

Abstract
Protein-protein interactions are required for many biological functions. Previous work has demonstrated an interaction between the human cytomegalovirus DNA polymerase subunit UL44 and the viral replication factor UL84. In this study, glutathione S-transferase pulldown assays indicated that residues 1 to 68 of UL84 are both necessary and sufficient for efficient interaction of UL84 with UL44 in vitro. We created a mutant virus in which sequences encoding these residues were deleted. This mutant displayed decreased virus replication compared to wild-type virus. Immunoprecipitation assays showed that the mutation decreased but did not abrogate association of UL84 with UL44 in infected cell lysate, suggesting that the association in the infected cell can involve other protein-protein interactions. Further immunoprecipitation assays indicated that IRS1, TRS1, and nucleolin are candidates for such interactions in infected cells. Quantitative real-time PCR analysis of viral DNA indicated that the absence of the UL84 amino terminus does not notably affect viral DNA synthesis. Western blotting experiments and pulse labeling of infected cells with [35S]methionine demonstrated a rather modest downregulation of levels of multiple proteins and particularly decreased levels of the minor capsid protein UL85. Electron microscopy demonstrated that viral capsids assemble but are mislocalized in nuclei of cells infected with the mutant virus, with fewer cytoplasmic capsids detected. In sum, deletion of the sequences encoding the amino terminus of UL84 affects interaction with UL44 and virus replication unexpectedly, not viral DNA synthesis. Mislocalization of viral capsids in infected cell nuclei likely contributes to the observed decrease in virus replication.
INTRODUCTION
Most biological processes require protein-protein interactions. We have been studying human cytomegalovirus (HCMV) UL44 with the hypothesis that association of viral and cellular proteins with this protein is required for viral DNA synthesis. UL44 is the presumptive processivity subunit of the HCMV DNA polymerase and shares notable structural homology with the eukaryotic DNA polymerase processivity factor PCNA (3, 4). Multiple proteins required for DNA synthesis and repair associate with PCNA as the need for the function of these proteins arises (31, 32). Based on the structure of UL44 (3) and the interaction of UL44 with the catalytic subunit of the HCMV DNA polymerase UL54 (4), we hypothesized that, like PCNA, UL44 might interact with multiple viral and cellular proteins during viral DNA synthesis (4). In several proteomic studies we catalogued a large number of viral and cellular proteins that associate with UL44 in infected cell lysate (47, 50). These included the cellular protein nucleolin (47) and the viral proteins IRS1, TRS1 (49), and UL84 (47, 50). UL44 has also been found to associate with UL84 in a proteomic screen for interaction partners of UL84 in infected cell lysate (15).
UL84 has been described as necessary for virus replication of HCMV laboratory strains AD169 and Towne (13, 16, 55) and for viral DNA synthesis of AD169 (16). Chromatin immunoprecipitation and proteomic studies have shown that UL84 is present at the viral origin of replication (7, 26). It has been reported that UL84 has UTPase activity, and it has been suggested that UL84 functions as a viral origin binding protein (7, 9). However, sequence analysis of UL84 and related proteins indicates that UL84 is more related to dUTPase, not UTPase (11). Additionally, UL84 can interact with the viral transcriptional transactivator IE2 (44), potentially to modulate IE2-mediated transcription (19). Interaction of UL84 with IE2, via amino acids 68 to 105 of UL84 is necessary to maintain UL84 protein levels in the infected cell (37, 38). However, a recent report has indicated that in at least one strain of HCMV (TB40/E), UL84 is not required for viral replication (45).
It is unknown what purpose the association of UL44 and UL84 serves in the infected cell and what regions of the two proteins are required for protein-protein interaction. UL84 does not bind to UL44 at the site used by UL54, the catalytic subunit of the viral DNA polymerase (50). Here, we sought to identify a region of UL84 that mediates UL44-UL84 interaction in vitro. We then went on to assay the effects of deletion of sequences encoding that region on virus replication, which led us to uncover an unusual capsid localization phenotype.
MATERIALS AND METHODS
GST pulldown assays.
Glutathione S-transferase (GST) pulldown assays were carried out essentially as previously described (50). UL84 expression vectors are described elsewhere (38), except for UL84(1–68) which was generated by inserting a UL84 fragment generated from pZIP13 (33) using PCR primers UL84(1–68)Fw and UL84(1–68)Rv (see supplemental Table 1 posted at https://coen.med.harvard.edu) into the expression vector pRSET-A (Invitrogen). Radiolabeled protein (45 μl) was used in each pulldown reaction mixture. Input (5 μl) and eluted proteins were visualized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and autoradiography.
Cells and viruses.
Human foreskin fibroblast (HFF) cells (Hs29, obtained from the American Type Culture Collection [ATCC], Manassas, VA) were maintained in complete Dulbecco's modified Eagles medium (DMEM) (Gibco) containing 5% fetal bovine serum (FBS) (Gibco) and used in all experiments. Virus AD169rv is derived from AD169-BAC (where BAC is bacterial artificial chromosome) (21).Viruses expressing FLAG-tagged IRS1 or TRS1, also derived from AD169-BAC, have been described elsewhere (49).
BACs and reconstitution of virus.
A BAC in which a cassette encoding green fluorescent protein (GFP) under the control of the HCMV major immediate early promoter (MIEP) was derived from a bacmid encoding virus pp28-Luc (where Luc is luciferase) (27) (a kind gift from Gloria Komazin-Meredith, Harvard Medical School). pp28-Luc is a derivative of bacmid pAD/Cre (56) (kindly provided by Dong Yu and Tom Shenk, Princeton University). Briefly, using primers EGFP-KanF and EGFP-KanR (see supplemental Table 1 posted at https://coen.med.harvard.edu), a PCR product was generated from pEP-EGFP-in (a kind gift from Nikolaus Osterrieder, Freie Universität Berlin) to produce a cassette containing a kanamycin (Kan) resistance gene and I-Sce-1 site within the sequence encoding enhanced GFP (EGFP). This cassette was inserted into the sequence encoding GFP in plasmid pEGFPN1 at restriction enzyme recognition sites BglII and NotI to produce plasmid pEGFP-Kan. pEGFP-Kan was used as a template for PCR primers EGFPF and EGFPR (see supplemental Table 1 posted at the URL mentioned above) to generate a PCR product which was then used in two-step Red recombination (52) with bacmid pp28-Luc in Escherichia coli strain GS1783 (a kind gift from Gregory Smith, Northwestern University Medical School). Thus, the sequence encoding pp28-Luc was replaced with sequence encoding the HCMV MIEP and GFP. The resulting bacmid was termed pBADGFP. Mutation of the UL84 coding sequence within BADGFP virus was performed using two-step Red recombination. Repair of UL84 sequences was also performed using two-step Red recombination. Here, a kanamycin resistance gene and I-Sce-1 site were inserted into the UL84 coding sequence in plasmid pZIP13 (33) using primers RepairUL84Fw and RepairUL84Rv. A PCR product generated from the modified pZIP13 plasmid using primers Rev-UL84stop fw and Rev-UL84stop rv was used to repair sequences in the appropriate bacmids. Primer sequences are shown in supplemental Table 1 posted at the URL mentioned above. To generate viruses, BACs were electroporated into HFF cells with plasmids pCGN71 (5) and pBRep-Cre (21), as described previously (52).
Virus replication assays.
To assess HCMV replication, 1 × 105 HFF cells per well were seeded in 12-well plates 24 h before infection. At the time of infection the indicated virus (0.2 ml) was added to each well at the multiplicity of infection (MOI) indicated in the text. After incubation for 1 h at 37°C, virus supernatant was removed and replaced with 1 ml of complete Dulbecco's modified Eagle's medium (DMEM) (Gibco) containing 5% fetal bovine serum (FBS) (Gibco). At the time points indicated in the legends, the medium from a well (virus supernatant) was taken from the cells and stored at −80°C until required. Dilutions of each virus supernatant were titrated simultaneously on monolayers of HFF cells to determine virus titer.
Fluorescence microscopy.
Cells were electroporated as described previously (52) with bacmid and plasmids pCGN71 (5) and pBRep-Cre (21). Images were acquired using a Nikon TS100 microscope and Q Capture Pro, version 6.0, software (Q Imaging).
IP of proteins from infected cell lysate.
Preparation of cell lysate, treatment with Benzonase, and immunoprecipitation (IP) using monoclonal antibodies (MAbs) recognizing UL44 (MAb ICP36; Virusys) or UL84 (8) or FLAG-tagged proteins were performed as described elsewhere (47, 50).
Western blotting.
Western blotting of proteins separated on 10% or 4 to 20% polyacrylamide gels was carried out as described elsewhere (51), using antibodies recognizing UL44, UL57, pp28, UL84, IE1-72 and IE2-86 (all from Virusys), β-actin (Sigma), FLAG (Sigma), nucleolin (Abcam), eIF2α (Santa Cruz), eIF2α phosphorylated at Ser51 ([eIF2α-P] Cell Signaling Technology), UL112-113(p84) (2) (kindly provided by Gary Hayward, Johns Hopkins University School of Medicine), and IRS1 (35) and TRS1 (6) (both kindly provided by Tom Shenk, Princeton University), plus UL85 and UL86 (both a kind gift from Wade Gibson, Johns Hopkins University School of Medicine) (30, 53) (all at a 1:1,000 dilution). All primary antibodies were raised in mice, except for antibodies recognizing UL112-113(p84), eIF2α-P, UL85, and UL86, which were raised in rabbits. Primary antibodies were detected using anti-mouse or anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (Southern Biotech), except where HRP-conjugated Tru Blot antibody (eBioscience) was used to detect protein from immunoprecipitation. Chemiluminescence solution (Pierce) was used in each case to detect secondary antibodies. Where indicated in the figure legends, Quantity One software (Bio-Rad) was used to analyze band intensity.
Real-time quantitative PCR analysis of viral DNA synthesis.
Briefly, DNA was isolated from infected cells using a NucleoSpin Tissue Kit (Macherey-Nagel) according to the manufacturer's instructions. Viral genomes were quantified with a primer pair (pp549s and pp812as) to UL83 (18), and the number of viral genomes was normalized to cellular copies of adipsin (for primer sequences, see supplemental Table 1 posted at https://coen.med.harvard.edu). Unknown sample values were determined on the basis of standard curves of known copy numbers of UL83 (AD169-BAC) and adipsin gene (from uninfected cell DNA). PCRs for UL83 and adipsin gene were carried out on a StepOnePlus machine using SYBR green PCR Master Mix (Applied Biosystems) as per the manufacturer's instructions. Linear regression analysis of UL83 and adipsin gene standards in triplicate yielded R2 values of 0.996 ± 0.001 and 0.991 ± 0.002 (means ± standard deviations [SD]) and efficiencies of 87% ± 0.1% and 100% ± 0.1% [calculated as 10(−1/slope) − 1], respectively.
Analysis of protein synthesis.
Uninfected HFF cells or HFF cells infected at a multiplicity of infection (MOI) of 1 were incubated with [35S]methionine (PerkinElmer) as described previously (47). A total of 1 × 105 cells were harvested in 100 μl of 2× Laemmli buffer and boiled at 95°C for 5 min. Ten microliters of each sample was analyzed using 4 to 20% SDS-PAGE. Gels were then dried and exposed to a phosphor screen for 24 h. The signal from phosphor screens was detected using Quantity One software (Bio-Rad) on a Bio-Rad phosphor imager.
Electron microscopy.
Cells infected at an MOI of 1 were incubated for 1 h at room temperature in fixative (2.5% glutaraldehyde, 1.25% paraformaldehyde, and 0.03% picric acid in 0.1 M sodium cacodylate buffer [pH 7.4]) at 72 h postinfection. Cells were provided to the Harvard Medical School Electron Microscope Facility; they were washed in 0.1 M sodium cacodylate buffer (pH 7.4), then postfixed for 30 min in 1% osmium tetroxide (OsO4)–1.5% potassium ferrocyanide (KFeCN), washed in water three times, and incubated in 1% aqueous uranyl acetate for 30 min. This step was followed by two washes in water and subsequent dehydration in ethanol (5 min each; at 50%, 70%, 95%, and twice at 100%). Cells were removed from the dish in propylene oxide, pelleted at 3,000 rpm for 3 min, and infiltrated for 2 h in a 1:1 mixture of propylene oxide and TAAB Epon (Marivac Canada, Inc., St. Laurent, Canada). The samples were subsequently embedded in TAAB Epon and polymerized at 60°C for 48 h. Ultrathin sections (about 60 nm) were cut on a Reichert Ultracut-S microtome and picked up onto copper grids stained with lead citrate. All samples were examined, and images were recorded using a JEOL 1200EX transmission electron microscope and an AMT 2k charge-coupled-device (CCD) camera, respectively.
RESULTS
Identification of a region in UL84 required for association of UL44 in vitro.
To identify a region of UL84 required for the association of UL84 and UL44 in vitro, we used glutathione S-transferase (GST) pulldown assays. In these experiments we assayed the binding of radiolabeled UL84 and UL84 mutants (Fig. 1A) expressed by in vitro transcription/translation to either GST or a GST-UL44 fusion protein purified from bacteria (Fig. 1B and C) that lacks the carboxyl-terminal segment of UL44 (GST-UL44ΔC290) (3). Confirming our previous results (50), radiolabeled UL84 bound to GST-UL44ΔC290 (Fig. 1C, lane 2) but not detectably to GST (Fig. 1C, lane 1). Deletion of residues 1 to 68 of UL84 (UL84Δ68) or larger deletions of the N terminus were sufficient to prevent efficient binding of UL84 to GST-UL44ΔC290 (Fig. 1C, lanes 3 to 5). Deletion of UL84 carboxyl-terminal residues 513 to 586 (UL84Δ513) had no obvious effect on the binding of UL84 to GST-UL44ΔC290 (Fig. 1C, lane 6). Thus, the amino terminus of UL84 (residues 1 to 68), but not the carboxyl terminus of UL84, was necessary for the efficient association of UL84 and UL44 in vitro.
Fig 1.
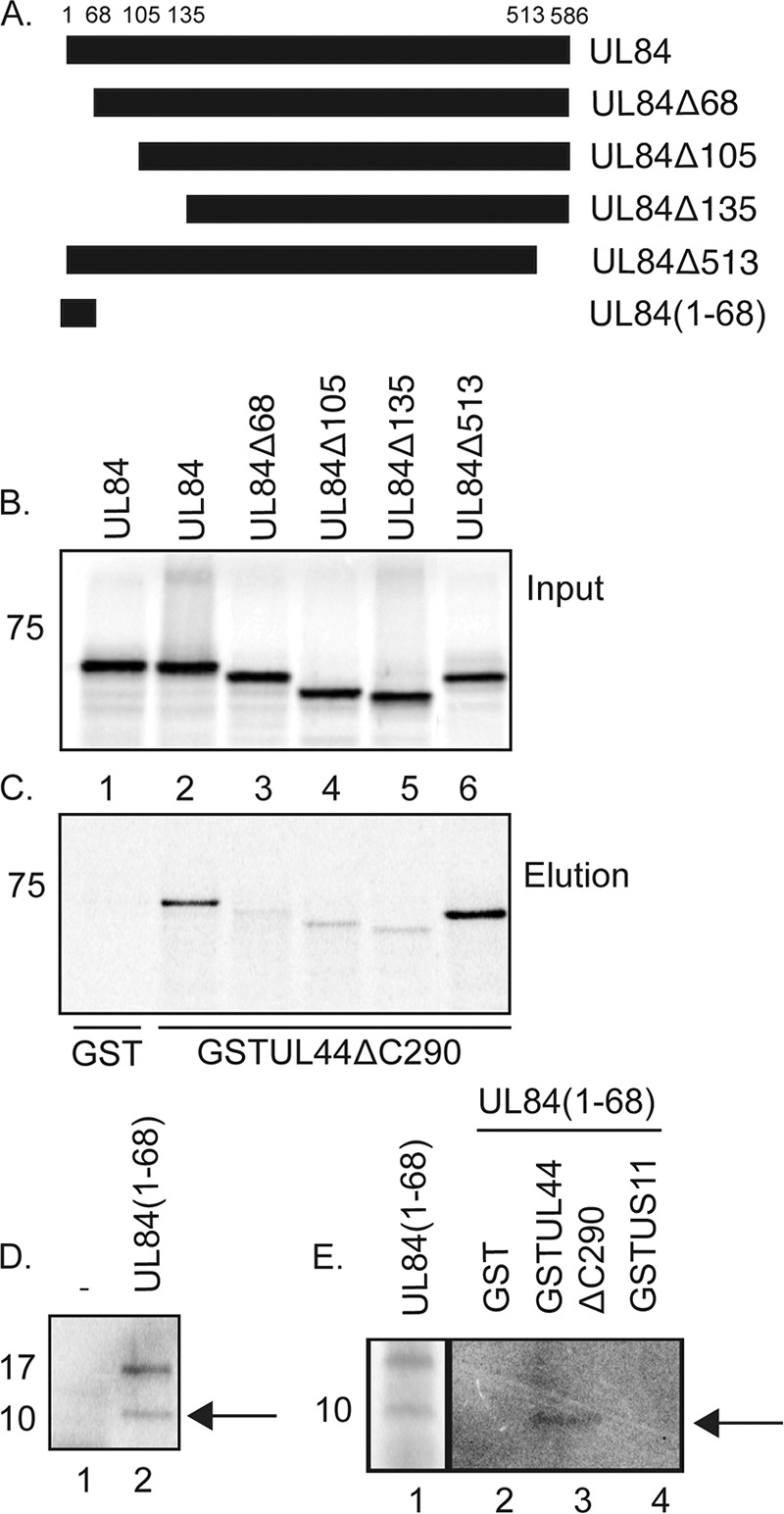
Binding of UL84 and UL84 mutants to UL44 in vitro. (A) Schematic of UL84 and UL84 mutants used in GST pulldown assays. The residues of UL84 at which truncations have been made are noted above the figure. The name of each mutant is noted to the right of the figure. (B and C) GST pulldown assays were performed where GST or GST-UL44ΔC290 fusion proteins were incubated with radiolabeled UL84 or UL84 mutants shown in panel A produced by in vitro transcription/translation and passed over a glutathione column. The radiolabeled and GST proteins used in each reaction are noted above and below the figure, respectively. The input (B) and protein eluted by glutathione (C) from each reaction are shown. (D) Protein products of in vitro transcription/translation in reaction mixtures containing no protein expression vector (lane 1) or an expression vector encoding UL84(1–68) (lane 2) (from panel A). (E) GST pulldown assays were performed where GST, GST-UL44ΔC290, or GST-US11 fusion protein was incubated with radiolabeled UL84(1–68) and passed over a glutathione column. The GST proteins used in each reaction mixture are noted above the figure. Lane 1, input protein; lanes 2 to 4, protein eluted by glutathione from each reaction. The position of the approximately 10-kDa form of UL84(1–68) is indicated with an arrow in panels D and E. The position of molecular mass markers in panels B to E are indicated to the left of the figure.
To assay whether UL84 residues 1 to 68 are sufficient to bind GST-UL44ΔC290 (Fig. 1D and E), we created a vector to express UL84 residues 1 to 68 (Fig. 1A) and expressed this protein [UL84(1-68)] using in vitro transcription/translation in the presence of a radiolabel (Fig. 1D). In a control in vitro transcription/translation reaction using a mixture that contained no protein expression vector, no bands could be detected when the protein from the reaction was resolved by SDS-PAGE (Fig. 1D, lane 1). In contrast, in a reaction mixture containing the expression vector for UL84(1-68), we observed two distinct bands of approximately 10 and 17 kDa (Fig. 1D, lane 2). UL84(1-68) is predicted to have a molecular mass of approximately 7 kDa. Therefore, these bands are most likely specifically produced from the UL84(1-68) expression vector and may represent posttranslationally modified (approximately 17 kDa) and less modified (approximately 10-kDa) forms of UL84(1-68).
We tested the ability of radiolabeled UL84(1-68) produced by in vitro transcription/translation to bind to purified GST, GST-UL44ΔC290 and, as an additional control, GST-US11 (containing the herpes simplex virus US11 protein) fusion proteins in GST pulldown assays (Fig. 1E). We found binding of the approximately 10-kDa UL84(1-84) protein to GST-UL44ΔC290 (Fig. 1E, lane 3) but not GST or GST-US11 (Fig. 1E, lanes 2 and 4, respectively) in these assays. These data indicate that the UL84(1-68) protein binds to UL44ΔC290 in a specific manner. Therefore, the amino terminus of UL84 (residues 1 to 68) is necessary and sufficient to bind UL44 (residues 1 to 290) efficiently in vitro.
Generation and analysis of mutant viruses.
We sought to determine if the interaction of the UL84 N-terminal segment with UL44 is important for virus replication. We hypothesized that mutation of the sequence encoding UL84 in the viral genome would prevent virus replication. Therefore, similar to the strategy utilized by Xu et al. (54) to analyze replication of a virus with an insertion in the UL84 open reading frame, we created a virus expressing a fluorophore so that the presence of virus in the cells and its spread from cell to cell could be monitored. We inserted a cassette in which green fluorescent protein (GFP) could be expressed under the control of the HCMV major immediate-early promoter (MIEP) into a bacmid containing the HCMV genome (pAD/Cre) (56) to create pAD/CreGFP (Fig. 2A). We analyzed replication of the virus derived from pAD/CreGFP (BADGFP virus) compared to that of virus from pAD/Cre (BAD) and found that insertion of the GFP expression cassette had no obvious effect on virus replication (Fig. 2B). Red two-step recombination (52) was then used to insert either three sequential stop codons into the UL84 open reading frame to create pAD/CreGFPUL84n or to delete the sequence encoding amino acids 2 to 68 of UL84 to create pAD/CreGFPUL84Δ68 (Fig. 2A). Wild-type and mutant bacmids were electroporated into human foreskin fibroblast (HFF) cells. Cells electroporated with wild-type bacmid pAD/CreGFP exhibited green fluorescence that spread through the entire culture (Fig. 2C, frame i). However, disruption of the UL84 open reading frame with stop codons appeared to prevent virus replication and spread as only single cells exhibited green fluorescence and as no viral plaques could be observed over time (over 21 days) in cells electroporated with BADGFPUL84n (Fig. 2C, frame ii). This finding is consistent with previous work using strains AD169 and Towne (13, 54, 55) but in contrast to a recent work by Spector and Yetming using strain TB40/E (45). Replication-competent virus was found in cells electroporated with bacmid pAD/CreGFPUL84Δ68. Virus generated from these cells (BADGFPUL84Δ68) replicated less well than virus generated from electroporation of cells with the bacmid BADGFP (BADGFP virus) (cells infected at an MOI of 1) (Fig. 2D). From 3 days postinfection (dpi) there was approximately 10-fold less virus produced by BADGFPUL84Δ68 than by BADGFP at all time points. The mutations made in bacmids BADGFPUL84n and BADGFPUL84Δ68 were repaired to generate viruses BADGFPUL84nrev and BADGFPUL84Δ68rev. These viruses replicated with efficiencies similar to BADGFP efficiency (Fig. 2E). Therefore, the mutations made in the UL84 coding sequences outlined above are responsible for the replication phenotypes. In sum, UL84 appears to be necessary for virus replication, at least in this strain, and deletion of the region encoding the UL84 amino terminus from the viral genome, identified as necessary and sufficient for association of UL84 and UL44 in vitro (Fig. 1), causes a defect in virus replication.
Fig 2.

Generation and characterization of virus expressing mutant UL84 proteins. (A) A schematic of the HCMV genome encoded in bacmid BADGFP is shown in the center of the figure. The unique long (UL) and unique short (US) segments of the genome are indicated. The internal and terminal repeats of UL and US are shown as black and white boxes, respectively. Dotted lines lead to magnified regions of the HCMV genome. Indicated below the HCMV genome is the intergenic region between open reading frames encoding US9 and US10 in which a cassette containing sequence encoding GFP (in gray) under the control of the major immediate-early promoter (MIEP) has been inserted. Indicated above the HCMV schematic of the genome is the region that contains the UL84 open reading frame. The insertion of stop codons into the UL84 open reading frame to create BADGFPUL84n is indicated with a vertical line. Also indicated is the truncation that creates BADGFPUL84Δ68. (B, D, and E) Viral replication. HFF cells were infected at an MOI of 1 with the indicated viruses. Virus supernatant was harvested at the indicated day postinfection (dpi). Virus titer is represented as PFU/ml (PFU/ml) on HFF cells. In each panel each data point represents the mean value from two independent experiments. In panel D, the error bars represent the standard error of the mean of the values used to calculate the data points. The titers of the virus stocks used in the assay were measured at the time the assays were performed to ensure that the correct inocula were used in each case. (C) Green fluorescence in cells electroporated with the indicated bacmid. Images are presented in grayscale.
Association of UL44 and UL84 in infected cell lysate.
We next assessed the association of UL44 and UL84Δ68 in infected cells. We performed IP of proteins from cells infected with BADGFP or BADGFPUL84Δ68 or uninfected cells using a monoclonal antibody (MAb) recognizing UL44 or a control antibody of the same isotype as UL44 MAb in the presence of the nuclease Benzonase. Benzonase was used to prevent nonspecific association due to binding of proteins to adjacent regions of nucleic acid. Agarose gel electrophoresis confirmed that Benzonase efficiently degraded nucleic acid (see supplemental Fig. 1 posted at https://coen.med.harvard.edu/). Immunoprecipitated proteins were examined by Western blotting using MAbs recognizing UL44 or UL84 (Fig. 3A). UL44 or UL84 protein was not found in protein immunoprecipitated using an isotype control antibody (lanes 1, 3, and 5) or in protein immunoprecipitated from uninfected cell lysate (lanes 1 and 2). Similar levels of UL44 were immunoprecipitated from BADGFP- or BADGFPUL84Δ68-infected cell lysate (lanes 4 and 6). Although similar levels of UL84 and UL84Δ68 were found in BADGFP- and BADGFPUL84Δ68-infected cell lysates (lanes 8 and 9, respectively), less UL84Δ68 coimmunoprecipitated with UL44 than UL84 (lanes 6 and 4, respectively). Similar results to those shown in Fig. 3A were observed when protein was immunoprecipitated from BADGFP- and BADGFPUL84Δ68-infected cell lysates using a MAb recognizing UL84 in the presence of Benzonase (data not shown). Thus, UL84Δ68 associates with UL44 in BADGFPUL84Δ68-infected cell lysate, albeit at a reduced level. Therefore, removal of the amino terminus of UL84 inhibits, but does not completely abrogate, association of UL84 with UL44 in infected cell lysate.
Fig 3.

IP of UL44 and UL84 from infected cell lysate. (A) Lysates from uninfected HFF cells or HFF cells infected with the indicated viruses (MOI of 3) were prepared and precleared with immunoglobulin. IP was then carried out with a monoclonal antibody (MAb) recognizing UL44 or a control antibody of the same isotype as the MAb used (Ig). Immunoprecipitated proteins were analyzed by Western blotting using MAb recognizing UL84 or UL44, as indicated to the right of the figure. Lanes 1 and 2, uninfected cells immunoprecipitated with Ig and MAb, respectively; lanes 3 and 4, BADGFP-infected cells immunoprecipitated with Ig and MAb, respectively; lanes 5 and 6, BADGFPUL84Δ68-infected cells immunoprecipitated with Ig and MAb, respectively; lanes 7 to 9, uninfected and infected cell lysate. Where indicated, high and low exposures of blot to film are shown. (B) Lysate from cells infected with BAD (lane 4), HCMV-IRSF (IRSF) (lane 5), and HCMV-TRSF (TRSF) (lane 6) and proteins immunoprecipitated using an anti-FLAG antibody from those lysates (lanes 1, 2, and 3, respectively) were separated on a 10% polyacrylamide gel. Proteins in each lane were examined by Western blotting for the presence of FLAG-tagged IRS1, FLAG-tagged TRS1, UL44, UL84, UL86, UL57, and nucleolin (Ncl) using antibodies recognizing these proteins, as indicated to the right of the figure. The positions of molecular weight mass (kDa) are indicated to the left of each figure.
Association of viral and cellular proteins with UL44 and UL84 in infected cell lysate.
Because residues 1 to 68 of UL84 are required for efficient interaction with UL44 in vitro (Fig. 1) but less so in infected cells (Fig. 3A), we hypothesized that some interactions between UL44 and UL84 in the infected cell can be bridged by another viral or cellular protein. We have previously demonstrated that UL84, the viral proteins IRS1 and TRS1, and the cellular protein nucleolin can each be specifically immunoprecipitated from infected cell lysate with UL44 in the presence of Benzonase (47, 49, 50). Furthermore, UL84, IRS1, TRS1, and nucleolin can each associate with UL44 in in vitro GST pulldown assays (47–50). It is possible that either IRS1, TRS1, or nucleolin or a combination of these proteins may bridge the interaction between UL44 and UL84 in infected cell lysate.
We, therefore, investigated whether these proteins could be found together in protein complexes immunoprecipitated from infected cell lysate. Cells were infected with virus expressing either FLAG-tagged IRS1 or FLAG-tagged TRS1 (49), and protein was immunoprecipitated using a MAb recognizing FLAG in the presence of Benzonase to degrade nucleic acids. Immunoprecipitated protein was analyzed by Western blotting (Fig. 3B). As noted previously, FLAG-tagged IRS1 and FLAG-tagged TRS1 plus a number of smaller FLAG-tagged proteins could be found in immunoprecipitated protein (Fig. 3B, lanes 2 and 3) (49). UL44, UL84, and nucleolin were found in protein immunoprecipitating with either FLAG-tagged IRS1 or FLAG-tagged TRS1 (lanes 2 and 3). In contrast, the viral single-stranded DNA binding protein UL57 (Fig. 3B), viral capsid protein UL86 (Fig. 3B), viral capsid protein UL85 (data not shown), and the cellular protein β-actin (data not shown) were not detected. Thus, it is likely that the association of UL44, UL84, and nucleolin with FLAG-tagged IRS1 or TRS1 is specific and not dependent upon the presence of nucleic acid during immunoprecipitation. IRS1 and TRS1 do not simultaneously associate with UL44 in infected cell lysate (49). Therefore, IRS1, TRS1, or nucleolin or a combination of these may bridge the association of UL44 and UL84 in infected cell lysate, which, in part, may explain the association of UL84Δ68 with UL44 shown in Fig. 3A.
We also considered the possibility that IE2-86, known to interact directly with UL84 (44), might bridge the association of UL44 and UL84. However, in none of our previous proteomic studies examining the association of viral and cellular proteins with UL44 (47, 50) could IE2-86 be found with UL44 although UL84 was readily detected. To investigate this possibility further, protein was immunoprecipitated with an antibody recognizing FLAG from lysate of cells infected with AD169rv, as a negative control, or a virus expressing FLAG-tagged UL44 (47). Immunoprecipitated protein was examined by Western blotting using antibodies recognizing FLAG, UL84, and an antibody recognizing both IE1-72 and IE2-86. Very low levels of IE1-72 and IE2-86 could be detected by Western blotting from both lysates, indicating that there was no specific association of either protein with FLAG-tagged UL44-UL84 complexes (see supplemental Fig. 2 posted at https://coen.med.harvard.edu). Therefore, it is unlikely that IE2-86 has a role in bridging the association of UL44 and UL84 in infected cell lysate.
The N terminus of UL84 is not required for viral DNA synthesis.
Because it has been reported that UL84 is involved in viral DNA synthesis (33, 34, 54), we speculated that the defect in virus replication displayed by BADGFPUL84Δ68 compared to BADGFP (Fig. 2C) is due to a defect in viral DNA synthesis. Viral DNA synthesis in BADGFP-, BADGFPUL84Δ68-, and BADGFPUL84Δ68rev-infected cells was assayed using quantitative real-time PCR (Fig. 4). The number of viral genomes present in each sample was determined by normalizing the copy number of a viral gene (UL83) to the copy number of a cellular gene (adipsin). We observed little difference between the rates of viral DNA accumulation in BADGFP-, BADGFPUL84Δ68-, and BADGFPUL84Δ68rev-infected cells, other than a 2-fold decrease in viral DNA found in BADGFPUL84Δ68-infected cells compared to levels in cells infected with the other two viruses at 96 h postinfection (hpi) (infection at an MOI of 1) (Fig. 4). We observed similar levels of viral DNA accumulation in BADGFP- and BADGFPUL84Δ68-infected cells in two other experiments (data not shown). Therefore, the 10-fold defect in virus production exhibited by BADGFPUL84Δ68 is unlikely to be due to a defect in viral DNA synthesis of this virus.
Fig 4.
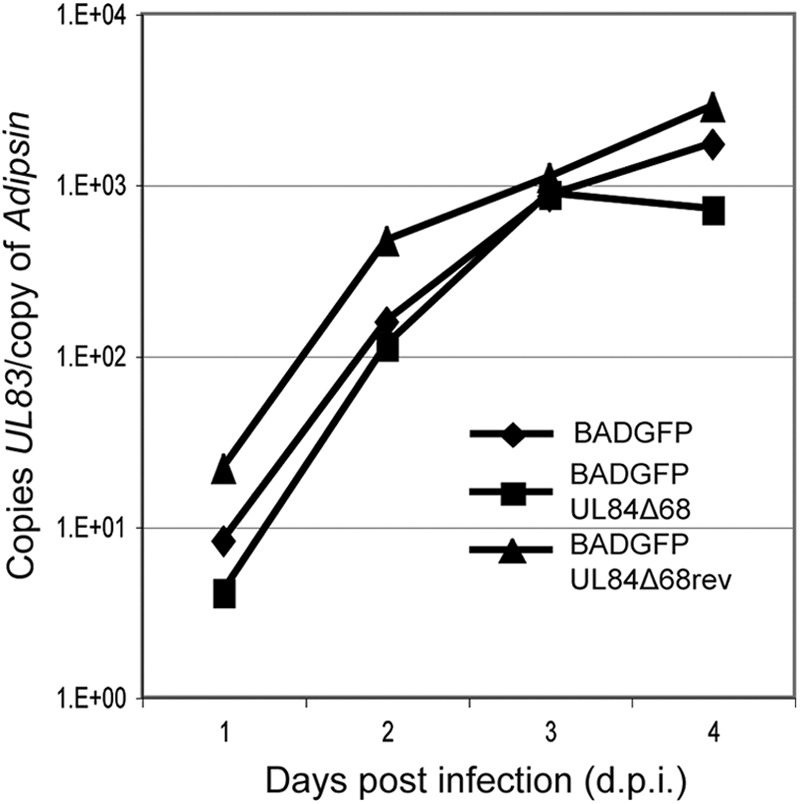
Levels of viral DNA synthesis in infected cells. Viral DNA synthesis in each experiment was determined by quantitative real-time PCR at each of the time points indicated. The amount of viral DNA assayed is represented as copies of the viral gene UL83 per copy of the cellular adipsin gene.
The deletion affects levels of viral and cellular proteins in infected cells.
We next assayed the levels of certain viral and cellular proteins in BADGFP- and BADGFPUL84Δ68-infected cells compared to levels in uninfected cells by Western blotting (Fig. 5A). Similar levels of IE1-72, IE2-86, UL112-113(p84), and UL57 were found in BADGFP- and BADGFPUL84Δ68-infected cells (Fig. 5A). As also shown in Fig. 3A, UL84Δ68 accumulation in BADGFPUL84Δ68-infected cells was similar to that of UL84 in BADGFP-infected cells (Fig. 5A). Interestingly, although UL44 accumulated to substantial levels at 72 hpi in cells infected with BADGFP and BADGFPUL84Δ68, accumulation of UL44 protein was compromised at early times in BADGFPUL84Δ68-infected cells compared to levels in BADGFP-infected cells (Fig. 5A, compare lanes 2 and 5). Analysis of band intensity in Fig. 5A and Western blotting of dilutions of BADGFP-infected cell lysate (Fig. 5B) indicate that there was an approximately 2-fold difference in UL44 protein levels in BADGFP- and BADGFPUL84Δ68-infected cells at 24 hpi. We also assayed levels of UL44 in BADGFPUL84Δ68rev-infected cells compared to levels in BADGFPUL84Δ68-infected cells (Fig. 5C). UL44 levels were increased in BADGFPUL84Δ68rev-infected cells at early time points compared to those found in BADGFPUL84Δ68-infected cells. Thus, the data displayed in Fig. 5A and B are unlikely to be the result of a spurious mutation in the bacmid from which BADGFPUL84Δ68 was generated.
Fig 5.

Levels of viral and cellular proteins in infected cells. (A and C) Western blotting of infected cell lysate. HFF cells were infected at an MOI of 1 with the indicated viruses, and cell lysates were prepared for Western blotting at the indicated time points (indicated above the figure). (B) Determination of relative protein levels in cells. A 2-fold dilution series of protein from lane 2 of panel A (lanes 1 to 3) was analyzed by Western blotting using antibodies recognizing UL44 compared to undiluted protein from lane 5 of panel A. Proteins recognized by the antibodies used in each experiment are indicated to the right of each figure. The positions of molecular mass markers (kDa) are indicated to the left of each figure. In some panels the numbers below bands represent the percent adjusted volume of the signal from the particular bands measured within that panel using Quantity One software.
We also assayed the levels of proteins (IRS1, TRS1, and nucleolin) that are associated with UL44 in infected cell lysate (47, 49). Levels of IRS1, TRS1, and nucleolin decreased to the same levels (approximately 2- to 3-fold) in BADGFPUL84Δ68-infected cells and BADGFP-infected cells (Fig. 5A). Levels of a protein related to IRS1, IRS1263 (35), which can be detected by the MAb used here, were also decreased.
Finally, we assayed the levels of a viral tegument protein, pp28, expressed late in infection. pp28, whose production is linked to viral DNA replication (12), is required for production of infectious virus (39–41). A defect in the accumulation of pp28 was observed in BADGFPUL84Δ68-infected cells compared to levels in BADGFP-infected cells early, but by 72 hpi there was little difference in pp28 signal (Fig. 5A).
We considered that there might be widespread differences in protein production in BADGFP- and BADGFPUL84Δ68-infected cells. Uninfected and infected cells were radiolabeled with [35S]methionine to radiolabel newly translated protein (see supplemental Fig. 2A and B posted at https://coen.med.harvard.edu/). We found a slight (nearly 2-fold) decrease in the production of multiple labeled proteins produced in BADGFPUL84Δ68-infected cells compared to levels in BADGFP-infected cells. Thus, there appears to be widespread, but rather modest, downregulation of protein synthesis in BADGFPUL84Δ68- compared to that in BADGFP-infected cells. Similar results were observed comparing protein production in BADGFPUL84Δ68- and BADGFPUL84Δ68rev-infected cells (data not shown).
In HCMV-infected cells, shutoff of protein synthesis during the innate immune response results in inactivation of eIF2α by phosphorylation at residue serine 51 (eIF2α-P) by protein kinase R (PKR) (22). We also analyzed eIF2α levels in BADGFP- and BADGFPUL84Δ68-infected cell lysates by Western blotting using MAbs recognizing eIF2α and eIF2α phosphorylated at serine residue 51 (see supplemental Fig. 2C posted at https://coen.med.harvard.edu/). Slightly higher levels of phosphorylated eIF2α were observed in BADGFPUL84Δ68- than in BADGFP-infected cells. Western blotting of dilutions of infected cell lysate indicated that there was less than a 2-fold difference in eIF2α-P levels in BADGFP- and BADGFPUL84Δ68-infected cells (data not shown). Similar results were observed comparing eIF2α to eIF2α-P in BADGFPUL84Δ68- and BADGFPUL84Δ68rev-infected cells (data not shown).
Therefore, the deletion that removes the amino terminus of UL84 also results in decreased levels of certain viral and cellular proteins (Fig. 5), plus a rather modest, but widespread, downregulation of protein production in infected cells and a slight increase in the phosphorylation of eIF2α (see supplemental Fig. 2 posted at the URL mentioned above). None of these differences, however, is likely to result in the 10-fold decrease in BADGFPUL84Δ68 virus production observed in the experiments shown in Fig. 2.
The deletion affects UL85 and UL86 levels.
It has been reported (36) that the transcripts encoding UL85 and UL86 (also known as the minor and major capsid proteins, respectively) are 3′ coterminal with RNA encoding UL84 (Fig. 6A). We hypothesized that deletion of the sequences encoding the N terminus of UL84 might adversely affect the production of UL85 and UL86 proteins in the infected cells.
Fig 6.

Levels of capsid proteins in infected cells. (A) Schematic of locus in the HCMV virus genome from which UL83 to UL85 are produced. Filled arrows above the genome represent proteins encoded within the genome. Black lines below the genome represent transcripts from which those proteins are produced. pA, polyadenylation signal. (B) Western blotting of infected cell lysate. HFF cells were infected at an MOI of 1 with the indicated viruses, and cell lysates were prepared for Western blotting at the indicated time points (indicated above the figure). (C) Determination of relative protein levels in cells. A 2-fold dilution series of protein from lane 4 of panel B (lanes 1 to 3) was analyzed by Western blotting compared to undiluted protein from lane 7 of panel B (lane 4). Proteins recognized by the antibodies used in each experiment are indicated to the right of each figure. The positions of molecular mass markers (kDa) are indicated to the left of each figure.
We assayed the levels of UL83, UL84, UL85, UL86, and UL44 proteins in BADGFP- and BADGFPUL84Δ68-infected cells by Western blotting (Fig. 6B). Levels of UL44 in BADGFP- and BADGFPUL84-infected cells were similar to those seen in the Western blotting data presented in Fig. 5. In BADGFP-infected cells compared to BADGFPUL84Δ68-infected cells at 72 hpi, there was no obvious difference in the levels of UL83 and UL84 proteins and only a slight difference in the levels of UL86. However, we observed a more notable decrease in UL85 levels in BADGFPUL84-infected cells than in BADGFP-infected cells at 72 hpi. We also examined serial dilutions of infected cell lysate by Western blotting (Fig. 6C). Levels of UL86 and UL85 were reduced by approximately 2-fold and greater than 4-fold, respectively, in BADGFP-infected cells compared to levels BADGFPUL84Δ68-infected cells. Thus, deletion of the sequence encoding the N terminus of UL84 from the viral genome modestly affects levels of UL86 but has a greater effect on levels of UL85.
Mislocalization of viral capsids in BADGFPUL84Δ68-infected cells.
Based on the above results, we examined capsid production and localization in cells infected with our mutant virus. Cells infected with either BADGFP or BADGFPUL84Δ68 were analyzed by electron microscopy (Fig. 7A). We observed viral capsids distributed throughout the nuclei of cells infected with BADGFP (Fig. 7A, frame i), similar to the distribution of capsids we have previously observed in nuclei of cells infected with wild-type HCMV (29). Strikingly, in cells infected with BADGFPUL84Δ68, most capsids were found concentrated around the nucleolus (Fig. 7A, frame iv). To better visualize the distribution of capsids, the position of each capsid in frames i and iv of Fig. 7A is represented by a white dot in frames ii and v, respectively. Inspection of capsid localization in infected cell nuclei at higher magnification indicated that there was no obvious physical interaction between capsids and the nucleolus in BADGFP- or BADGFPUL84Δ68-infected cells (Fig. 7A, frames iii and vi, respectively). We found that capsid localization in BADGFPUL84Δ68rev-infected cells was similar to that in BADGFP-infected cells (Fig. 7B). Therefore, mislocalization of capsids in BADGFPUL84Δ68-infected cells is most likely due to the deletion of the sequence encoding the N terminus of UL84, not to a spurious mutation within the BADGFPUL84Δ68 genome.
Fig 7.

Electron microscopy of infected cells. Cells were infected with either BADGFP or BADGFPUL84Δ68 (A) or BADGFPUL84Δ68rev (B) were prepared for analysis by electron microscopy at 72 hpi. Panels in the left-hand column show microscopy images. Each white dot in panels in the middle column indicates the positions of a capsid in panels in the left-hand column. The microscopy images in the right-hand column are enlarged areas of the images in the left-hand column. (C) Electron microscopy image of a BADGFP-infected cell nucleus containing a nuclear dense body (NDB). The magnification of each image is indicated below the figure. The white scale bar represents 500 nm. (D) Representative examples of A, B, and C capsids (frames i to iii, respectively). NM, nuclear membrane.
It has been reported that murine gammaherpesvirus-68 (MHV-68) capsids localize around virally induced nuclear dense bodies (10) in much the same way that we find BADGFPUL84Δ68 capsids localize around nucleoli. Consistent with a report by Severi and coworkers (42), we found no association of capsids with nuclear dense bodies in HCMV-infected cells (an example of a nuclear dense body in BADGFP-infected cells is shown in Fig. 7C).
We also sought to analyze the number of capsids in infected cell nuclei. Three capsid forms of viral capsids are found in the nuclei of herpesvirus-infected cells (representative images of each is shown in Fig. 7C): A capsids (frame i), nonproductive forms thought to result from failed packaging of viral genomes; B capsids (frame ii), productive intermediates that contain a scaffolding protein; and C capsids (frame iii), assembled forms in which the scaffolding protein has been removed and replaced with viral DNA. The number of each of these capsids was counted in whole-cell sections from six different cells infected with either BADGFP or BADGFPUL84Δ68. B capsids were the most prevalent capsids in nuclei of cells infected with either virus (data not shown). We found no statistically significant difference in the total number of capsids (Fig. 8A) or the distribution of capsid morphologies (see supplemental Table 2 posted at https://coen.med.harvard.edu) found in nuclei of cells infected with either BADGFP or BADGFPUL84Δ68.
Fig 8.
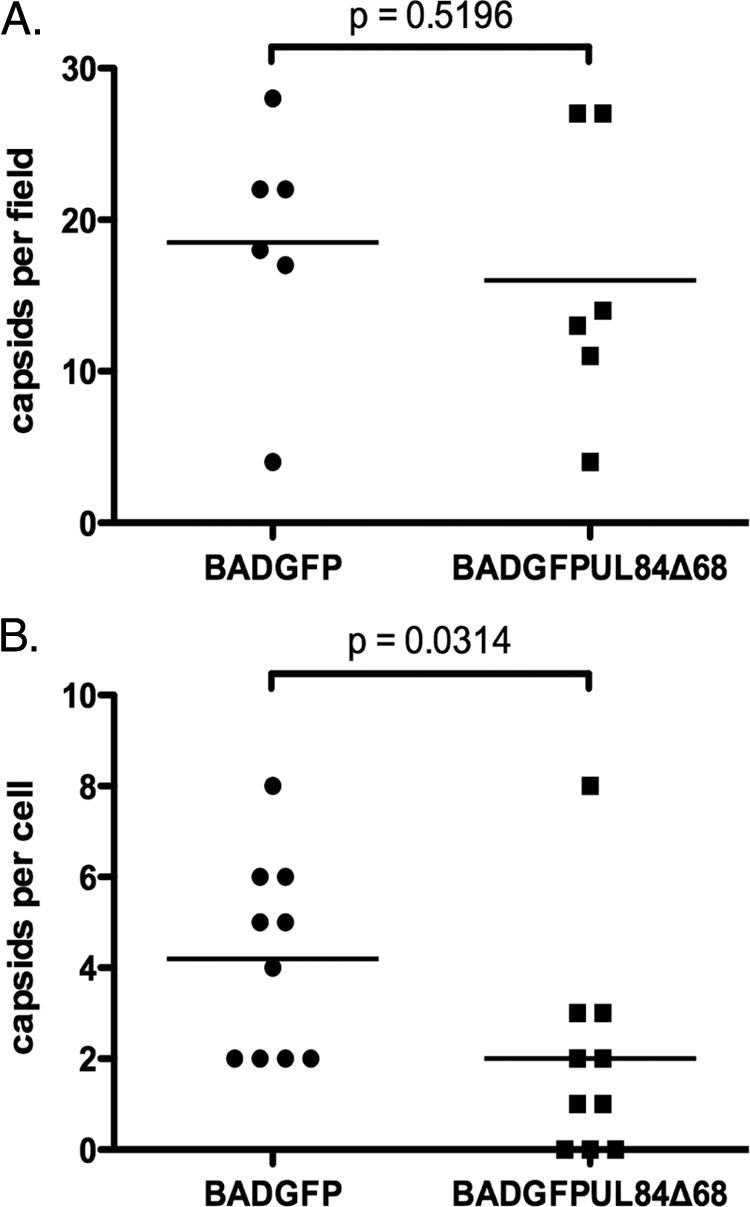
Capsids detected in infected cells. (A) The total numbers of A, B, and C capsids in the nuclei of infected cells were counted. An image of infected cell nuclei taken at a magnification of ×9,600 was chosen at random from each infected cell analyzed in two independent experiments. (B) The total number of enveloped and nonenveloped capsids in the entire cytoplasm of an infected cell was counted at a magnification of ×9,600. The result of a Mann-Whitney test applied in each experiment is shown in each figure.
Using electron microscopy, we also counted the number of cytoplasmic capsids in whole-cell sections of 10 different cells infected with each virus and detected fewer cytoplasmic capsids in BADGFPUL84Δ68-infected cells than in BADGFP-infected cells (Fig. 8B). This difference was statistically significant (Fig. 8B). Indeed, cytoplasmic capsids could not be found in several of the BADGFPUL84Δ68-infected cells analyzed, while all of the BADGFP-infected cells analyzed did contain cytoplasmic capsids (Fig. 8B). We did not observe an obviously aberrant clustering or mislocalization of cytoplasmic capsids in BADGFPUL84Δ68-infected cells compared to BADGFP-infected cells (data not shown). Thus, in BADGFPUL84Δ68-infected cells capsid mislocalization is associated with a defect in the nuclear egress of capsids.
In sum, the production of viral capsids in BADGFPUL84Δ68-infected cells did not appear to be compromised; however, there was a striking mislocalization of these capsids. This may result in their retention in the infected cell nuclei and, thus, contribute to the observed detect in virus production from BADGFPUL84Δ68-infected cells.
DISCUSSION
Here, we found that residues 1 to 68 of UL84 are necessary and sufficient for association of UL84 with a GST-UL44 fusion protein in GST pulldown assays. To our knowledge, no function has been previously ascribed to this region of UL84. Deletion of sequences encoding the amino terminus of UL84 impaired but did not fully abrogate interaction between UL84 and UL44 in the infected cell and did not result in a notable defect in viral DNA synthesis. However, the mutation did cause a modest widespread downregulation of protein production in the infected cell, a striking mislocalization of viral capsids in the nucleus, and a 10-fold defect in virus production.
In agreement with Xu and coworkers (54), we also found that UL84 is required for replication of virus derived from HCMV strain AD169. This contrasts with data from Spector and Yetming (45), who found that UL84 is not required for replication of HCMV strain TB40/E. The discrepancies among these studies remain unresolved. We suggest that strain-specific differences between the genomes of AD169 and TB40/E allow UL84-independent replication of TB40/E.
Studies of the role of UL84 in AD169-infected cells remain challenging as it is not currently possible to propagate AD169-derived UL84-null mutants. By using a BAC that expresses GFP, Xu et al. showed that cells electroporated with such a BAC in which UL84 was inactivated due to an insertion in the UL84 coding sequence did not exhibit normal HCMV replication compartments, as assayed by immunofluorescence for UL44 or detectable viral DNA synthesis (54). We note that it can be difficult to detect viral DNA synthesis in the initial electroporated cells (43). However, the distribution of UL44 in the UL84-null BAC electroporated cells (54) is likely to be due to a defect in viral DNA synthesis as we have previously demonstrated that UL44 is dispersed from viral replication compartments in the presence of an inhibitor of the viral DNA polymerase (46).
It is unclear what causes association of UL44 with truncated UL84 in infected cell lysate. One possibility is that a viral or cellular protein bridges the association of UL44 and truncated UL84 in infected cell lysate. We found that UL44 and UL84 can be part of protein complexes in the infected cell that also contain nucleolin and either IRS1 or TRS1. It is possible that either IRS1, TRS1, or nucleolin or a combination of these proteins bridges the association of UL44 and UL84 in the infected cell. The role of the association of these proteins within infected cells remains unknown. Recently, it has been reported that UL44 and UL84 are part of protein complex in infected cells that also contains the viral proteins IE2-86 and UL112-113(p84) (28). Thus, either IE2 or UL112-113(p84) might also bridge the association of UL44 and UL84 in infected cells. However, we found no evidence that IE2-86 bridges the association of UL44 and UL84 in this study. Also, in proteomic analyses performed in our laboratory as well as in another, UL112-113(p84) was not found to be associated with UL44 and UL84 (15, 47, 50). Alternatively, it is possible that regions of UL84 in addition to the amino terminus of UL84 mediate UL44-UL84 association in the infected cell although there was little evidence for this in our in vitro GST pulldown assays.
The small amount of UL84Δ68 protein able to associate with UL44 in infected cells appeared to be sufficient to promote viral DNA synthesis during infection. As viral DNA synthesis was not notably compromised, there must be other factors responsible for the clear defect in virus production we observe from BADGFPUL84Δ68-infected cells. One possible factor was the modest decrease in expression of multiple proteins in mutant infected cells. Some of the proteins downregulated—UL44, IRS1, TRS1, and nucleolin—associate with UL84; therefore, truncation of UL84 may affect stability of these proteins in the infected cell. Changes in the stability of UL44, IRS1, or TRS1 could conceivably affect levels of other proteins. UL44 has been reported to have a role in transcription from the viral genome (23–25). However, as UL44 is required for transcription of only certain classes of viral transcripts (23–25), it is unlikely that deregulation of UL44 levels is associated with the widespread decrease in protein levels we observe. It has been reported that UL84 binds IRS1 mRNA, which is necessary for accumulation of this mRNA in the cytoplasm (16). Low levels of IRS1 or TRS1 in the infected cell might contribute to widespread downregulation of protein production by compromising the ability of the virus to antagonize eIF2α inactivation by the PKR pathway as part of the innate immune response to infection (20, 22). However, as only a slight increase in eIF2α-P was observed in BADGFP-infected cells compared to BADGFPUL84Δ68-infected cells, it is unlikely that inactivation of the innate immune response is a major factor in preventing virus production in cells infected with our mutant virus.
Our analysis of capsid localization may explain at least part of the block to virus replication in BADGFPUL84Δ68-infected cells. However, consistent with previous observations from our laboratory (29), relatively few cytoplasmic capsids can be observed even in cells infected with wild-type virus. Thus, while we observed a statistically significant difference in the number of cytoplasmic capsids in BADGFP- and BADGFPUL84Δ68-infected cells, we only cautiously infer that mislocalization of capsids results in a defect in nuclear egress. Our finding that there was little or no difference in the number of capsids in the nucleus is consistent with the behavior of mutants that exhibit an even more profound defect in nuclear egress (29) than does BADGFPUL84Δ68.
To our knowledge, a phenotype where capsids appear to concentrate around the nucleolus has not been reported elsewhere in the study of HCMV or any other herpesvirus. It is unclear what influences HCMV capsid movement within replication compartments and nuclei and how capsid proteins might interact with these factors. However, it has been reported that movement of herpes simplex virus capsids within the nucleus requires myosin and actin (14). The location of the UL84Δ68 mutation leads us to speculate on three possible explanations for how the mutation affects capsid localization. The first is that UL84 somehow has a role in this process. The second is that the effects of the mutation on expression of neighboring genes that encode capsid proteins somehow affect capsid localization. The third is that products encoded from low-abundance transcripts antisense to this region of the genome (17) somehow could influence capsid localization. Regardless, the unusual localization of capsids in the mutant infected cells seems likely to point to hitherto unknown features of HCMV biology.
In summary, the defect in virus production we observe from BADGFPUL84Δ68-infected cells could be, at least, the result of both low levels of proteins required for virus replication and reduced nuclear egress. As levels of pp28 and TRS1, both of which are required for virus assembly and egress from the cell (1, 6, 39–41), were reduced in BADGFPUL84Δ68-infected cells, there may be as yet unappreciated late defects in virus replication in BADGFPUL84Δ68-infected cells.
ACKNOWLEDGMENTS
We gratefully acknowledge Gloria Komazin-Meredith, Nikolaus Osterrieder, Greg Pari, Gary Hayward, Dong Yu, Tom Shenk, and Wade Gibson for providing reagents. We also acknowledge Maria Ericsson and the staff of the Harvard Medical School Electron Microscope Facility for all their invaluable assistance. We thank Natalia Reim for assistance during assay development and Kevin Bryant and Frazer Rixon for helpful comments. We also thank all members of the Coen laboratory for their support.
This work was supported by NIH grants RO1 A0I19838 and RO1 A0I26077 to D.M.C. and RO1 CA034729 to D.H.S.
Footnotes
Published ahead of print 1 August 2012
REFERENCES
- 1. Adamo JE, Schroer J, Shenk T. 2004. Human cytomegalovirus TRS1 protein is required for efficient assembly of DNA-containing capsids. J. Virol. 78:10221–10229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahn JH, Jang WJ, Hayward GS. 1999. The human cytomegalovirus IE2 and UL112-113 proteins accumulate in viral DNA replication compartments that initiate from the periphery of promyelocytic leukemia protein-associated nuclear bodies (PODs or ND10). J. Virol. 73:10458–10471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Appleton BA, et al. 2006. Crystal structure of the cytomegalovirus DNA polymerase subunit UL44 in complex with the C terminus from the catalytic subunit. Differences in structure and function relative to unliganded UL44. J. Biol. Chem. 281:5224–5232 [DOI] [PubMed] [Google Scholar]
- 4. Appleton BA, Loregian A, Filman DJ, Coen DM, Hogle JM. 2004. The cytomegalovirus DNA polymerase subunit UL44 forms a C clamp-shaped dimer. Mol. Cell 15:233–244 [DOI] [PubMed] [Google Scholar]
- 5. Baldick CJ, Jr, Marchini A, Patterson CE, Shenk T. 1997. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J. Virol. 71:4400–4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blankenship CA, Shenk T. 2002. Mutant human cytomegalovirus lacking the immediate-early TRS1 coding region exhibits a late defect. J. Virol. 76:12290–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colletti KS, Smallenburg KE, Xu Y, Pari GS. 2007. Human cytomegalovirus UL84 interacts with an RNA stem-loop sequence found within the RNA/DNA hybrid region of oriLyt. J. Virol. 81:7077–7085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colletti KS, Xu Y, Cei SA, Tarrant M, Pari GS. 2004. Human cytomegalovirus UL84 oligomerization and heterodimerization domains act as transdominant inhibitors of oriLyt-dependent DNA replication: evidence that IE2-UL84 and UL84-UL84 interactions are required for lytic DNA replication. J. Virol. 78:9203–9214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Colletti KS, Xu Y, Yamboliev I, Pari GS. 2005. Human cytomegalovirus UL84 is a phosphoprotein that exhibits UTPase activity and is a putative member of the DExD/H box family of proteins. J. Biol. Chem. 280:11955–11960 [DOI] [PubMed] [Google Scholar]
- 10. Dai W, et al. 2008. Unique structures in a tumor herpesvirus revealed by cryo-electron tomography and microscopy. J. Struct. Biol. 161:428–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davison AJ, Stow ND. 2005. New genes from old: redeployment of dUTPase by herpesviruses. J. Virol. 79:12880–12892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Depto AS, Stenberg RM. 1992. Functional analysis of the true late human cytomegalovirus pp28 upstream promoter: cis-acting elements and viral trans-acting proteins necessary for promoter activation. J. Virol. 66:3241–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dunn W, et al. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forest T, Barnard S, Baines JD. 2005. Active intranuclear movement of herpesvirus capsids. Nat. Cell Biol. 7:429–431 [DOI] [PubMed] [Google Scholar]
- 15. Gao Y, Colletti K, Pari GS. 2008. Identification of human cytomegalovirus UL84 virus- and cell-encoded binding partners by using proteomics analysis. J. Virol. 82:96–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao Y, Kagele D, Smallenberg K, Pari GS. 2010. Nucleocytoplasmic shuttling of human cytomegalovirus UL84 is essential for virus growth. J. Virol. 84:8484–8494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gatherer D, et al. 2011. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. U. S. A. 108:19755–19760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gault E, et al. 2001. Quantification of human cytomegalovirus DNA by real-time PCR. J. Clin. Microbiol. 39:772–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gebert S, et al. 1997. The UL84 protein of human cytomegalovirus acts as a transdominant inhibitor of immediate-early-mediated transactivation that is able to prevent viral replication. J. Virol. 71:7048–7060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hakki M, Marshall EE, De Niro KL, Geballe AP. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 80:11817–11826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 74:7720–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Isler JA, Skalet AH, Alwine JC. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79:6890–6899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Isomura H, et al. 2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 82:1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Isomura H, et al. 2007. The late promoter of the human cytomegalovirus viral DNA polymerase processivity factor has an impact on delayed early and late viral gene products but not on viral DNA synthesis. J. Virol. 81:6197–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Isomura H, et al. 2011. The human cytomegalovirus gene products essential for late viral gene expression assemble into prereplication complexes before viral DNA replication. J. Virol. 85:6629–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kagele D, Rossetto CC, Tarrant MT, Pari GS. 2012. Analysis of the interactions of viral and cellular factors with human cytomegalovirus lytic origin of replication, oriLyt. Virology 424:106–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kamil JP, et al. 2009. Human papillomavirus 16 E7 inactivator of retinoblastoma family proteins complements human cytomegalovirus lacking UL97 protein kinase. Proc. Natl. Acad. Sci. U. S. A. 106:16823–16828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim YE, Ahn JH. 2010. Role of the specific interaction of UL112-113 p84 with UL44 DNA polymerase processivity factor in promoting DNA replication of human cytomegalovirus. J. Virol. 84:8409–8421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krosky PM, Baek MC, Coen DM. 2003. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J. Virol. 77:905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loveland AN, Chan CK, Brignole EJ, Gibson W. 2005. Cleavage of human cytomegalovirus protease pUL80a at internal and cryptic sites is not essential but enhances infectivity. J. Virol. 79:12961–12968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maga G, Hubscher U. 2003. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 116:3051–3060 [DOI] [PubMed] [Google Scholar]
- 32. Moldovan GL, Pfander B, Jentsch S. 2007. PCNA, the maestro of the replication fork. Cell 129:665–679 [DOI] [PubMed] [Google Scholar]
- 33. Pari GS, Anders DG. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979–6988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pari GS, Kacica MA, Anders DG. 1993. Open reading frames UL44, IRS1/TRS1, and UL36-38 are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA synthesis. J. Virol. 67:2575–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Romanowski MJ, Shenk T. 1997. Characterization of the human cytomegalovirus irs1 and trs1 genes: a second immediate-early transcription unit within irs1 whose product antagonizes transcriptional activation. J. Virol. 71:1485–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rudolph SA, Stamminger T, Jahn G. 1990. Transcriptional analysis of the eight-kilobase mRNA encoding the major capsid protein of human cytomegalovirus. J. Virol. 64:5167–5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sanders RL, Del Rosario CJ, White EA, Spector DH. 2008. Internal deletions of IE2 86 and loss of the late IE2 60 and IE2 40 proteins encoded by human cytomegalovirus affect the levels of UL84 protein but not the amount of UL84 mRNA or the loading and distribution of the mRNA on polysomes. J. Virol. 82:11383–11397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sanders RL, Spector DH. 2010. Human cytomegalovirus IE2 86 and IE2 40 proteins differentially regulate UL84 protein expression posttranscriptionally in the absence of other viral gene products. J. Virol. 84:5158–5170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seo JY, Britt WJ. 2007. Cytoplasmic envelopment of human cytomegalovirus requires the postlocalization function of tegument protein pp28 within the assembly compartment. J. Virol. 81:6536–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seo JY, Britt WJ. 2008. Multimerization of tegument protein pp28 within the assembly compartment is required for cytoplasmic envelopment of human cytomegalovirus. J. Virol. 82:6272–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seo JY, Britt WJ. 2006. Sequence requirements for localization of human cytomegalovirus tegument protein pp28 to the virus assembly compartment and for assembly of infectious virus. J. Virol. 80:5611–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Severi B, Landini MP, Cenacchi G, Zini N, Maraldi NM. 1992. Human cytomegalovirus nuclear and cytoplasmic dense bodies. Arch. Virol. 123:193–207 [DOI] [PubMed] [Google Scholar]
- 43. Silva LA, Loregian A, Pari GS, Strang BL, Coen DM. 2010. The carboxy-terminal segment of the human cytomegalovirus DNA polymerase accessory subunit UL44 is crucial for viral replication. J. Virol. 84:11563–11568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Spector DJ, Tevethia MJ. 1994. Protein-protein interactions between human cytomegalovirus IE2-580aa and pUL84 in lytically infected cells. J. Virol. 68:7549–7553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spector DJ, Yetming K. 2010. UL84-independent replication of human cytomegalovirus strain TB40/E. Virology 407:171–177 [DOI] [PubMed] [Google Scholar]
- 46. Strang BL, et al. 2012. Human cytomegalovirus UL44 concentrates at the periphery of replication compartments, the site of viral DNA synthesis. J. Virol. 86:2089–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Strang BL, Boulant S, Coen DM. 2010. Nucleolin associates with the human cytomegalovirus DNA polymerase accessory subunit UL44 and is necessary for viral replication J. Virol. 84:1771–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Strang BL, Boulant S, Kirchhausen T, Coen DM. 2012. Host cell nucleolin is required to maintain the architecture of human cytomegalovirus replication compartments. mBio 3(1):e00301–311 doi:10.1128/mBio.00301-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Strang BL, Geballe AP, Coen DM. 2010. Association of the human cytomegalovirus DNA polymerase subunit UL44 with the viral proteins IRS1 and TRS1 J. Gen. Virol. 91:2167–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Strang BL, Sinigalia E, Silva LA, Coen DM, Loregian A. 2009. Analysis of the association of the human cytomegalovirus DNA polymerase subunit UL44 with the viral DNA replication factor UL84. J. Virol. 83:7581–7589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Strang BL, Stow ND. 2005. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J. Virol. 79:12487–12494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 53. Wood LJ, Baxter MK, Plafker SM, Gibson W. 1997. Human cytomegalovirus capsid assembly protein precursor (pUL80.5) interacts with itself and with the major capsid protein (pUL86) through two different domains. J. Virol. 71:179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu Y, Cei SA, Huete AR, Pari GS. 2004. Human cytomegalovirus UL84 insertion mutant defective for viral DNA synthesis and growth. J. Virol. 78:10360–10369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]