosubtypic cross-protection against lethal influenza A H5N1 virus infection. J. Virol. 75:5141–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Uematsu S, et al. 2008. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 9:769–776 [DOI] [PubMed] [Google Scholar]
- 43. Yamada T, et al. 2010. Poly(I:C) induces BLyS-expression of airway fibroblasts through phosphatidylinositol 3-kinase. Cytokine 50:163–169 [DOI] [PubMed] [Google Scholar]
- 44. Yamada T, Zhang K, Yamada A, Zhu D, Saxon A. 2005. B lymphocyte stimulator activates p38 mitogen-activated protein kinase in human Ig class switch recombination. Am. J. Respir. Cell Mol. Biol. 32:388–394 [DOI] [PubMed] [Google Scholar]
- 45. Yamaya M, et al. 2010. Clarithromycin inhibits type A seasonal influenza virus infection in human airway epithelial cells. J. Pharmacol. Exp. Ther. 333:81–90 [DOI] [PubMed] [Google Scholar]