

Abstract
Arenaviruses are responsible for acute hemorrhagic fevers with high mortality and pose significant threats to public health and biodefense. These enveloped negative-sense RNA viruses replicate in the cell cytoplasm and express four proteins. To better understand how these proteins insinuate themselves into cellular processes to orchestrate productive viral replication, we have identified and characterized novel cytosolic structures involved in arenavirus replication and transcription. In cells infected with the nonpathogenic Tacaribe virus or the attenuated Candid#1 strain of Junín virus, we find that newly synthesized viral RNAs localize to cytosolic puncta containing the nucleoprotein (N) of the virus. Density gradient centrifugation studies reveal that these replication-transcription complexes (RTCs) are associated with cellular membranes and contain full-length genomic- and antigenomic-sense RNAs. Viral mRNAs segregate at a higher buoyant density and are likewise scant in immunopurified RTCs, consistent with their translation on bulk cellular ribosomes. In addition, confocal microscopy analysis reveals that RTCs contain the lipid phosphatidylinositol-4-phosphate and proteins involved in cellular mRNA metabolism, including the large and small ribosomal subunit proteins L10a and S6, the stress granule protein G3BP1, and a subset of translation initiation factors. Elucidating the structure and function of RTCs will enhance our understanding of virus-cell interactions that promote arenavirus replication and mitigate against host cell immunity. This knowledge may lead to novel intervention strategies to limit viral virulence and pathogenesis.
INTRODUCTION
Viruses are wholly reliant on the host cell for replication. Viral proteins must access cellular systems to establish a productive environment for generating progeny virions while also evading the innate antiviral response. Viruses have therefore evolved diverse strategies and multifunctional proteins to coopt the host cell infrastructure. Elucidating these virus-cell interactions may suggest novel targets for antiviral intervention.
Arenaviruses can cause acute hemorrhagic fevers with high mortality (34, 43). These viruses are endemic in rodent populations worldwide (47) and can be transmitted to humans by contact. Prototype arenavirus species include Lassa fever virus (LASV) in western Africa and Junín virus (JUNV) in the Pampas region of Argentina. These and other hemorrhagic fever arenaviruses pose ongoing threats to public health and raise concerns for biodefense planners. In the absence of licensed vaccines and specific antiviral therapies, these viruses are recognized as NIH category A priority pathogens (39). A critical understanding of the interactions of arenaviral proteins with the host cell will provide insight toward the development of effective therapies against arenavirus hemorrhagic fevers.
The arenaviruses are a diverse family of enveloped negative-sense RNA viruses that replicate entirely in the cell cytoplasm (9). The S and L RNA segments of the bipartite arenavirus genome each encode two proteins using an ambisense coding strategy (Fig. 1). The essential replicative proteins (27), the RNA-dependent RNA polymerase (L) and nucleoprotein (N), are present in the infecting virion and subsequently produced from mRNAs transcribed from the two genomic-sense viral RNAs (L and S, respectively). Progeny genomes are replicated from newly synthesized antigenomic-sense RNAs that also serve as the templates for mRNAs encoding the matrix (Z) and envelope (GPC) proteins, which, respectively, promote virus budding from the cell surface and entry by the progeny virion into the new host cell. Full-length genomic- and antigenomic-sense RNAs are replicated using a “prime and align” strategy, whereas mRNA transcription is initiated by short m7G-capped oligonucleotides derived from cellular mRNAs (15). Viral mRNAs terminate within intergenic hairpin regions (IGRs) that separate the two opposite-sense open reading frames on the genomic RNAs (Fig. 1) and are not polyadenylated (23). Based on a growing body of work describing the cytoplasmic life cycles of a number of RNA viruses (14, 21, 22, 25, 26, 38), arenavirus replication is likely to be compartmentalized to specialized virus-induced organelles referred to as replication-transcription complexes (RTCs).
Fig 1.

Arenavirus gene organization and expression. The top line depicts the S segment of the bisegmented TCRV genome, and the L segment (encoding Z and L) is similarly organized. GPC and N coding sequences and the intervening intergenic region (IGR) are shown. The TCRV IGR is thought to form two hairpin loops (23). Arrows represent L-mediated replication and transcription, and antigenomic-sense sequences are shown in gray. mRNAs contain a 5′ m7G cap and terminate heterogeneously within the IGR (3′ termini are arbitrarily depicted in the drawing) (23). Brackets on the full-length RNAs indicate the approximate location of the strand-specific RNA hybridization probes. The drawing is not to scale.
As a starting point for understanding the viral and cellular requirements for arenavirus replication, we have determined the intracellular localization and composition of RTCs generated during infection by the attenuated vaccine strain of JUNV (Candid#1) (31) and a closely related nonpathogenic species, Tacaribe virus (TCRV). We find that viral RNA synthesis takes place in discrete cytosolic puncta whose formation is induced in part by the virus nucleoprotein (N). Based on their low buoyant density and sensitivity to nonionic detergent, we suggest that RTCs also include a membrane component. In addition, phosphatidylinositol-4-phosphate (PI4P) was found to colocalize with RTCs and may play a role in defining this specialized membrane structure. We also find that genomic and antigenomic viral RNAs associate with N-containing RTCs in flotation and immunoprecipitation studies, whereas viral mRNAs were largely excluded, consistent with export to cellular ribosomes for translation. In keeping with the characteristic inclusion of host ribosomes in arenavirus particles, RTCs were also found to contain the ribosomal proteins L10a and S6, as well as the translation initiation factors eIF4G and eIF4A. Colocalization of the stress granule (SG) protein G3BP1 in RTCs points to additional interactions with the mRNA metabolism machinery of the cell. Collectively, these observations provide an initial description of the unique assortment of cellular factors assembled at the cytosolic sites of arenavirus replication.
MATERIALS AND METHODS
Cells, viruses, and molecular clones.
The attenuated strain of JUNV (Candid#1) (31) was kindly provided by Robert Tesh (WHO Reference Center for Arboviruses, University of Texas Medical Branch) and the Tacaribe virus (TRVL 11573) by Sean Amberg (6) (SIGA Technologies) (6). Both viruses were propagated in Vero cells grown in Dulbecco's modified Eagle medium (Life Technologies) supplemented with 10% fetal bovine serum. The molecularly cloned nucleoprotein gene of the pathogenic MC2 strain of JUNV (16) was obtained from Victor Romanowski (Universidad Nacional de la Plata, Argentina) and expressed in Vero cells by Lipofectamine 2000 (Life Technologies)-mediated transfection using a pCAGGS-based vector (37). A plasmid containing mouse β-actin cDNA (Ambion) was used to generate an RNA probe for blot hybridization.
Antibodies.
Monoclonal antibodies directed to JUNV N (AG12, BE06, AF08, BF08, and AA10) (48) were obtained through the NIAID Biodefense and Emerging Infections Research Resources Repository. Commercial antibody reagents directed to the following cellular markers were used in confocal microscopy to assess colocalization with N protein-containing puncta: Dcp1a (ab57654), Giantin (ab37266), Rab5 (ab13253), Rab 7 (ab77993), and TIA-1 (ab2712) from Abcam (Cambridge, MA); EEA1 (610457), GBF (612116), and Rab 4 (610889) from BD Biosciences (San Jose, CA); Giantin (PRB-114C) from Covance (Princeton, NJ); PI4P (Z-004) from Echelon (Salt Lake City, UT); eIF2α (ADI-KAP-CP130) from Enzo Life Sciences (Farmingdale, NY); GM130 (1837-1) and tubulin (2971-1) from Epitomics (Burlingame, CA); PDI (S34200), mitochondrial ATP synthetase (A21355), and TfR1 (13-6800) from Life Technologies (Carlsbad, CA); sec16 (NB100-1799) from Novus Biologicals (Littleton, CO); and COX1 (sc-58347), clathrin (sc-12734), eIF3 (sc-16377, sc-74507, sc-16376), eIF4G (sc-136142, sc-11373, sc-100730), eIF4A (sc-137147) and eIF4E (sc-9976, sc-13963), G3BP1 (sc-81940), HSP70 (sc-59571), PABP (sc-166027), RPL10a (sc-100827), RPS6 (sc-74459), sec61α (sc-12322), TGN38 (sc-166224), tubulin (sc-5286), and Xrn1 (sc-165985) from Santa Cruz Biotechnology (CA). Appropriate Alexa Fluor 488 F(ab′)2 secondary antibodies were used for detection, and colocalization by murine monoclonal antibodies (MAbs) was confirmed using a DyLight 488 Fab′ secondary antibody (Jackson ImmunoResearch). Marker-specific labeling of the cells was judged by comparison with vendor and published images. Intracellular membranes were stained using BODIPY TR methyl ester (Invitrogen). The autophagy marker LC3 (microtubule-associated protein 1 light chain 3) was detected by transient expression as an enhanced green fluorescent protein (EGFP) fusion protein (pEGFP-LC3; Addgene).
Confocal microscopy.
Vero cells were infected with TCRV or Candid#1 using an inoculum predetermined to infect ∼20% of the cells under the conditions of the experiment. Typically, cells were incubated with virus at 37°C for 8 to 12 h and subsequently washed. In some experiments, contact with the inoculum was limited to 45 min in order to produce a pseudosynchronous infection. Cells were then resuspended with trypsin and plated onto CELLview glass bottom dishes (Greiner Bio One, Monroe, NC) pretreated with poly-d-lysine. For detecting newly synthesized RNA, cells were metabolically labeled for 3 h in medium containing 2 mM ethynyl uridine (EU; Life Technologies). In most experiments, cultures were treated with 2 μM actinomycin D (ActD; Sigma) for 30 min prior to and during EU labeling to suppress cellular RNA synthesis. At appropriate times, cultures were fixed with −20°C methanol, air dried, and rehydrated in phosphate-buffered saline (PBS) containing 0.1% Triton X-100. Following two washes in PBS, cells were incubated with Alexa Fluor 488 azide in Click-iT buffer (Life Technologies) for 30 min in the dark. Cultures were washed sequentially using PBS, PBS containing 3% bovine serum albumin (PBS-BSA), and PBS-BSA containing 0.1% Triton X-100. N protein was subsequently labeled using Alexa Fluor 568-conjugated MAb AG12 (labeling kit; Life Technologies). Following three washes in PBS containing 0.1% Triton X-100, coverslips were covered with SlowFade Gold reagent DAPI (4′,6-diamidino-2-phenylindole; Life Technologies) for viewing.
For confocal microscopy of cellular markers, cells were fixed with PBS-buffered ice-cold 4% paraformaldehyde (Electron Microscopy Sciences) for 1 h and processed as described above, with the exception that those stained with Bodipy TR methyl ester (Life Technologies) were permeabilized using digitonin (1) to retain membrane labeling. In all the experiments shown, the monovalent DyLight 488-conjugated Fab′ fragment was used to detect murine MAbs directed to cellular markers. The majority of confocal images were acquired using a Zeiss LSM 710 microscope (×63 and ×100 objectives) and ZEN software, kindly provided by Sonja Best (NIH Rocky Mountain Labs, Hamilton, MT), or an Olympus FluoView 1000 microscope (×60 objective) and FluoView software (University of Montana Molecular Histology and Fluorescence Imaging Core). Colocalization of cellular components with discrete N puncta was determined by the coincidence of localized peaks of green and red fluorescence along linear transects of confocal images (29). Statistical tools for colocalization are insensitive to correlations between discrete puncta and more diffuse staining patterns (7), as is the case with many of the cellular markers.
Immunopurification of RTCs.
TCRV-infected Vero cells were harvested by scraping and resuspended in diethylpyrocarbonate (DEPC)-treated homogenization buffer containing 50 mM MOPS (morpholinepropanesulfonic acid; pH 7.1), 150 mM NaCl, and 2 mM MgCl2, to which the following were added: 2 mM dithiothreitol (DTT), 1 μg/ml each of the protease inhibitors aprotinin, leupeptin, and pepstatin, 400 U/ml RNaseOUT (Life Technologies), and 250 U/ml SUPERase·In (Life Technologies). Cells were disrupted on ice using a Dounce homogenizer and tight pestle (50 strokes). In other experiments, the homogenization buffer was first made to 1% Nonidet P-40 (NP-40) (lysis buffer) and TCRV-infected cells were incubated for 30 min prior to Dounce homogenization. Centrifugation was omitted, as N-containing particles were found to pellet even at low speeds. Both homogenization (no NP-40) and lysis (with NP-40) buffers contained 500 μg/ml heparin sulfate (Sigma).
N-containing material was immunopurified using protein G Dynabeads (Life Technologies) and a mixture of four N-directed MAbs (BE06, AF08, BF08, and AA10) (48) belonging to distinct complementation groups (J. York and J. H. Nunberg, unpublished data). Beads were pretreated with 0.1 M NaOH to minimize RNase activity (as recommended for streptavidin Dynabeads by the vendor), and MAbs were loaded in PBS containing 0.01% Tween 20. Excess MAb was removed after binding. Cell homogenates and lysates were incubated with the loaded beads for 2 h at 4°C, and bound and supernatant fractions were separated magnetically. Beads were washed extensively in the appropriate buffer prior to SDS-PAGE analysis (NuPAGE 4 to 12% Bis-Tris; Life Technologies), whereas input and supernatant fractions were concentrated by precipitation in 10% trichloroacetic acid and subsequently dissolved in SDS-PAGE sample buffer containing 2 mM MgCl2 and 0.1U/μl Benzonase (Novagen). Resolved proteins were transferred to a polyvinylidene difluoride (PVDF) membrane and probed for N using MAb AG12, with a secondary horseradish peroxidase-conjugated antibody and ECL Plus chemiluminescence reagent (Amersham). Western blots were quantitated using a Fuji FLA-3000G imager and Image Gauge software.
RNA was isolated directly from washed Dynabeads or from aqueous fractions using TRIzol LS (Life Technologies) by following the manufacturer's instructions and resuspended in DEPC-treated buffer containing 10 mM MOPS (pH 7.1), 1 mM EDTA, and 1 U/μl SUPERase·In.
OptiPrep density gradient analysis.
TCRV-infected Vero cells were disrupted by Dounce homogenization in an intracellular buffer (modified buffer B) (33) comprising 38 mM (each) the K+ salts of aspartic acid, glutamic acid, and gluconic acid, 20 mM MOPS (pH 7.1), 10 mM KHCO3, 2 mM MgCl2, 2 mM DTT, 10 μM ZnCl2, 1 μg/ml each of the protease inhibitors pepstatin, aprotinin, and leupeptin, 400 U/ml RNaseOUT, and 250 U/ml SUPERase·In. Alternatively, solubilized cell lysates were prepared using modified buffer B containing 2% NP-40 (lysis buffer). In order to abrogate the unexpected divalent cation-dependent RNase activity detected during equilibrium centrifugation (see Results), MgCl2 was replaced by 5 mM EDTA in some experiments (see Fig. 4B and D).
Fig 4.

OptiPrep density gradient analysis of RTCs. TCRV-infected cells were disrupted by Dounce homogenization (A and B) or solubilized using 1% NP-40 (C and D), and the mixtures were subjected to buoyant density centrifugation through 15 to 48% OptiPrep gradients (fraction numbers and a density graphic are shown at the bottom). Materials in panels A and C were prepared in buffers containing Mg2+, and those in panels B and D were prepared in EDTA (see Discussion). The histograms display the relative concentrations of N as determined by ELISA. The buoyant densities (ρ) of peak fractions of N are shown. Northern blot images below demonstrate the distributions of genomic RNA and GPC mRNA, antigenomic RNA and N mRNA, and cellular actin mRNA.
Cell homogenates and lysates were made to 50% OptiPrep (iodixanol) using a 60% (wt/vol) stock solution (Sigma), placed in Ultra-Clear centrifuge tubes (Beckman) and overlaid with a continuous 15 to 48% OptiPrep gradient prepared in modified buffer B. Samples were centrifuged to equilibrium in a Beckman MLS-50 rotor (100,000 × g for 20 h at 4°C), and 12 fractions were collected manually from the top. Densities were determined by refractometry and an empirically determined standard curve. N protein was quantitated by sandwich enzyme-linked immunosorbent assay (ELISA) using MAb AG12 for capture and biotinylated (EZ-Link Sulfo-NHS-LC-Biotin; Life Technologies) MAb AA10 for detection. RNA was extracted from gradient fractions using TRIzol LS (Life Technologies). Heparin sulfate could not be used for density separations because it rendered all forms of ribonucleoprotein complexes (RNPs) equally buoyant (∼1.16 g/cm3).
Strand-specific RNA probes.
A 524-nucleotide (nt) segment of the S segment of TCRV genomic RNA, including the intergenic region (IGR) and spanning the 3′-terminal sequences of the opposite-sense N and GPC mRNAs (Fig. 1) (nucleotides 1321 to 1844, accession number NC_004293), was amplified from infected Vero cells using the SuperScript III CellsDirect cDNA synthesis kit (Life Technologies) and Platinum Taq DNA polymerase (Life Technologies). The resulting PCR product was inserted into the pGEM-7Zf(+) vector (Promega) using restriction sites introduced via the PCR primers. Strand-specific RNA probes were synthesized using a MAXIscript kit (Ambion) with biotinylated UTP (Bio-16-UTP; Ambion) and either T7 or SP6 RNA polymerase.
RNAs isolated by immunopurification or density gradient centrifugation were analyzed by blotting using the NorthernMax kit (Ambion). RNAs were denatured with paraformaldehyde and resolved by agarose gel electrophoresis, transferred to a BrightStar membrane, and hybridized with the strand-specific RNA probes according to the kit instructions. Hybridization was detected using BrightStar BioDetect (Ambion) and a Fuji LAS imager.
RESULTS
Colocalization of newly synthesized arenavirus RNA with N in cytoplasmic puncta.
In order to investigate the intracellular sites of arenavirus replication, we made use of two New World clade B arenaviruses (11) that can be grown in a biosafety level 2 laboratory: the attenuated Candid#1 vaccine strain of JUNV and the closely related nonpathogenic Tacaribe virus (TCRV). Vero cells infected with either virus were fixed and permeabilized 24 h after infection and stained for confocal immunofluorescence microscopy using an Alexa 568-conjugated MAb directed to JUNV N (MAb AG12) (48). N was found to accumulate in cytosolic puncta that varied in size, number, and granularity in the infected cells (Fig. 2A, shown in red). The time course for development of these punctate structures was examined throughout a single infection cycle. N-containing puncta were typically first observed by 9 h postinfection (hpi) and were small and numerous by 12 hpi (Fig. 2B, shown in red). The centers appeared to coalesce into larger (1- to 5-μm) clusters by 24 hpi, and the secondary infection of neighboring cells was detected. The time course observed for puncta formation is consistent with the ∼24-h eclipse period of virus replication (York and Nunberg, unpublished).
Fig 2.

Cytoplasmic sites of arenavirus replication. Newly synthesized viral RNA was visualized for confocal microscopy using click chemistry (Click-iT; Life Technologies). Cells were metabolically labeled for 3 h with ethynyl uridine (EU), and incorporation was detected with Alexa Fluor 488 azide (green). Actinomycin D (ActD) was added to minimize EU uptake into cellular RNA. The N protein of TCRV and JUNV was detected using the directly conjugated Alexa Fluor 568 MAb AG12 (red) (48). Sites of EU labeling without corresponding N likely represent cellular RNAs that escape the ActD block. (A) TCRV and Candid#1 infection induces RTCs. Vero cells were infected with TCRV or Candid#1 (Cand) and metabolically labeled in the presence (+) or absence (no) of ActD (24 hpi). RTCs are defined by colocalization of EU-labeled RNA (green) and N protein (red). The merged image is shown on the right. (B) Time course of RTC formation. TCRV-infected Vero cells were metabolically labeled with EU in the presence of ActD and fixed with 4% paraformaldehyde at the indicated times after infection. An enlarged image of a TCRV-infected cell (12 hpi) is shown with flanking orthogonal views to demonstrate colocalization in the z direction. All images were taken under identical conditions, and mature puncta vary in size from 1 to 5 μm, as judged by linear traces shown in Fig. 5. (C) Recombinant N induces formation of cytosolic puncta. Vero cells were transfected to ectopically express JUNV N, and N-containing puncta were visualized. These puncta were often larger than those induced during virus infection and do not bind newly synthesized cellular RNAs nonspecifically (no ActD).
Puncta were also observed in Vero cells transfected with a plasmid expressing JUNV N (Fig. 2C, red). This observation suggests that the formation of punctate structures represents an intrinsic property of N, either through its inherent ability to self-aggregate or in conjunction with cellular protein(s).
Intracellular sites of viral RNA replication were detected by metabolically labeling cells for 3 h using ethynyl uridine (EU). Newly synthesized RNA was subsequently visualized for confocal microscopy using an azide-conjugated Alexa 488 fluorophore and click chemistry (5). In most studies, cellular RNA synthesis was suppressed by the addition of 2 μM actinomycin D (ActD) for 30 min prior to and during EU uptake. Short-term ActD treatment had no effect on infection and did not alter the appearance of N puncta (not shown) (30). The pattern of EU labeling in TCRV- and Candid#1-infected cells is shown in green in Fig. 2A. Inspection of red N puncta revealed a coincident accumulation of EU; in many cases, discrete EU puncta could be discerned in the green, and these invariably colocalized with foci of N protein. Diffuse EU staining present in all cells likely reflects cellular RNAs that escape the ActD block. Moreover, the localized accumulation of EU followed the same time course as the development of N puncta in TCRV-infected cells (Fig. 2B). EU-stained puncta could also be discerned in the absence of ActD treatment, albeit overlaid with a higher background of cellular RNA synthesis (Fig. 2A). The orthogonal views of infected cells demonstrated that EU was concentrated at the center of the punctate structure (Fig. 2B).
Because a primary function of N is to bind RNA, it was possible that nonspecific binding of residual cellular RNA might account for the localized accumulation of EU in infected cells. Therefore, we performed similar EU-labeling studies in Vero cells transiently expressing recombinant JUNV N. Even in the absence of ActD (Fig. 2C), EU staining in these cells was diffuse and did not colocalize with N-containing puncta. We conclude that the focal accumulation of EU with N in infected cells is from newly synthesized viral RNA and therefore that these puncta represent authentic arenavirus RTCs.
Isolated RTCs contain viral RNAs.
In order to identify viral RNA species associated with RTCs, we isolated N-containing ribonucleoprotein complexes (RNPs) from TCRV-infected cells by magnetic-bead immunopurification using N-directed MAbs (48). Pilot studies suggested that RTCs are more efficiently captured by N-directed MAbs from nonionic-detergent cell lysates than from cell homogenates produced without detergents (Fig. 3A and B, top panel). Therefore, we isolated RTCs in the presence of 1% NP-40 and determined the distribution of viral RNAs by Northern blot analysis using strand-specific RNA probes that span the IGR and include the 3′ termini of the N and GPC mRNAs. Full-length genomic and antigenomic RNAs, as well as GPC and N mRNAs of the respective senses, could thus be distinguished (Fig. 3A, bottom panels). We found that only the full-length genomic and antigenomic RNAs were associated with immunopurified N. Viral mRNAs were largely in the supernatant. A cellular mRNA control (actin) was likewise found only in the supernatant fraction.
Fig 3.

Immunopurified RTCs contain predominantly full-length viral RNAs. (A) TCRV-infected Vero cells were solubilized in the presence of 1% NP-40 (+ NP40) and N-containing material was collected using protein G magnetic Dynabeads bearing a mixture of four N-directed MAbs. Equal aliquots of the total cell lysate (Total), the immunopurified protein (Bead), and the supernatant fraction (Supt) were analyzed by immunoblotting to determine the efficiency of N protein collection (top panel). The MAb heavy chains (HC) are coincidentally detected via the secondary antibody. RNA was extracted and subjected to Northern blot analysis using strand-specific RNA probes (bottom panels). (B) TCRV-infected cells were disrupted by Dounce homogenization (no NP-40) prior to immunopurification in the continued absence of nonionic detergent. Fractions were analyzed as described for panel A.
To examine the possibility that viral mRNAs are extracted from RTCs during solubilization in detergent, we performed similar immunopurification studies using cell homogenates (Fig. 3B). We found that full-length and messenger viral RNAs retained their asymmetrical distribution in the absence of nonionic detergent. If viral mRNAs are inadvertently lost from RTCs during purification, this loss is not due to detergent solubilization. These results indicate that RTCs contain predominately genomic and antigenomic RNAs and suggest that transcribed viral mRNAs may be rapidly transported from RTCs, as has been described for vesicular stomatitis virus (VSV) mRNAs (21).
Membrane association of arenavirus RTCs.
Enveloped and nonenveloped viruses frequently make use of cellular membranes to support efficient replication (12, 41). To investigate whether arenaviruses utilize an analogous strategy, we determined the buoyant density of N-containing RTCs. Typically, enveloped viruses float at ∼1.1 g/cm3 in OptiPrep (iodixanol) density medium while nonenveloped viruses band at ∼1.2 g/cm3 (2). In our experiments, homogenates of TCRV-infected cells prepared in the absence of detergent were made to 50% OptiPrep and overlaid with a 15 to 48% gradient of the density medium. On equilibrium centrifugation, soluble proteins are retained at the bottom of the tube while membrane-associated structures and nonmembranous RNPs float to their respective densities. Fractions were then collected and assayed for N protein content by ELISA. As depicted in Fig. 4A (top panel), the peak concentration of N was found at ∼1.13 g/cm3, suggesting that the majority of N is associated with membrane.
We then examined the distribution of viral RNAs. Full-length genomic and antigenomic RNAs were found to coincide with membrane-bound N protein (Fig. 4A). In contrast, the N mRNA was skewed to a higher density and peaked at ∼1.21 g/cm3, similar to that of cellular actin mRNA. Thus, the RNP form of N mRNA is physically distinct from membrane-associated RTCs bearing full-length viral RNAs, consistent with our findings from immunopurification studies (Fig. 3). The buoyant density of GPC mRNA was similar to that of the membrane-bound N protein, likely due to its association with rough endoplasmic reticulum (rER).
Upon solubilization of the infected cells in NP-40 (Fig. 4C), the densities of N protein and full-length viral RNAs increased to ∼1.18 g/cm3, consistent with nonmembranous RNPs. Both N and GPC mRNAs now coincided with full-length viral RNAs and with actin mRNA. Based on the low buoyant density of RTCs and their sensitivity to nonionic detergent, we conclude that these structures are likely associated with cellular lipids or membranes.
In these gradients, we noted an unexpected loss of RNA due to degradation when lysates were prepared in the presence of NP-40, despite the addition of commercial RNase inhibitors. Fractions prepared from solubilized cell lysates also revealed novel RNA species that were larger than GPC mRNA and smaller than N mRNA (Fig. 4C, bottom panels). This pattern is consistent with the endonucleolytic cleavage of the full-length viral RNA at the IGR hairpin (Fig. 1). For instance, the cleavage of the genomic strand would generate one species coincident with GPC mRNA and another species similar in size but opposite in sense to the larger N mRNA. This cleavage was not seen in immunopurification studies, possibly due to the relatively short incubation period. The biological significance of this RNase activity is unclear.
Fortuitously, this RNase activity was found to be dependent on divalent cations. Therefore, in the presence of EDTA, we could determine the distribution of viral RNAs in the absence of extraneous cleavage. EDTA did not affect RTC immunopurification (not shown) and had only a minimal effect on buoyant densities, suggesting that RNP structure is not disrupted. In these studies, we confirmed the association of full-length RNAs with membrane-associated N protein, as well as the physical separation of mRNA-containing RNPs (Fig. 4B). Under these conditions, RNPs containing GPC mRNA are released from the rER (53), allowing for clear separation of N and GPC mRNAs from the membrane-associated RTCs, demonstrating again the relative exclusion of viral mRNAs from RTCs. In the presence of EDTA and nonionic detergent (Fig. 4D), all RNA species formed dense, nonmembranous RNPs. The difference in density between viral and cellular mRNAs relative to viral genomic RNAs observed in Fig. 4D may reflect differential effects of divalent cation extraction on RNPs comprising cellular versus viral nucleoproteins. Collectively, our findings indicate that RTCs associate with cellular lipids or membranes. The dearth of viral mRNAs in RTCs suggests that newly synthesized mRNAs are likely exported to cellular ribosomes for translation (21).
RTCs are enriched in PI4P.
The phosphatidylinositide PI4P is typically found on the cytoplasmic leaflet of Golgi membranes, where it promotes recruitment of cellular proteins required for vesicular budding and membrane dynamics (17). Recently, PI4P and its associated PI4 kinases have also been implicated in membrane remodeling events associated with replication sites of picornaviruses and hepatitis C virus (HCV) (18, 22, 45). To determine whether PI4P might be involved in arenavirus RTCs, we examined its intracellular localization in Candid#1-infected Vero cells by using confocal microscopy. Despite the diffuse distribution of PI4P in cells, localized accumulations could be detected in RTCs of infected cells (Fig. 5). This colocalization is best visualized by the spatial coincidence of discrete peaks of PI4P (green) and N (red) fluorescence intensity (29), as shown in two representative transects to the right of the image. The accumulation of PI4P is consistent with the involvement of membranes in RTC formation. Interestingly, PI4P is also enriched in puncta formed by ectopic expression of the recombinant JUNV N protein (Fig. 5).
Fig 5.
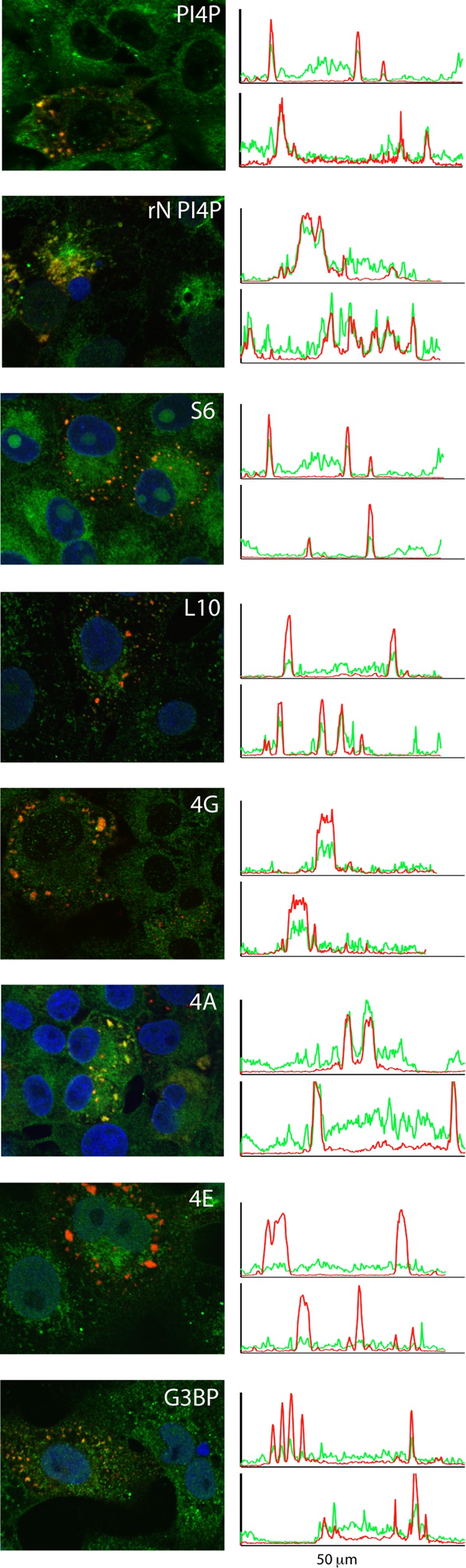
Colocalization of cellular components in RTCs. TCRV-infected Vero cells were fixed using ice-cold 4% paraformaldehyde, permeabilized in PBS containing 0.1% Triton X-100, and stained using the following antibodies: PI4P (Z-004), G3BP1 (sc-81940), small ribosomal subunit protein S6 (sc-74459), large ribosomal protein L10a (sc-100827), eIF4G (sc-11373), eIF4A (sc-137147), and eIF4E (sc-9976). DyLight 488-conjugated Fab′ fragment was used to detect bound murine MAbs. N protein was detected with Alexa Fluor 568-conjugated MAb AG12. Each panel was chosen to include both infected (red puncta) and uninfected (green only) cells. Other than the localized accumulation in RTCs, no differences were observed in the distribution of these and other cellular markers in comparing infected and uninfected cells. In the image labeled rN PI4P, recombinant N protein was expressed via transfection. Colocalization is visualized in representative 50-μm transects across the cell (right), from which green and red fluorescence intensities can be spatially correlated (29). As shown, mature puncta typically range from 1 to 5 μm in diameter. Colocalization was determined based on at least two independent experiments and inspection of numerous cells; multiple transects of eIF4E staining using two independent antibodies failed to reveal any examples of colocalization.
Although PI4P is typically associated with Golgi membranes, RTCs did not colocalize with the Golgi proteins giantin, GM130, Golgi-specific brefeldin A resistance factor (GBF1), or TGN38 (not shown). These results suggest a specific accumulation of PI4P with RTCs and exclude the intact Golgi as a site of viral replication. An initial scan of other membrane-associated markers did not find colocalization of RTCs with ER (protein disulfide isomerase, sec61α, and sec16) or with several classes of endosomal vesicles (EEA1, Rab 4, Rab5, and Rab 7). No association was observed with TfR1 (the entry receptor for the virus) or with clathrin and tubulin (not shown). Nor did RTCs colocalize with mitochondria (ATP synthetase and COX1), peroxisomes (PMP70), or autophagosomes (LC3) (not shown). Indeed, staining with Bodipy TR methyl ester, a marker for generic intracellular membranes, revealed no evidence for bulk accumulation of membranes at RTCs (not shown). Together, these findings suggest that the membrane contribution to arenavirus RTCs may be distinct from the large multivesicular assemblages found at the replication sites of some positive-sense RNA viruses (4, 55).
RTCs contain elements of the translational machinery.
The sandy (Latin, arena) appearance of the arenavirus interior in electron micrographs is attributed to the presence of cellular ribosomes (46), although the role of these ribosomes is unknown (28). In light of this long-standing mystery, we probed RTCs for evidence of ribosomes. Using two ribosomal proteins as markers for the small 40S and large 60S subunits (RPS6 and RPL10a, respectively), we identified strong colocalization with RTCs (Fig. 5). The presence of two arbitrarily chosen ribosomal proteins is consistent with the notion that ribosomal subunits are associated with RTCs and suggests a possible explanation for the incorporation of ribosomes into virion particles.
To investigate the potential role of these ribosomes, we probed for the presence of translation initiation factors. Prior to recruiting mRNA, the 40S subunit binds a series of initiation factors (eIF1, eIF1A, eIF2-GTP, and eIF3) to assemble the 43S preinitiation complex containing the initiator methionine-tRNA (35). Despite the presumed presence of the 40S ribosome subunit in RTCs, eIF2α, eIF3b, and eIF3c did not colocalize (not shown). mRNA is subsequently recruited to the 43S preinitiation complex via interaction with the m7G cap-binding eIF4E subunit of the eIF4F complex, which also comprises the eIF4G scaffold protein and the eIF4A helicase (35). Interestingly, we found that both eIF4G and eIF4A colocalized with RTCs (Fig. 5). Unexpectedly, the eIF4E protein could not be detected using either an eIF4E MAb (Fig. 5) or a specific polyclonal antibody (sc-13963, not shown). Collectively, these results describe a noncanonical collection of ribosomal proteins and translation initiation factors whose functional significance in RTCs is unclear.
RTCs contain a stress granule protein G3BP.
In addition to the historical association between ribosomes and arenaviruses, many RNA viruses utilize elements of the host cell RNA metabolic machinery to further replication. Processing bodies (PBs) and stress granules (SGs) are two related nonmembranous cytosolic structures that are intimately involved in cellular mRNA regulation and metabolism (reviewed in references 3, 13, 24, and 51). PBs mediate mRNA decay through removal of the m7G cap and poly(A) tail and subsequent 3′-exonucleolytic activity, whereas SGs serve as temporary stores for mRNAs when translation is stalled in response to endogenous or exogenous stressors. Hantaviruses make use of PBs as a source of m7G caps for their mRNAs (36), and other viruses will induce translational arrest and SG formation to favor translation of their own mRNAs (54).
To determine whether these elements of cellular RNA metabolism are involved in arenavirus RNA replication, we examined the colocalization of specific SG and PB proteins with RTCs. No association was seen with PB markers Dcp1a or Xrn1, or with SG markers TIA1, PABP, or HSP70 (not shown). In contrast, the SG-related RasGAP-associated endoribonuclease (G3BP1) demonstrated strong colocalization with RTCs (Fig. 5). This protein has also been identified in replication centers of rubella virus (32), again in the absence of TIA1 and PABP, and in those of HCV (56). G3BP1 is involved in recruiting proteins to SGs (52) and might play an analogous role in RTC formation. Because the large 60S ribosome is specifically excluded from conventional SGs, the presence of RPL10a and G3BP1 in arenavirus RTCs is indicative of a unique collection of cellular proteins.
DISCUSSION
In this report, we describe for the first time the cytosolic structures in which arenavirus replication and transcription take place. We demonstrate that ongoing viral RNA synthesis occurs within discrete cytoplasmic bodies induced by the viral nucleoprotein (N). Biochemical analyses of RTCs reveal the presence of full-length genomic and antigenomic RNA species, as well as a notable dearth of viral mRNAs. We suggest that both full-length and messenger RNAs are generated locally by the viral RNA-dependent RNA polymerase (L) and that mRNAs are rapidly exported for translation on cellular ribosomes.
The biochemical composition of these virus-induced RTCs is complex and appears to include elements of multiple cellular processes. The low buoyant density of RTCs (∼1.13 g/cm3), their sensitivity to nonionic detergent, and their association with PI4P strongly suggest that cellular membranes are mobilized for RTC formation. Although it is formally possible that our biochemical methods inadvertently contribute to changes in RTC buoyant density, identical findings are obtained regardless of the homogenization method (Dounce or tungsten ball), the presence or absence of divalent metal ions, or the nonionic detergent (Triton X-100 or NP-40) (not shown). We therefore believe that our results are biologically relevant and indicative of membrane involvement. It is significant that PI4P and its kinases have also been implicated in RTC formation by picornaviruses and HCV (22, 49, 58). Consistent with its role in cells, PI4P may serve to establish specialized membrane platforms for recruiting cellular proteins to RTCs. Despite the apparent membrane association, arenavirus RTCs did not colocalize with markers of any of the canonical membrane compartments tested or with the generic intracellular membrane stain Bodipy TR. These findings suggest that membrane involvement in arenavirus RTCs may differ from that in some positive-sense RNA viruses, where large multivesicular structures are formed (4, 55).
Collectively, our studies suggest that arenavirus RTCs contain a range of cellular lipids and proteins, including select translation initiation factors, large and small ribosomal subunit proteins, and the SG-associated endoribonuclease G3BP1. The failure to detect colocalization with other cellular proteins may be technical in nature and should not be taken to establish their absence. In this regard, we have thus far been unable to demonstrate the presence of colocalizing protein markers in the immunopurified RTCs isolated from cells directly or following reversible cross-linking with formaldehyde (not shown) (40). The failure to isolate colocalizing proteins may reflect the limitations of our biochemical methods. It is also possible that cellular and viral protein interactions that regulate RTC activity during arenavirus infection may be indirect or transient.
The presence of ribosomes in arenaviral RTCs is unexpected, notwithstanding the historical finding of host cell ribosomes in mature arenavirus particles (46). Our finding that viral mRNAs do not appear to accumulate in RTCs argues that colocalizing ribosomes are not involved in the translation of viral proteins, which likely occurs on bulk cellular ribosomes. The apparent absence of eIF2 and eIF3 further suggests that RTC ribosomes are not present as canonical preinitiation complexes (35). However, it remains possible that viral mRNAs are lost during purification, despite mild isolation conditions. Additional research is required to determine the role of cellular ribosomes in arenavirus RTCs and virion particles.
If translation is not local, what significance can be placed on the presence of a subset of initiation factors in arenavirus RTCs? Strategies to reprogram the host cell translational machinery are common among viruses and differ in detail from one virus to the other (reviewed in reference 54). It is possible that cellular translational elements are recruited to the centers of arenavirus transcription and associate with viral mRNAs prior to export in order to prime translation. Alternatively, translational elements may be sequestered in arenavirus RTCs to disadvantage the translation of cellular mRNAs.
Despite the presence of eIF4G and eIF4A in arenavirus RTCs, we were unable to detect the eIF4E subunit with monoclonal and polyclonal antibodies. This negative result does not exclude the presence of eIF4E, but it does raise the possibility that the cap-binding function of eIF4E may be superfluous for the translation of capped arenaviral mRNAs. Interestingly, X-ray crystallographic studies of N reveal a potential cap-binding motif in the N-terminal domain (44). Although cap-binding activity has not been directly demonstrated (8, 20), it is tempting to speculate that N may replace eIF4E in recruiting eIF4G and eIG4A to viral mRNAs. Indeed, the hantavirus cap-binding nucleocapsid protein has been shown to interact directly with the small ribosomal protein RPS19 to bypass the eIF4F complex entirely (10, 19).
It is now appreciated that viral infection induces a global reprogramming of cellular processes, including alterations in messenger and small RNA transcription, in metabolic pathways and lipid biosynthesis, and in the organization of intracellular membranes and organelles (22, 38, 42, 50, 57, 59). These changes subsume both proviral functions of the virus and antiviral responses of the cell and are initiated in part by viral proteins. Characterization of the interactions of these multifunctional proteins may suggest novel intervention strategies that go beyond targeting enzymatic functions while maintaining therapeutic specificity to the virus.
ACKNOWLEDGMENTS
We are grateful to Sangeeta Rojanala (Life Technologies) for providing EU and technical guidance prior to product launch and to Sonja Best (NIH Rocky Mountain Labs) for access to a confocal microscope during pilot studies. We also thank Lou Herritt and the Molecular Histology and Fluorescence Imaging Core (The University of Montana) for additional assistance with confocal microscopy. MAbs directed to the JUNV N protein were provided by the CDC and obtained through the NIAID Biodefense and Emerging Infections Research Resources Repository. We are grateful to Meg Trahey and Mark Grimes (University of Montana), Victor Romanowski (Universidad Nacional de la Plata, Argentina), Robert Tesh (University of Texas Medical Branch), and Sean Amberg (SIGA Technologies) for sharing advice and critical reagents.
The Molecular Histology and Fluorescence Imaging Core is supported by U.S. Public Health Service award P20RR017670. Funding for the project was provided by U.S. Public Health Service research grants R01AI074818, R21AI099870, and U54AI065357 (Rocky Mountain Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (principal investigator, J. Belisle).
Footnotes
Published ahead of print 8 August 2012
REFERENCES
- 1. Agnihothram SS, York J, Trahey M, Nunberg JH. 2007. Bitopic membrane topology of the stable signal peptide in the tripartite Junín virus GP-C envelope glycoprotein complex. J. Virol. 81:4331–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Axis-Shield 2011. OptiPrep application sheets for isolation of viruses. Axis-Shield, Dundee, Scotland [Google Scholar]
- 3. Beckham CJ, Parker R. 2008. P bodies, stress granules, and viral life cycles. Cell Host Microbe 3:206–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Belov GA, et al. 2012. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 86:302–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Best MD. 2009. Click chemistry and bioorthogonal reactions: unprecedented selectivity in the labeling of biological molecules. Biochemistry 48:6571–6584 [DOI] [PubMed] [Google Scholar]
- 6. Bolken TC, et al. 2006. Identification and characterization of potent small molecule inhibitor of hemorrhagic fever New World arenaviruses. Antiviral Res. 69:86–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bolte S, Cordelières FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224:213–232 [DOI] [PubMed] [Google Scholar]
- 8. Brunotte L, et al. 2011. Structure of the Lassa virus nucleoprotein revealed by X-ray crystallography, small-angle X-ray scattering, and electron microscopy. J. Biol. Chem. 286:38748–38756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buchmeier MJ, de la Torre JC, Peters CJ. 2007. Arenaviridae: the viruses and their replication, p 1791–1828 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 10. Cheng E, et al. 2011. Characterization of the interaction between hantavirus nucleocapsid protein (N) and ribosomal protein S19 (RPS19). J. Biol. Chem. 286:11814–11824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clegg JCS. 2002. Molecular phylogeny of the arenaviruses. Curr. Top. Microbiol. Immunol. 262:1–24 [DOI] [PubMed] [Google Scholar]
- 12. den Boon JA, Ahlquist P. 2010. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 64:241–256 [DOI] [PubMed] [Google Scholar]
- 13. Erickson SL, Lykke-Andersen J. 2011. Cytoplasmic mRNP granules at a glance. J. Cell Sci. 124:293–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fontana J, López-Montero N, Elliott RM, Fernández JJ, Risco C. 2008. The unique architecture of Bunyamwera virus factories around the Golgi complex. Cell Microbiol. 10:2012–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garcin D, Kolakofsky D. 1990. A novel mechanism for the initiation of Tacaribe arenavirus genome replication. J. Virol. 64:6196–6203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghiringhelli PD, Rivera-Pomar RV, Lozano ME, Grau O, Romanowski V. 1991. Molecular organization of Junin virus S RNA: complete nucleotide sequence, relationship with other members of the Arenaviridae and unusual secondary structures. J. Gen. Virol. 72:2129–2141 [DOI] [PubMed] [Google Scholar]
- 17. Graham TR, Burd CG. 2011. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 21:113–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greninger AL, Knudsen GM, Betegon M, Burlingame AL, DeRisi JL. 2012. The 3A protein from multiple picornaviruses utilizes the Golgi adaptor protein ACBD3 to recruit PI4KIIIβ. J. Virol. doi:10.1128/JVI.06778–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haque A, Mir MA. 2010. Interaction of hantavirus nucleocapsid protein with ribosomal protein S19. J. Virol. 84:12450–12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO. 2011. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. U. S. A. 108:2396–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog. 6:e1000958 doi:10.1371/journal.ppat.1000958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsu NY, et al. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iapalucci S, López N, Franze-Fernández MT. 1991. The 3′ end termini of the Tacaribe arenavirus subgenomic RNAs. Virology 182:269–278 [DOI] [PubMed] [Google Scholar]
- 24. Kedersha N, Anderson P. 2007. Mammalian stress granules and processing bodies. Methods Enzymol. 431:61–81 [DOI] [PubMed] [Google Scholar]
- 25. Knoops K, et al. 2012. Ultrastructural characterization of arterivirus replication structures: reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 86:2474–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kopek BG, Perkins G, Miller DJ, Ellisman MH, Ahlquist P. 2007. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 5:e220 doi:10.1371/journal.pbio.0050220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee KJ, Novella IS, Teng MN, Oldstone MB, de La Torre JC. 2000. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J. Virol. 74:3470–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leung WC, Rawls WE. 1977. Virion-associated ribosomes are not required for the replication of Pichinde virus. Virology 81:174–176 [DOI] [PubMed] [Google Scholar]
- 29. Lindquist ME, Lifland AW, Utley TJ, Santangelo PJ, Crowe JE., Jr 2010. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J. Virol. 84:12274–12284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. López R, Grau O, Franze-Fernández MT. 1986. Effect of actinomycin D on arenavirus growth and estimation of the generation time for a virus particle. Virus Res. 5:213–220 [DOI] [PubMed] [Google Scholar]
- 31. Maiztegui JI, et al. 1998. Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever. AHF Study Group. J. Infect. Dis. 177:277–283 [DOI] [PubMed] [Google Scholar]
- 32. Matthews JD, Frey TK. 2012. Analysis of subcellular G3BP redistribution during rubella virus infection. J. Gen. Virol. 93:267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCaffrey G, Welker J, Scott J, van der Salm L, Grimes ML. 2009. Resolution of signalling endosomes containing different receptors. Traffic 10:938–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCormick JB, Fisher-Hoch SP. 2002. Lassa fever. Curr. Top. Microbiol. Immunol. 262:75–109 [DOI] [PubMed] [Google Scholar]
- 35. Merrick WC. 2010. Eukaryotic protein synthesis: still a mystery. J. Biol. Chem. 285:21197–21201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mir MA, Duran WA, Hjelle BL, Ye C, Panganiban AT. 2008. Storage of cellular 5′ mRNA caps in P bodies for viral cap-snatching. Proc. Natl. Acad. Sci. U. S. A. 105:19294–19299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miyazaki J, et al. 1989. Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene 79:269–277 [DOI] [PubMed] [Google Scholar]
- 38. Nagy PD, Pogany J. 2011. The dependence of viral RNA replication on co-opted host factors. Nat. Rev. Microbiol. 10:137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. NIAID 2009. NIAID category A, B, and C priority pathogens. NIAID, Bethesda, MD [Google Scholar]
- 40. Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA. 2002. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26:182–190 [DOI] [PubMed] [Google Scholar]
- 41. Novoa RR, et al. 2005. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol. Cell 97:147–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perera R, et al. 2012. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 8:e1002584 doi:10.1371/journal.ppat.1002584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peters CJ. 2002. Human infection with arenaviruses in the Americas. Curr. Top. Microbiol. Immunol. 262:65–74 [DOI] [PubMed] [Google Scholar]
- 44. Qi X, et al. 2010. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 468:779–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reiss S, et al. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rowe WP, et al. 1970. Arenoviruses: proposed name for a newly defined virus group. J. Virol. 5:651–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Salazar-Bravo J, Ruedas LA, Yates TL. 2002. Mammalian reservoirs of arenaviruses. Curr. Top. Microbiol. Immunol. 262:25–63 [DOI] [PubMed] [Google Scholar]
- 48. Sanchez A, et al. 1989. Junin virus monoclonal antibodies: characterization and cross-reactivity with other arenaviruses. J. Gen. Virol. 70:1125–1132 [DOI] [PubMed] [Google Scholar]
- 49. Sasaki J, Ishikawa K, Arita M, Taniguchi K. 2012. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 31:754–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smith JL, Grey FE, Uhrlaub JL, Nikolich-Zugich J, Hirsch AJ. 2012. West Nile virus induction of the cellular microRNA, Hs_154, contributes to viral-mediated apoptosis through repression of anti-apoptotic factors. J. Virol. doi:10.1128/JVI.06883–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thomas MG, Loschi M, Desbats MA, Boccaccio GL. 2011. RNA granules: the good, the bad and the ugly. Cell Signal. 23:324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tourrière H, et al. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160:823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53. Vecchio G, Tsuchida N, Shanmugam G, Green M. 1973. Virus-specific messenger RNA and nascent polypeptides in polyribosomes of cells replicating murine sarcoma-leukemia viruses. Proc. Natl. Acad. Sci. U. S. A. 70:2064–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Walsh D, Mohr I. 2011. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 9:860–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Welsch S, et al. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yi Z, et al. 2011. Hepatitis C virus co-opts Ras-GTPase-activating protein-binding protein 1 for its genome replication. J. Virol. 85:6996–7004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu Y, Clippinger AJ, Alwine JC. 2011. Viral effects on metabolism: changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 19:360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang L, et al. 2012. ARF1 and GBF1 generate a PI4P-enriched environment supportive of hepatitis C virus replication. PLoS One 7:e32135 doi:10.1371/journal.pone.0032135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhao H, Dahlö M, Isaksson A, Syvänen AC, Pettersson U. 2012. The transcriptome of the adenovirus infected cell. Virology 424:115–128 [DOI] [PubMed] [Google Scholar]