

Abstract
Assembly of the bacteriophage T4 head structure occurs at the cytoplasmic face of the inner membrane of Escherichia coli with the formation of proheads. The proheads contain an internal scaffolding core that determines the size and the structure of the capsid. In a mutant where the major shell protein gp23 was compromised, core structures without a shell had been detected. Such core structures were also found in the mutant T4am20am23. Since the mutation in gene 20 is at the N terminus of gp20, it was assumed that these core structures assemble in the absence of gp20. However, sequencing showed that the mutation introduces a new ribosome binding site that leads to a restart at codon 15. Although the mutant protein gp20s lacks the very N-terminal sequence, we found that it still binds to the membrane of the host cell and can initiate prohead assembly. This explains its activity to allow the assembly of core structures and proheads at the membrane surface. With a cross-linking approach, we show here that gp20 and gp20s are escorted by the chaperones DnaK, trigger factor, and GroEL and dock on the membrane at the membrane protein YidC.
INTRODUCTION
Viruses infecting bacteria are the most abundant life forms on earth, with around 1031 particles (27). Enterophage T4 is one of the seven T-type phages isolated in the 1940s (8). From the beginning, it was used as a model system for lytic phages. Particularly, it proved helpful in understanding the principles of assembly and morphogenesis of macromolecular complexes (3, 7, 30). The assembly of the phage T4 is divided into three distinct major pathways. The head, the baseplate with the tail, and the tail fibers are first independently assembled inside the infected cell (29). The head is made at the inner surface of the inner membrane of Escherichia coli. At first, a membrane-bound initiator complex is built by gp20, gp40, and yet-unknown E. coli proteins (4, 21). Around this small structure, core and shell proteins (gpalt, IPI, IPII, IPIII, gp20, gp21, gp22, gp23, gp24, gp67, and gp68) assemble to form the prohead (3, 28). The internal zymogen gp21 is activated to become the T4-own prohead protease gp21*, which then degrades most scaffolding proteins into small peptides (13, 22, 25), which are expelled from the head structure. Next, the head detaches from the inner cellular membrane and is filled with DNA from a DNA-concatemer by activation of the terminase gp16/gp17 (3). Finally, the DNA-filled head is connected to the tail and the tail fibers to complete the assembly of the virion (29).
The portal ring protein gp20 plays a crucial role for this assembly process. This 61-kDa protein lacks defined transmembrane segments, but it appears to be membrane bound (21). It is assumed that the membrane binding is the starting point of the normal head assembly process (3). In the mature phage virion, the portal ring is found at one special vertex as a dodecamer with 12-fold rotational symmetry connecting the head to the tail (9). The portal has a diameter of 17 nm and a thickness of 7 nm with an internal cavity of 14 nm by 7 nm. The dodecamer is completely buried inside the mature virion and exposed at the shell surface in proheads (10, 17). It is most likely that the N terminus of each monomer is displayed outside the head, while the C terminus is inside the head structure (2). The N-terminal region of gp20 might therefore be important to bind to the membrane and, later, in the DNA-filled heads to the tail structures. The role of gp20 in the initiation and maturation of the T4 bacteriophage has been studied with several gene 20 mutant phages. Two mutations, am20E208 and am20E481, have been mapped to the very beginning of the gene, and another mutant, am20B8, encodes a protein lacking the last 33 residues. The am20B8 and am20E481 mutants allow the generation of very few phage progeny (2). For the am20E481 mutant, it has been suggested that an in-frame restart leads to an amino-terminal truncated protein that might be partially functional (2).
To study the function of the am20E481 protein in detail, we expressed the protein from a plasmid with an N-terminal His tag. The protein, gp20s, is ∼1.7 kDa smaller than the wild-type gp20 and lacks the amino-terminal 14 residues. gp20s was found at the cell periphery when expressed as a green fluorescent protein (GFP) fusion protein, as was the wild-type protein. Therefore, it is possible that gp20s participates in the head assembly process. We found that gp20 and gp20s can be cross-linked to Tig, DnaK, GroE, and the membrane protein YidC. We hypothesize that YidC most likely functions to recruit gp20 at the membrane surface, thus nucleating prohead assembly.
MATERIALS AND METHODS
Bacterial strains, bacteriophage, and culture conditions.
E. coli B was used as the nonpermissive strain, and E. coli CR63 F− λ− supD60 lamB63 (1) was used as the permissive strain for amber mutants. All phage mutants were from our collection. Medium preparation and bacterial manipulations were performed according to standard methods (19). When appropriate, ampicillin (200 μg/ml, final concentration) was added to the medium. Plasmid-encoded protein expression of gp20 was carried out with E. coli BL21(DE3) (26). T4D and the amber mutants in gene 20 (amE208, amE481), gene 21 (tsN8), and gene 23 (amH11) were from our collection.
Genetic manipulations.
Genes g20 and g40 were amplified from pT20-40 plasmid DNA (kind gift of L. W. Black [2]) using the primers forward 5′-CGG GGA TCC GAT GAA ATT TAA TGT ATT AAG TTT GTT TGC and reverse 5′-AAT GGG ATC CGA ATA ATT TCT ACC ACA CTT ACT CC introducing BamHI cleavage sites (underlined). The digested PCR fragment was ligated into pET16b (Novagen) to obtain pET20-40, and the correct orientation and sequence were checked by sequencing. To introduce the amber mutation (underlined) in plasmids pT20-40 and pT20gfp-40, primers forward, 5′ GTT TGC TCC ATA GGC TAA AAT GGA CG, and reverse, 5′ CGT CCA TTT TAG CCT ATG GAG CAA AC, were used. To construct pT20s-40, a BamHI restriction site was introduced by PCR with the primers forward, 5′-CGG GGA TCC GAT GGA CGA ACG AAA TTT TAA AGA CC, and reverse, 3′-AAT GGG ATC CGA ATA ATT TCT ACC ACA CTT ACT CC. The PCR amplicon was digested with BamHI and ligated into pET16b. Correct orientation and sequence were verified by sequencing.
Plaque assay.
To determine phage titers, dilutions of the various phages used in this study were plated. Three milliliters of melted Hershey top agar (47°C) were mixed with 1 ml prewarmed 0.01 M Tris, pH 7.5, to ensure better phage diffusion. A 300-μl volume of plating bacteria (E. coli B as the nonpermissive strain and E. coli CR63 as the permissive strain) grown to a cell density of 4 × 108 CFU/ml and appropriate phage dilutions were added. The mixture was poured onto the agar plates and incubated overnight at 37°C. Plaques were counted, and dilutions were plated three times to obtain a mean value. For a rapid determination of phage titers, aliquots of dilutions were pipetted directly onto the hardened Hershey top agar containing the plating bacteria.
Complementation of T4D am20 mutants.
To complement the assembly defect of T4D am20 mutants, different mutants of g20-expressing plasmids were used. Cells bearing those plasmids were grown at 37°C to an optical density at 600 nm (OD600) of 0.6 and induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After a short time allowing expression of gp20 (∼30 min), the cells were infected at a cell density of 4 × 108 cells/ml with a multiplicity of infection (MOI) of 5 with phage am20E481. Thirty minutes later, the infection cycle was stopped by adding a few drops of chloroform. The lysate titers were determined on agar plates.
NaCl extraction.
A fresh overnight culture of E. coli BL21(DE3) harboring pET20-40 was diluted 1:100 and shaken at 37°C to an OD600 of 0.6. The culture was shifted to 18°C and continued for 16 h. The cells were harvested and lysed by three passages through a French pressure cell at 8,000 lb/in2. Cell debris was removed by centrifugation, and the membranes were collected at 160,000 × g for 60 min at 4°C. The membranes were resuspended in buffer containing 0.05 M Tris (pH 7.5) and 10% glycerol, NaCl was added as indicated (0.1 M, 0.3 M, 0.6 M, 0.9 M, and 2 M), and the suspensions were incubated for 30 min on ice. The extracted protein was separated by an additional centrifugation step at 160,000 × g for 60 min at 4°C. Pellet and supernatant were trichloroacetic acid (TCA) precipitated and analyzed by SDS-PAGE and Western blotting.
Electrophoresis.
SDS-PAGE and subsequent staining with Coomassie brilliant blue (R250) or silver and Western blotting were performed according to standard protocols (19). For standard analysis of proteins, 12% minigels with a length of 7 cm were used. To identify the amber fragment of mutant am20E481, a 40-cm-long 7.5% SDS gel was run for 9 h at 4°C.
Localization of His-gp20 and His-gp20s.
Five hundred milliliters of LB medium was inoculated 1:100 with a fresh culture of BL21(DE3) harboring plasmid pET20-40 or pT20s-40. Cells were incubated at 37°C to an OD of 0.6, and protein expression was induced with 1 mM IPTG. The culture was continued for 5 h at 30°C and harvested by centrifugation at 8,000 × g for 20 min at 4°C. The cell pellet was resuspended in buffer (0.05 M Tris [pH 7.5], 0.05 M NaCl, 10% glycerol), and cells were lysed by three passages through the French pressure cell at 8,000 lb/in2. Cell debris was removed from the sample by a low-speed centrifugation step (10,000 × g, 20 min, 4°C). The membranes were centrifuged at 160,000 × g for 60 min at 4°C. Pellet and supernatant were TCA precipitated and analyzed by SDS-PAGE and Western blotting using antibodies to GroEL and YidC as indicators for a cytoplasmic and a membrane protein, respectively. The resuspended pellet fraction was then loaded on a 3-step sucrose gradient (35%, 58%, and 78%) and run for 16 h at 112,000 × g at 4°C to purify the membrane vesicles.
In vivo protease mapping.
One-milliliter volumes of BL21(DE3) cultures harboring the respective plasmids were grown to a cell density of 2 × 108 cells/ml. The expression of His-gp20 and His-gp20s was induced with 1 mM IPTG for 1 h. The cells were centrifuged at 7,000 × g for 2 min at 4°C, resuspended in ice-cold spheroplast buffer (40% sucrose, 33 mM Tris-acetate, pH 8.0), and treated with 0.05 mg/ml lysozyme (in spheroplast buffer) and 1 mM EDTA, pH 8.0, on ice for 15 min. An aliquot was treated with 0.75 mg/ml proteinase K (in spheroplast buffer) for 1 h on ice. Another aliquot was treated with 0.75 mg/ml proteinase K in the presence of 3% Triton X-100 for 1 h on ice. All samples were TCA precipitated and analyzed by SDS-PAGE and Western blotting.
Purification of His-gp20 and His-gp20s and cross-linking.
E. coli BL21(DE3) harboring plasmid pET20-40 was grown in LB medium (4 liters) supplemented with 200 μg/ml ampicillin at 37°C to an optical density (OD600) of 0.6 and induced with 1 mM IPTG. The culture was shifted to 18°C and continued overnight. Cells were harvested by centrifugation at 5,000 × g for 15 min at 4°C and resuspended in 0.05 mM Tris (pH 7.5), 0.05 M NaCl, and 10% glycerol. The cells were lysed by three passages through a French pressure cell at 8,000 lb/in2. After removal of cell debris at 8,000 × g for 20 min at 4°C, the membranes were collected by centrifugation at 160,000 × g for 60 min at 4°C and solubilized in buffer (0.05 mM Tris [pH 7.5], 0.05 M NaCl, and 10% glycerol) containing 1% N-lauroylsarcosine for 1 h at 4°C. After centrifugation at 160,000 × g for 60 min at 4°C, the solubilized protein was added to a Ni-nitrilotriacetic (NTA) matrix (Qiagen) supplemented with 0.02 M imidazole and incubated for 1 h at 4°C. The matrix was washed with buffer (0.04 M imidazole, 0.05 mM Tris [pH 7.5], 0.05 M NaCl, and 10% glycerol). Elution was performed with 0.2 M imidazole. Purification was checked by SDS-PAGE and Coomassie brilliant blue R250 staining. The cross-linking experiments were done as described in reference 12.
Isolation of proheads.
A 500-ml volume of LB medium was inoculated 1:100 with a fresh E. coli B overnight culture and incubated at 37°C to a cell density of 4 × 108 cells per ml. Cells were infected with phage T4D am20E481 at an MOI of 5 for 7 min and superinfected for an additional 53 min. The culture was centrifuged at room temperature at 6,000 × g for 30 min in a prewarmed rotor (37°C) to minimize lysing effects due to temperature stress. The cell pellet was transferred to a 15-ml test tube, resuspended in buffer (0.05 M Tris [pH 6.0], 2 mM MgCl2, 0.02 M NaCl, 0.5 mM CaCl2, and 1 unit Benzonase), and kept overnight. The cells were lysed by three passages through a French pressure cell at 8,000 lb/in2. Cell debris was removed at 14,000 × g for 10 min at 4°C, and phage particles and prohead structures were collected by centrifugation at 19,000 × g for 30 min at 4°C. The pellet was resuspended in buffer (0.05 M Tris [pH 6.0], 2 mM MgCl2, 0.02 M NaCl, 0.5 mM CaCl2) and loaded onto a 0 to 35% glycerol gradient. The run was for 90 min at 130,000 × g at 15°C. Fractions of 1 ml were collected and used immediately for the preparation of transmission electron microscopy (TEM) grids. Nine hundred fifty microliters was TCA precipitated and analyzed by SDS-PAGE and subsequent Western blotting.
Fluorescence microscopy.
Fresh overnight cultures of E. coli BL21(DE3) harboring plasmid pT20gfp-40 (kind gift of L. W. Black) or its derivative pT20sgfp-40 were diluted 1:100 in LB medium and grown at 30°C to an OD600 of 0.5. The expression of gp20-GFP and gp20s-GFP was induced with 1 mM IPTG and continued as indicated. Samples of the different cultures were placed onto poly-l-lysine-coated cover glasses (Sigma-Aldrich) and analyzed immediately by a Zeiss AXIO Imager M1 fluorescence microscope.
Liquid chromatography-electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS).
To identify proteins, the cross-linked samples were loaded onto a 10% SDS gel and silver stained (6). The bands of interest were cut out with a sterile knife and transferred to a test tube. The gel pieces were washed with acetonitrile for 5 min at room temperature. For reduction of the disulfide bonds, 10 mM dithiothreitol was applied for 30 min at 56°C followed by a washing step with acetonitrile. For alkylation, 100 mM iodoacetamide was used for 20 min at room temperature in the dark. The gel pieces were washed with acetonitrile and then digested overnight at 37°C by trypsin (1 ng/μl). To inactivate the enzyme, a 10% trifluoroacetic acid (TFA) solution was mixed with the supernatant. The peptide fragments were analyzed with a Nano LC-ESI.
Electron microscopy.
Seven-microliter volumes of the glycerol gradient fractions were adsorbed to carbon-coated copper TEM grids (Type 483; Plano GmbH, Wetzlar, Germany) for 2 min. Unbound protein material was removed by touching filter sheets, and the grids were dried for 2 min. Negative staining was done by adsorption of 7 μl 2% phosphotungstic acid (pH 7) for 1 min. Most of the staining solution was removed by filter sheets again, and the grids were dried for 2 min. Electron microscopy was performed with a TECNAI G2, FEI at 120 kV. All pictures were taken with a magnification of ×19,000.
RESULTS
Phage T4 am20 mutants encode a truncated gp20 protein.
To investigate the functionality of the am20 mutants of T4D for the assembly of the T4 proheads, the two mutants am20E208 and am20E481 were cloned into the plasmid pUC19 to exactly map the amber mutations in the phage genome. DNA sequencing verified that the two mutants have the identical amber mutation (TGG to TAG) at amino acid codon position 12. For both mutants, the amber mutation had created a new Shine-Dalgarno sequence that matches with an ATG codon at codon position 15 in a 5-base distance (Fig. 1). When the expression of gp20 was analyzed after phage infection of E. coli CR63 or B cells, two proteins of different sizes were respectively expressed (Fig. 1C). In the suppressor strain, the full-length 61-kDa protein was detected. Nonpermissive phage infections showed a shorter protein of about 59 kDa on a Western blot, termed gp20s (Fig. 1C, lanes 4 and 6), suggesting that it was initiated at the ATG at position 15. The expression rate of gp20s was lower than that of gp20 expressed under permissive conditions.
Fig 1.

Sequence analysis of gp20 in T4D and the T4 am20 mutants E208 and E481. The first 16 codons of gp20 of the wild-type T4D (A) and the identical codons of the mutants E208 and E481 (B) are shown. The amber mutation (codon 12) is highlighted in bold, and the ribosome binding sequence (RBS) of gp20s is underlined. The start codon of gp20s is at codon 15. (C) The expression of gp20 and gp20s was analyzed in infected E. coli CR (lanes 1, 3, and 5) and E. coli B (lanes 2, 4, and 6) with an antibody to gp20. To resolve the difference between gp20 and gp20s, a 40-cm-long 7.5% SDS-polyacrylamide gel was used.
Complementation of am20 phages.
In a next step, we analyzed whether the plasmid-encoded gp20 and gp20s can complement various amber phage infections (Fig. 2). E. coli BL21(DE3) cells were transformed with pT20-40 (2) encoding gp20 and gp40. In parallel, pT20s-40, which is identical to pT20-40 but has the amber mutation at the codon position 12 in gene 20, was transformed. The cells were grown to exponential phase, expression was induced with 1 mM IPTG, and the cells were infected 30 min later. Wild-type and two T4 mutants were tested for phage propagation by a spot test. As a positive control, wild-type T4 (T4wt), showing efficient plaque formation on all plates, was used. In the absence of a suppressor (E. coli B lawn), the mutant am20E481 was complemented by gp20 but not by am20, showing that the expression of gp20s is not functional for phage propagation. In addition, His-gp20 and gp20-GFP fully complemented am20E481 phage growth. Also, as a control, phage am23H11 was not complemented on all the E. coli B plates.
Fig 2.
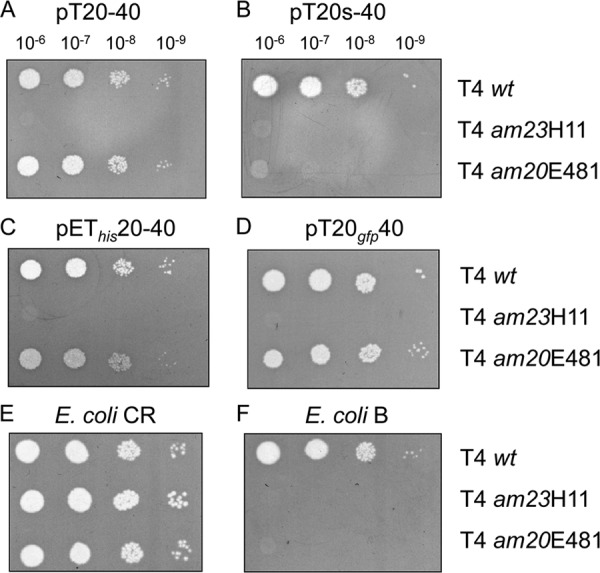
Complementation of the T4 am20 mutants by plasmid-borne expression. Diluted phage stocks were spotted onto top agar containing a lawn of E. coli B cells bearing the following plasmids: pT20-40 (A), pT20s-40 (B), pET20-40 (C), and pT20gfp-40 (D). For a control, E. coli CR and B with no plasmid were tested as plating bacteria (E, F).
Cellular localization of gp20 and gp20s.
To analyze the localization of the plasmid-encoded gp20 in cells, a fusion protein of gp20 and GFP was used. Plasmid pT20gfp-40 (2) encoding the gp20-GFP fusion protein and gp40 was transformed into E. coli BL21(DE3) cells and analyzed by fluorescence microscopy (Fig. 3). Typically, the fluorescence signal was distributed at the cell periphery (Fig. 3A through C). Clusters of the fluorescent dots were observed at the membrane with an apparently regular spacing between them. Also, in phage am20E481-infected cells, the fluorescent clusters were observed at 15 min postinfection (Fig. 3B). We conclude that gp20 is membrane bound, either by itself or in an aggregated form. Expression of GFP alone appeared to be distributed in the cytoplasm (Fig. 3D and E).
Fig 3.

Cellular localization of gp20 in uninfected and T4-infected cells. gp20-GFP fusion proteins were expressed in E. coli B for 5 h and examined with fluoromicroscopy (A through C). The plasmid-expressed gp20 (A, C) and the gp20s expressed from E481-infected cells (B) are shown. For a control, GFP alone was expressed under the same conditions (D, E).
To examine the location of gp20 and gp20s in more detail, His10-tagged versions of gp20 and gp20s were generated by subcloning genes 20 and 40 into pET16b. E. coli BL21(DE3) cells expressing His-gp20 or His-gp20s were grown overnight at 18°C, and the proteins were analyzed by cell fractionation (Fig. 4). Whereas gp40 was mainly in the cytoplasmic fraction, gp20 and gp20s were found only in the membrane fraction. For controls, YidC was analyzed as a membrane protein and GroEL as a cytoplasmic protein (Fig. 4A). The binding to membrane vesicles was verified by a sucrose gradient (Fig. 4B). Inner and outer membrane vesicles were collected in fractions and analyzed for their content of gp20 (Fig. 4B, middle panel) and gp20s (lower panel). The results clearly show that the membrane binding of gp20 is not caused by its N-terminal 14 residues (Fig. 1).
Fig 4.
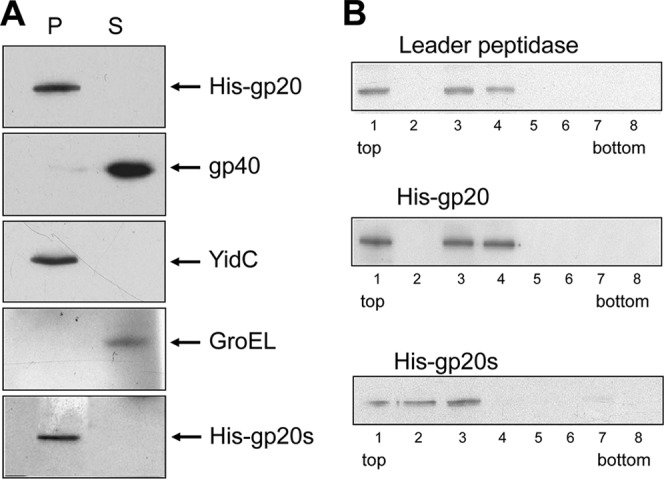
Plasmid-encoded gp20 is found in the membrane fraction, whereas gp40 is in the supernatant. E. coli BL21 cultures expressing His-gp20 with gp40 and His-gp20s with gp40 were lysed by a French press and analyzed for soluble and membrane-bound protein (A). The soluble fraction (S) and the membrane fraction (P) were collected by centrifugation and analyzed for their protein content on a Western blot using the respective antibodies. (B) The membrane fraction was placed onto a sucrose step gradient, and fractions 1 to 8 were analyzed for their content of leader peptidase (inner membrane marker protein, top panel), His-gp20 (central panel), and His-gp20s (lower panel) on Western blots.
Membrane interaction of gp20.
Since gp20 and gp20s were found to be firmly bound to the membrane, we tested their extraction from the membrane using different detergents. Only the ionic detergent laurylsarcosine was capable of efficiently solubilizing gp20, whereas other detergents like dodecyl maltoside, 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfate (CHAPS), Asb14, and 12-phosphocholine did not solubilize gp20 (data not shown). This suggests that gp20 is bound mainly by ionic interactions as a peripherally bound membrane protein. Therefore, extraction of the protein with NaCl was investigated (Fig. 5A). Indeed, at 900 mM NaCl, gp20 was efficiently released from the membrane, whereas at 300 mM it was still found in the membrane fraction.
Fig 5.
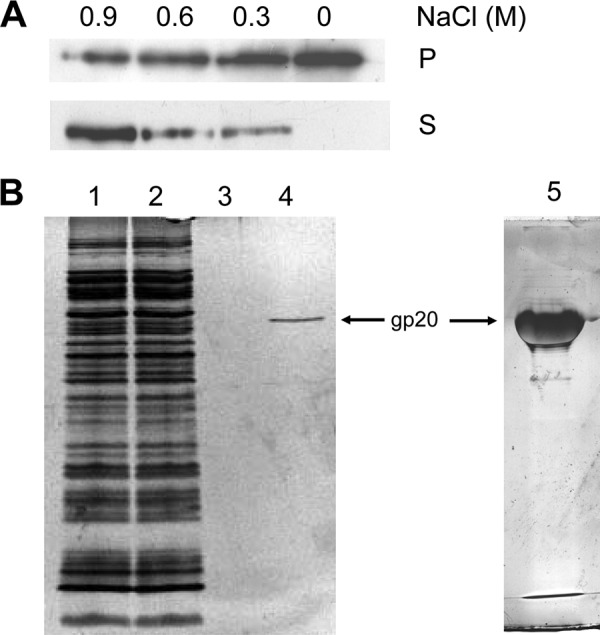
Membrane binding of gp20. The binding is sensitive to 900 mM NaCl (A). Membrane fractions containing His-gp20 were extracted with the indicated NaCl concentrations and collected by centrifugation. Supernatant (S) and pellet fractions (P) were analyzed. (B) Affinity purification of His-gp20 does not show any comigrating protein. Load fraction (lane 1), flowthrough (lane 2), wash (lane 3), and elution (lanes 4 and 5) are shown.
To identify potential partner proteins, His-gp20 was purified by affinity chromatography (Fig. 5B). The apparently pure protein was eluted and analyzed by PAGE. Even on an overloaded silver stained gel (lane 5), no comigrating protein was found. gp20-containing membranes were treated with 0.6% formaldehyde cross-linker for 30 min and affinity purified. The eluted fraction was heated to 95°C for 20 min to reverse the cross-links, and the polypeptides were separated by SDS-PAGE (Fig. 6). The protein bands were excised and analyzed by mass spectrometry. The identified proteins from the cross-linked band are presented in Table 1. The peptide sequences identified the E. coli chaperones Tig, DnaK, and GroEL. Moreover, the only identified integral membrane protein was the YidC membrane insertase. Other contacts were found (e.g., to EF-Tu). Taken together, these results suggest that gp20 may first interact with trigger factor at the ribosome, then with the chaperones DnaK and GroEL and gp40 in the cytoplasm, and finally with YidC at the membrane before the assembly of the head core and shell is initiated.
Fig 6.
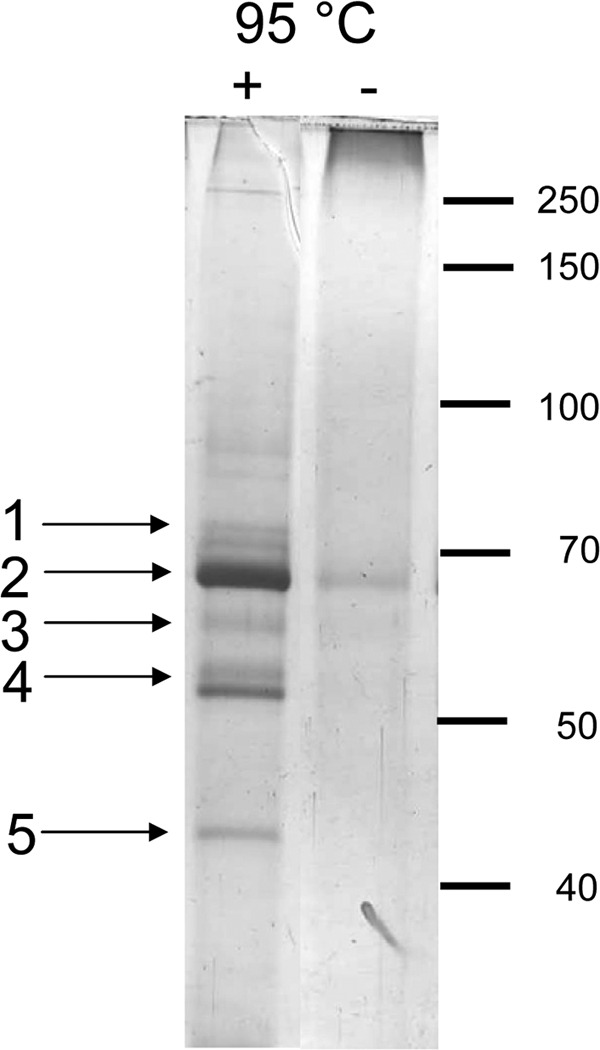
Cross-linking of His-gp20. E. coli BL21 cells expressing His-gp20 were lysed, and the membrane fraction was treated with 0.6% formaldehyde for 30 min. To resolve the cross-links, the sample was incubated at 95°C for 20 min. To identify the protein bands, mass spectrometry was performed and the peptides were identified. 1, DnaK; 2, YidC; 3, GroEL; 4, Tig; 5, EF-Tu.
Table 1.
Identified sequences from the cross-linked peptides
| Band no. | Assigned protein | Peptide sequence |
|---|---|---|
| 1 | DnaK | RDAEANAEADRK |
| KMQELAQVSQKL | ||
| 2 | YidC | RISQEMMALYKA |
| RGGDVEQALLPAYPKE | ||
| 3 | GroEL | RVEDALHATRA |
| KAVTAAVEELKA | ||
| 4 | Tig | KSELVNVAKKV |
| RELFEEQAKR | ||
| 5 | EF-Tu | RAGENVGVLLRG |
| KVGEEVEIVGIKE |
To test whether gp20 and gp20s when anchored at the membrane undergo deep penetration and are exposed to the periplasm, a protease-mapping experiment was conducted (Fig. 7). Cells expressing His-gp20 or His-gp20s, respectively, were converted to spheroplasts. Proteinase K was added to the outside for 1 h, and the proteins were acid precipitated. After solubilization, the gp20 proteins were visualized by Western blotting with the specific gp20 antibody. Both His-gp20 (Fig. 7A) and His-gp20s (Fig. 7B) appeared intact and were not digested by the protease (upper panels, lane 2) in contrast to the outer membrane protein A (OmpA), which was accessible in spheroplasts (lower panels). Therefore, the data do not support a transmembrane topology of gp20 and gp20s. We conclude that both proteins are most likely peripherally bound to the cytoplasmic surface of the inner membrane.
Fig 7.
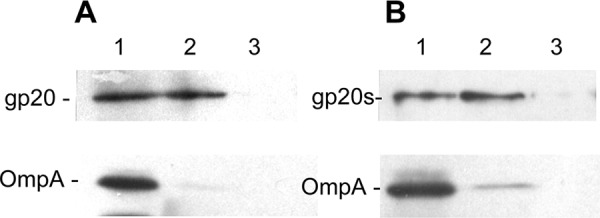
Protease mapping of His-gp20 and His-gp20s. E. coli BL21 cells expressing His-gp20 (A) or His-gp20s (B) were converted to spheroplasts and analyzed on a Western blot (lanes 1). To one aliquot, proteinase K (1 mg/ml) was added for 1 h (lane 2), and for another aliquot, the cells were lysed by 3% Triton X-100 and digested by proteinase K for 1 h (lane 3).
gp20s is incorporated into T4 proheads.
Our experiments have shown that the cellular location of gp20s was similar to that of gp20-GFP (Fig. 3B), suggesting that the defect in am20E481-infected cells occurs after the initiation of prohead assembly. Therefore, T4 am20E481-infected cells were analyzed for prohead formation on a glycerol gradient. Clearly, proheads were found and seemed to contain a partially degraded core structure (Fig. 8A). Most of the proheads presented a neck structure, and some had a filamentous material attached that could represent DNA (Fig. 9). The protein profile of these purified proheads showed that gp23 is in its processed form (Fig. 8B), since its SDS-PAGE migration was identical to that of gp23 from phage (lane 1). gp20s was clearly present in the purified prohead fraction of T4 am20E481-infected cells and in the purified prohead fraction, in contrast to the phage-derived gp20 (lane 5) on a Western blot. The protein found in the purified proheads was clearly smaller (lanes 3 and 4).
Fig 8.

Proheads isolated from T4 am20E481-infected cells. (A) E. coli B cells were infected with T4 amE481, and the lysate was analyzed by electron microscopy. Bar = 100 nm. (B) Phage heads (lanes 1 and 5) and proheads from T4 amE481-infected cells (lanes 2 to 4) were purified, and their proteins were analyzed by SDS-PAGE (lanes 1 and 2) and by Western blotting to gp20 (lanes 3 to 5). The gp23 and IPIII proteins were identified by their molecular weight (MW, in thousands) and by mass spectrometry. To resolve the difference between gp20 and gp20s, the Western blot was made from a high-resolution 40-cm SDS-PAGE. Two different amounts of T4 amE481 proheads were applied in lanes 3 and 4.
Fig 9.

Assembly intermediates accumulating in T4 am20E481-infected cells. A gallery of proheads showing connected neck structures. Bar = 100 nm.
DISCUSSION
The phage T4 head is assembled with a proteinaceous core structure and a icosahedric shell structure of the major head proteins gp23 and gp24 at the vertices. Presumably, the assembly of the core and the shell occurs simultaneously at the membrane (3). However, it has been shown that the assembly of the core and the shell can be dissected (28). Under conditions where the major capsid proteins gp23 (amH11) and gp20 (amE481) were compromised, core structures were found preferentially at the cytoplasmic periphery of the membrane. These cores have a defined size and shape, indicating that they basically determine the prolate capsid architecture of the phage. Clearly, the proximal vertex of these core structures is localized at the membrane. Since gp20 is localized at this vertex, one question was whether gp20 might be involved in the formation of these cores. Indeed, the N-terminal amber mutants in gene 20, E481 and E208, allowed the formation of cores (28). However, the sequencing of the E481 mutation showed that the amber mutation is located in the codon for residue 12, but it also showed that this mutation creates a possible translation start at codon 15 (2) (Fig. 1B). The identical mutation was observed in the E208 mutant of gp20.
The expression of gp20 from wild-type T4 and gp20 from T4 amE481 (termed gp20s) was then studied after cloning the respective genes on plasmids with an N-terminal His tag (Fig. 4A). The proteins were coexpressed with gp40, which was shown earlier to be required as a chaperone (21). We verified that gp20s was of smaller size than the wild-type protein. The plasmid-derived expression of gp20s did not allow T4 am20 or T4 am23 phages to form phage plaques (Fig. 2), clearly demonstrating that the N terminus of gp20 was essential for phage propagation. It has been suggested earlier that the N-terminal region of gp20 is exposed in the proheads but buried in the phage, whereas the C-terminal part is inside the capsid. This suggested that the N-terminal region of gp20 contains the membrane binding activity (2). To determine whether gp20s can normally bind to the membrane surface, we studied gp20 and gp20s as fusion proteins with GFP and followed their fluorescence in whole cells (Fig. 3). We have previously used this method to study the targeting of membrane proteins by the signal recognition particle (18). The plasmid-derived expression of gp20-GFP showed a localization at the membrane, which appeared as dots, often with regular spacing. We obtained a similar result with gp20s-GFP. To verify the membrane localization of gp20 and gp20s, cell fractionation was performed (Fig. 4) showing that gp20 and gp20s were found in the pellet fraction containing membrane proteins such as YidC. Surprisingly, gp40 remained in the supernatant, suggesting that it is released from gp20 after its membrane association. Previously, gp40 had been found in membrane vesicles, although preferentially in the outer membrane (21). Since gp20 lacks a hydrophobic stretch in its sequence, membrane binding might occur by electrostatic interactions with a membrane protein. Indeed, membrane extraction experiments showed that its fractionation with the membrane is sensitive to salt (Fig. 5A). We concluded from these experiments that gp20 is likely to be peripherally bound to a membrane protein and not directly to the membrane surface as had been suggested earlier (21).
To test participation of other proteins in the membrane binding process of gp20, cross-linking with formaldehyde was performed. The cross-linked products were identified by LC-ESI-MS/MS as trigger factor, DnaK, and GroE (Table 1). Since all these proteins have a chaperone function, an interaction with gp20 shortly after its synthesis is plausible. Alternatively, the binding to the chaperones could be due to misfolding of gp20. An interaction between a chaperone and gp20 does not indicate that this is absolutely necessary; it may be only supportive. For trigger factor, it was recently proposed that it has a function in assembling protein complexes (14, 20).
The integral membrane protein YidC was the only membrane protein identified as a cross-linking partner of gp20. YidC is dedicated to the insertion of newly synthesized membrane proteins into the lipid bilayer (16, 24). It is anchored with 6 transmembrane segments in the membrane and has a positively charged C-terminal domain. It is conceivable that YidC binds gp20 as it releases the chaperone gp40, allowing the initiation of the head assembly process of bacteriophage T4.
The N-terminally truncated gp20s that is synthesized in T4 amE481-infected cells normally binds to the membrane surface, as suggested by the GFP fusion proteins and by the fractionation studies with His-gp20s. Both proteins gp20 and gp20s bind to YidC as determined by several YidC-derived peptides in the cross-linked complexes. Interestingly, T4 infection by a double mutant of gp20amE481 and gp23amH11 allowed the assembly of core structures at the membrane surface (28). This clearly indicates that gp20s is still functional to initiate core assembly and therefore is blocked in a later step in the assembly pathway. Also, proheads were found in T4 amE481 infections (Fig. 8), consistent with the idea that shell assembly is not disturbed by gp20s. The proheads display an angular appearance, with a processed major head protein gp23 and with a digested core structure most likely resembling prohead II, which have previously been found in T4 cold-sensitive mutants defective in gene 20 (15) and are similar to the empty small particles (ESPs) found in am17 infections (5). The N-terminal portion of gp20 is located on the outer face of the head (11) that might contact gp17, the packaging motor (23, 31). Therefore, binding of gp17 to gp20s might be disturbed, as it lacks the first 14 residues. Another possibility is that the detached proheads bind to gp13/14 prematurely and thereby prevent DNA packaging. Taken together, our data suggest that the block in T4 amE481 infections takes place after the assembly of proheads, presumably at the initiation of the DNA packaging process.
Previously, it has been reported that in T4 amE481 infections about one-third of the proheads were found free in the cytoplasm (28). The authors concluded from this that the membrane binding of these proheads is disturbed. Our data presented here do not support this. Rather, membrane binding, processing, and detachment of the proheads from the membrane occur normally in T4 amE481-infected cells. This would also result in a fraction of detached proheads. The assembly block is more likely to occur at the step of the DNA packaging of the processed prohead, resulting in the accumulation of proheads in the cytoplasm of the infected cells. These proheads appear with unusual neck structures (Fig. 9) that might point to the specific block in the T4 head assembly pathway that the gp20s mutant causes.
ACKNOWLEDGMENTS
We thank Lindsay Black for his generous support providing plasmids and antisera and Stephan Nussberger for his help with the electron microscopy. Technical assistance by Gisela Nagler is greatly acknowledged.
Footnotes
Published ahead of print 1 August 2012
REFERENCES
- 1. Appleyard RK, McGregor JF, Baird KM. 1956. Mutation to extended host range and the occurrence of phenotypic mixing in the temperate coliphage lambda. Virology 2:565–574 [DOI] [PubMed] [Google Scholar]
- 2. Baumann RG, Mullaney J, Black LW. 2006. Portal fusion protein constraints on function in DNA packaging of bacteriophage T4. Mol. Microbiol. 61:16–32 [DOI] [PubMed] [Google Scholar]
- 3. Black LW, Showe MK, Steven AC. 1994. Morphogenesis of the T4 head, 218–258 In Karam JD. (ed), Molecular biology of bacteriophage T4. ASM Press, Washington, DC [Google Scholar]
- 4. Brown SM, Eiserling FA. 1979. T4 gene 40 mutants II. Phenotypic properties. Virology 97:77–89 [DOI] [PubMed] [Google Scholar]
- 5. Carrascosa JL, Kellenberger E. 1978. Head maturation pathway of bacteriophage T4 and T2. III. Isolation and characterization of particles produced by mutants in gene 17. J. Virol. 25:831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chevallet M, et al. 2008. Sweet silver: a formaldehyde-free silver staining using aldoses as developing agents, with enhanced compatibility with mass spectrometry. Proteomics 8:4853–4861 [DOI] [PubMed] [Google Scholar]
- 7. Coombs DH, Arisaka F. 1994. T4 tail structure and function, p 259–281 In Karam JD. (ed), Molecular biology of bacteriophage T4. ASM Press, Washington, DC [Google Scholar]
- 8. Demerec M, Fano U. 1945. Bacteriophage-resistant mutants in Escherichia coli. Genetics 30:119–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Driedonks RA, Caldentey J. 1983. Gene 20 product of bacteriophage T4 II. Its structure organization in prehead and bacteriophage. J. Mol. Biol. 166:341–360 [DOI] [PubMed] [Google Scholar]
- 10. Driedonks RA, Engel A, ten Heggeler B, van Driel R. 1981. Gene 20 product of bacteriophage T4. Its purification and structure. J. Mol. Biol. 152:641–662 [DOI] [PubMed] [Google Scholar]
- 11. Fokine A, et al. 2004. Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. U. S. A. 101:6003–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herzberg CL, et al. 2007. SPINE: a method for the rapid detection and analysis of protein-protein interactions in vivo. Proteomics 7:4032–4035 [DOI] [PubMed] [Google Scholar]
- 13. Hintermann E, Kuhn A. 1992. Bacteriophage T4 gene 21 encodes two proteins essential for phage maturation. Virology 189:474–482 [DOI] [PubMed] [Google Scholar]
- 14. Hoffmann A, Bukau B. 2009. Trigger factor finds new jobs and contacts. Nat. Struct. Mol. Biol. 16:1006–1008 [DOI] [PubMed] [Google Scholar]
- 15. Hsiao LC, Black LW. 1977. DNA packaging and pathway of bacteriophage T4 head assembly. Proc. Natl. Acad. Sci. U. S. A. 74:3652–3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuhn A, Stuart R, Henry R, Dalbey RE. 2003. The Alb3/OxaI/YidC protein family: membrane-localized chaperones facilitating membrane protein insertion? Trends Cell Biol. 13:510–516 [DOI] [PubMed] [Google Scholar]
- 17. Leiman PG, Chipman PR, Kostyuchenko VA, Mezyanzhinov VV, Rossmann MG. 2004. Three-dimensional rearrangement of protein in the tail of bacteriophage T4 on infection of its host. Cell 118:419–429 [DOI] [PubMed] [Google Scholar]
- 18. Maier K, et al. 2008. An amphiphilic region in the cytoplasmic domain of KdpD is recognized by SRP and targeted to the E. coli membrane. Mol. Microbiol. 68:1471–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maniatis T, Fritsch EF, Sambrook J. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 20. Martinez-Hackert E, Hendrickson WA. 2009. Promiscuous substrate recognition in folding and assembly activities of the trigger factor chaperone. Cell 138:923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Michaud G, Zachary A, Rao VB, Black LW. 1989. Membrane-associated assembly of phage T4 DNA entrance vertex structure studied with expression vectors. J. Mol. Biol. 209:667–681 [DOI] [PubMed] [Google Scholar]
- 22. Onorato L, Showe MK. 1975. Gene 21 protein-dependent proteolysis in vitro of purified gene 22 product of bacteriophage T4. J. Mol. Biol. 92:395–412 [DOI] [PubMed] [Google Scholar]
- 23. Rao VB, Black LW. 2010. Structure and assembly of bacteriophage T4 head. Virol. J. 7:356 doi:10.1186/1743-422X-7-356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Samuelson JC, et al. 2000. YidC mediates membrane protein insertion in bacteria. Nature 406:637–641 [DOI] [PubMed] [Google Scholar]
- 25. Showe MK, Isobe E, Onorato L. 1976. Bacteriophage T4 prehead proteinase I. Purification and properties of a bacteriophage enzyme which cleaves the capsid precursor proteins. J. Mol. Biol. 107:35–54 [DOI] [PubMed] [Google Scholar]
- 26. Studier FW, Moffat BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130 [DOI] [PubMed] [Google Scholar]
- 27. Suttle CA. 2005. Viruses in the sea. Nature 437:356–361 [DOI] [PubMed] [Google Scholar]
- 28. Traub F, Maeder M. 1984. Formation of the prohead core of bacteriophage T4 in vivo. J. Virol. 49:892–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wood WB. 1980. Bacteriophage T4 morphogenesis as a model for assembly of subcellular structure. Q. Rev. Biol. 55:353–367 [DOI] [PubMed] [Google Scholar]
- 30. Wood WB, Eiserling FA, Crowther RA. 1994. Long tail fibers: genes, proteins, structure, and assembly, p 282–290 In Karam JD. (ed), Molecular biology of bacteriophage T4. ASM Press, Washington, DC [Google Scholar]
- 31. Zhang Z, et al. 2011. A promiscuous DNA packaging machine from bacteriophage T4. PLoS Biol. 9:e1000592 doi:10.1371/journal.pbio.1000592 [DOI] [PMC free article] [PubMed] [Google Scholar]