

Abstract
A group of vaccinia virus (VACV) proteins, including A11, L2, and A6, are required for biogenesis of the primary envelope of VACV, specifically, for the acquisition of viral membrane precursors. However, the interconnection among these proteins is unknown and, with the exception of L2, the connection of these proteins with membranes is also unknown. In this study, prompted by the findings that A6 coprecipitated A11 and that the cellular distribution of A11 was dramatically altered by repression of A6 expression, we studied the localization of A11 in cells by using immunofluorescence and cell fractionation analysis. A11 was found to associate with membranes and colocalize with virion membrane proteins in viral replication factories during normal VACV replication. A11 partitioned almost equally between the detergent and aqueous phases upon Triton X-114 phase separation, demonstrating an intrinsic affinity with lipids. However, in the absence of infection or VACV late protein synthesis, A11 did not associate with cellular membranes. Furthermore, when A6 expression was repressed, A11 did not colocalize with any viral membrane proteins or associate with membranes. In contrast, when virion envelope formation was blocked at a later step by repression of A14 expression or by rifampin treatment, A11 colocalized with virion membrane proteins in the factories. Altogether, our data showed that A11 associates with viral membranes during VACV replication, and this association requires A6 expression. This study provides a physical connection between A11 and viral membranes and suggests that A6 regulates A11 membrane association.
INTRODUCTION
Vaccinia virus (VACV) is the prototypical member of the Orthopoxvirus genus of the Poxviridae, a family of large DNA viruses that replicate in the cytoplasm (17). VACV produces two major forms of infectious viruses: the intracellular mature virion (MV) and the extracellular enveloped virion (EV). The majority of the infectious viruses made by infected cells are MVs, which contain an envelope and stay within the cells until cell lysis. A fraction of the MVs acquire additional envelopes through wrapping with cellular organelles and are ultimately exocytosed as EVs (26). EVs have an additional envelope embedded with EV-specific membrane proteins but are otherwise structurally identical to MVs. Within the lipid envelope, an MV has a dumbbell-shaped core, containing a linear DNA genome of approximately 190 kbp. The elaborate process of assembling MVs from components, including the viral DNA genome, approximately 50 core proteins, and an envelope embedded with nearly 20 membrane proteins, occurs in viral factories, which are defined by cytoplasmic DNA staining under fluorescence microscopy. A series of intermediate stages of assembly have been revealed by electron microscopy (reviewed in reference 4). Viral factories appear first as areas of cytoplasm that largely exclude cellular organelles and have a lower electron density than the surrounding areas. Electron-dense viroplasms comprised of viral core proteins appear next in the factories. At the periphery of viroplasms, crescent-shaped membranes of a single lipid bilayer develop. The membranes are stabilized by an external scaffold of a honeycomb lattice comprised of D13 proteins (8, 29), which lack a transmembrane domain but interact with the integral membrane protein A17 (2). Crescent membranes engulf part of the viroplasm to form the spherical immature virions (IVs). The viral genome is encapsidated before the IV membrane closes off, forming IVs with an electron-dense nucleoid (IVN). The D13 lattice on the IVs is removed by proteolytic processing of A17 by the I7 protease (2), which also processes several virion core proteins, including A10 (1, 3). Concomitant with these proteolytic processing events, IVNs mature into brick-shaped MVs.
The origin and biogenesis of the MV envelope is one of the least understood aspects of poxvirus biology. The crescent membranes are believed to be derived from a cellular organelle, either the intermediate compartment between the endoplasmic reticulum and Golgi apparatus (ERGIC) (20, 27) or the endoplasmic reticulum (ER) (9). Consistent with this idea, several membrane proteins destined for the MV envelope are synthesized on the ER (9, 23), and a pathway exists for the trafficking of MV membrane proteins from the ER to IVs (9). The trafficking of MV membrane precursors or MV membrane proteins to viral factories appears to be an active, virus-mediated process, as repressing VACV A6 expression results in the accumulation of MV membrane proteins in secretory compartments outside the viral factories (13).
Through studies of conditional lethal VACV mutants, several VACV proteins have been identified to be essential for the formation of crescent membranes. F10 (30, 31), A11 (19), H7 (24), L2 (11), and A6 (13) are required at an early step prior to the appearance of viral membrane precursors in the factories, as repressing the expression of these genes results in the loss of discernible membrane structure in the factories and the accumulation of large viroplasm inclusions. A14 and A17 are required at a later step following the acquisition of viral membrane precursors, because repressing the expression of A14 (22, 32) or A17 (21, 34) results in the accumulation of vesicular or tubular membrane structures at the boundaries of large viroplasm inclusions. A14 and A17 are integral membrane proteins of the MV. In contrast, viral proteins participating in early steps of membrane biogenesis are largely not MV structural proteins and, with the exception of L2 and F10, have no known connection with membranes or lipids. L2 is an integral membrane protein that localizes to the ER (12). F10 has been reported to localize to either viral factories (30) or to the ER and ERGIC (18). It associates with membranes during infection and binds some lipids in vitro (30). Both A6 and H7 localize to the cytoplasm (14, 24). A11 localizes specifically to viral factories, but it is not incorporated by virions (19). In the current study, we found that A11 coprecipitated with A6 and that repression of A6 expression resulted in a diffuse cytoplasmic localization of A11. Further studies led us to the surprising finding that A11 associates with viral membranes during VACV replication, providing a physical connection between A11 and its purported site of action. Furthermore, we found that association of membranes by A11 requires viral replication and, in particular, the expression of A6, suggesting that A6 regulates the A11 membrane association.
MATERIALS AND METHODS
Cells and antibodies.
BHK, HeLa, and 293T cells were maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (FBS). Murine monoclonal antibody (MAb) against V5 was purchased from Sigma-Aldrich. MAbs against VACV proteins A10 (BG3), A13 (11F7), A14 (FE11), D8 (BD6), WR148 (HE7), and D13 (HD9) were described previously (16, 35). BG3 was previously reported to be of the IgG1 isotype (16), but it was determined to be of the IgG2a isotype in the current study. MAbs against A11 (10G11) and A6 (10F1) were developed from mice immunized with recombinant A11 or A6 protein, respectively. The hybridomas were generated as described previously (16).
Plasmids and viruses.
The plasmids for prokaryotic expression of A11 proteins were constructed by PCR amplifying the A11 open reading frame (ORF) from genomic DNA of WR VACV and subcloning the PCR product in frame into a modified pET-22b vector containing glutathione S-transferase (GST) or maltose-binding protein (MBP) at the 5′ end of the cloning sites. Several plasmids for mammalian expression of A11 were used in the study. One was constructed by PCR amplifying the A11 ORF from VACV DNA and subcloning the PCR product between NheI and XhoI sites of the pcDNA3.1(-) vector. This plasmid was used for T7 promoter-regulated expression of A11 in cells infected by vTF7.3 (6). However, it expressed A11 poorly in mammalian cells in the absence of VACV infection. Thus, a human codon-optimized A11 gene was synthesized commercially (Genscript) and subcloned between NheI and BamHI sites of pcDNA3.1(-). This plasmid was used in the experiments described in Fig. 7A. For expression of wild-type (WT) or truncated A11 in VACV-infected cells, the A11 coding sequence and 188 bp of its upstream sequence, including its native promoter, was PCR amplified from VACV DNA and subcloned into BamHI and XhoI sites of pYW31 (15), placed next to green fluorescent protein (GFP) regulated by the p11 promoter. Recombinant PCR was used to remove the coding sequence for the N-terminal 40 amino acids or C-terminal 31 amino acids of A11 from the plasmid.
Fig 7.
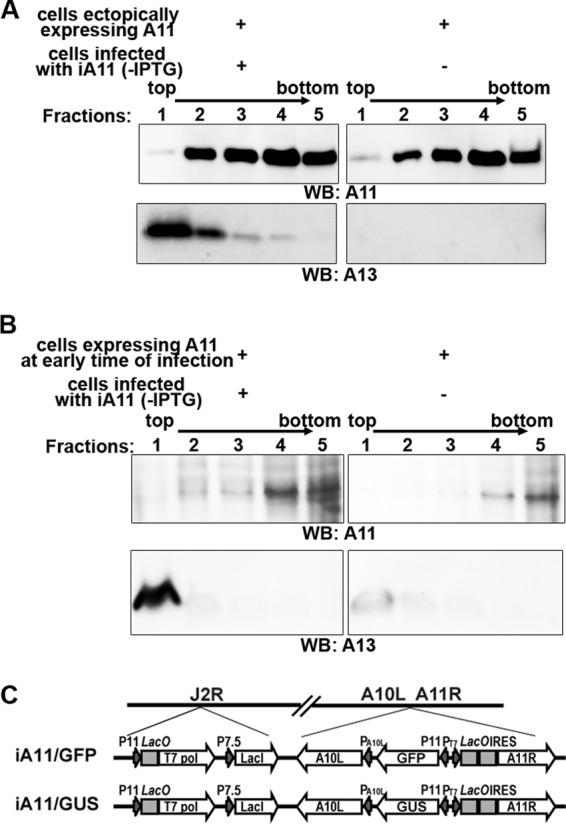
Association of membranes by A11 requires viral late gene expression. (A) 293T cells were transfected with a mammalian A11 expression plasmid for 48 h. (B) 293T cells were transfected with a plasmid containing a T7 promoter-regulated A11 and then infected with vTF7.3 in the presence of AraC for 12 h. A membrane flotation assay was performed as described for Fig. 5C. To provide viral membrane proteins as an internal control, cells infected with an inducible A11 VACV (iA11) in the absence of the inducer were added to one set of samples during the centrifugation step. (C). Schematic representation of the genome structure of iA11 (iA11/GFP and iA11/GUS). LacO, lac operator; T7 pol, bacteriophage T7 RNA polymerase; LacI, E. coli lac repressor; PT7, T7 promoter.
An inducible A14 virus (iA14/GFP) was constructed in two steps. An intermediate virus was first constructed by inserting an inducible copy of the A14L gene into the A56R locus of vT7LacOI (33). The A14 coding sequence was PCR amplified from VACV DNA and subcloned between NcoI and BamHI sites of pVote1 (33). This plasmid was used for homologous recombination with vT7LacOI, and the recombinant virus was isolated under selection for guanine phosphoribosyl transferase according to a standard protocol (5). The final inducible A14 virus was constructed by replacing the native A14L gene in the intermediate virus with GFP. A DNA sequence that included the following elements was assembled by recombinant PCR: (i) the 500-bp left flank of the A14 gene derived from VACV DNA (using primers 5′-TGCGCCGGCAGTCACATCTGTT-3′ and 5′-GCTGTACAAGTAAATTATTATCGTCCATATATCT-3′); (ii) coding sequence for GFP derived from pYW31 (using primers 5′-GACGATAATAATTTACTTGTACAGCTCGTCCATG-3′ and 5′-ATGGTGAACAAGGGCGAG-3′); (iii) the 500-bp right flank of the A14 gene, including the native A14 promoter derived from VACV DNA (using primers 5′-CTCGCCCTTGTTCACCATTTAACTAATAAAAATTTTAAATCG-3′ and 5′-GGTTGGGCGCGGCCATAACA-3′). Underlined in the primer sequences are those portions that are not present in the PCR templates but are added to create overlapping sequences between different PCR products. The recombinant PCR product was TA cloned into the pGEM vector (Promega), and the resultant plasmid was transfected into cells infected by the intermediate virus. Recombinant virus expressing GFP was plaque purified in the presence of 500 μM isopropyl-β-d-thiogalactopyranoside (IPTG).
An inducible A11 virus expressing GFP (iA11/GFP) was constructed from vT7LacOI by replacing the native promoter for A11 with an IPTG-inducible T7 promoter and the simultaneous insertion of a GFP cassette between the A10L gene and the T7 promoter. A DNA sequence that included the following elements was assembled by recombinant PCR: (i) the 500-bp left flank of the A11 gene, including the A10L promoter and part of the A10 coding sequence derived from VACV DNA (using primers 5′-GTTAAGATCTGGATATTTCTTTTC-3′ and 5′-GCTGTACAAGTAATTTAATACTAAATAAATGATGC-3′); (ii) GFP controlled by the P11 late promoter derived from pYW31 (15) (using primers 5′-ATTTAGTATTAAATTACTTGTACAGCTCGTGC-3′ and 5′-CGGGATCGAGATCGGCGCGCCTTTCATTTTG-3′); (iii) a T7 promoter, LacO sequence, and the encephalomyocarditis virus internal ribosome entry site (IRES) derived from pVote1 (using primers 5′-TGAAAGGCGCGCCGATCTCGATCCCGCGAAA-3′ and 5′-GTACGGTCGTCATATCCATGGTATTATCGTG-3′; (iv) 500 bp of the A11 ORF derived from VACV DNA (using primers 5′-AATACCATGGATATGACGACCGTACCAGTG-3′ and 5′-GATCGTTGATTTCTAAGATTAAC-3′). Underlined in the primers are sequences that are not present in the PCR templates and were added to create overlapping sequences between different PCR products. The recombinant PCR product was transfected into cells infected with vT7LacOI, and recombinant viruses expressing GFP were plaque purified in the presence of 25 μM IPTG. An inducible A11 virus expressing β-glucuronidase (GUS; iA11/GUS) was constructed similarly by using GUS instead of GFP as the selective marker.
The inducible viruses were confirmed by PCR analysis of the viral genomic DNA, the inducible expression of A14 or A11 confirmed by Western blotting with specific MAbs, and IPTG-dependent replication confirmed by plaque assay.
Recombinant VACV expressing a C-terminal V5 tagged A46 (vA46-V5) was constructed by appending the tag sequence before the stop codon of A46 and the insertion of a GFP cassette between A46R and A47L in VACV WR, similar to the construction of the VACV expressing V5-tagged K1L (15).
Immunoprecipitation and mass spectrometry.
HeLa cells were infected with VACV (vA6-V5 [14], vA46-V5, or iA6-GFP [13]) at a multiplicity of infection (MOI) of 5 PFU/cell. At 12 h postinfection (hpi), the cells were harvested and resuspended in lysis buffer (0.5% [wt/vol] Triton X-100, 25 mM Tris [pH 7.4], 150 mM NaCl) supplemented with protease inhibitor cocktail tablet (Roche Molecular Biochemicals). The clarified cell lysates were incubated with anti-V5–agarose beads (Sigma-Aldrich) for 1 h at 4°C. The beads were washed five times with wash buffer (0.1% [wt/vol] Triton X-100, 50 mM Tris [pH 7.4], 300 mM NaCl, 5 mM EDTA) and eluted with 100 mM glycine, pH 1.8. For Western blot analysis, the beads were added with SDS-PAGE loading buffer, and the samples were resolved by SDS-PAGE. For mass spectrometry (MS) analysis, the eluted proteins were precipitated with trichloroacetic acid (TCA), and the pellets were dissolved in urea. The samples were reduced, alkylated, digested with trypsin, and analyzed on a hybrid LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to a New Objectives PV-550 nanoelectrospray ion source and an Eksigent NanoLC-2D chromatography system. Ion masses were used to search a viral protein sequence database (the NCBInr_032810 database filtered for viral sequences; 628,255 entries) with Mascot (Matrix Science) and X! Tandem (www.thegpm.org). Scaffold (Proteome Software Inc.) was used to validate tandem MS (MS/MS)-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 80.0% probability as specified by the Peptide Prophet algorithm (10). Protein identifications were accepted if they could be established at greater than 95.0% probability and contained at least 2 identified peptides.
Subcellular fractionation.
Subcellular fractionation was performed as described previously (13). Briefly, HeLa cells in a 150-mm dish were infected with VACV at an MOI of 5 PFU/cell and harvested at 8 hpi. The postnuclear supernatant was loaded onto a 10.5-ml preformed 5-to-25% continuous iodixanol gradient (Axis Shield) and centrifuged at 200,000 × g for 2.5 h at 4°C. Fractions were collected and precipitated with TCA. Precipitated proteins were analyzed by Western blotting.
Sedimentation of membranes.
Subcellular fractions containing A11 were mixed with Triton X-100 at a final concentration of 1% or with an equal volume of H2O, layered on top of a 0.5 M sucrose cushion, and centrifuged in an SW55Ti rotor at 35,000 rpm for 2 h. The pellets and TCA-precipitated supernatants were analyzed by Western blotting.
Limited proteinase K digestion.
Subcellular fractions containing A11 were mixed with Triton X-100 at a final concentration of 1% or with an equal volume of H2O and incubated with 50 μg/ml of proteinase K for 5 min at 25°C. The reaction was stopped by adding phenylmethanesulfonylfluoride (PMSF) to 1 mM. TCA-precipitated samples were analyzed by Western blotting.
Membrane flotation assay.
The membrane flotation assay was performed similarly to methods described previously (25). Samples were untreated or incubated for 30 min with Triton X-100 at a final concentration of 1%, or Na2CO3 (pH 11.5) at a final concentration of 100 mM, or an equal volume of H2O. The iodixanol concentration in the samples was adjusted with 60% iodixanol to a final concentration of 30%. One milliliter of the sample was placed at the bottom of a centrifuge tube and overlaid with 3 ml of 25% iodixanol and 0.5 ml of 5% iodixanol. The tubes were centrifuged in an SW55Ti rotor at 46,000 rpm for 2 h. After the centrifugation, the gradient was harvested as 5 fractions collected from the top of the tube. Membranes were floated to the 5%/25% interface, which was harvested in the first fraction. All fractions were precipitated with TCA and analyzed by Western blotting.
Triton X-114 phase separation.
HeLa cells in a 100-mM dish were infected with WT VACV at an MOI of 5 PFU/cell. After 20 h, the cells were harvested, resuspended in 1 ml of lysis buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-114), and incubated on ice for 1 h. The clarified cell lysate was incubated at 37°C for 3 min and centrifuged to separate the aqueous and detergent phases. A 100-μl volume of fresh cold lysis buffer was added to the aqueous phase, and phase separation was repeated as described above. The detergent phases from the two extractions were pooled and kept as the “detergent phase” after a wash at room temperature with 1 ml of a buffer without detergent (10 mM Tris [pH 7.4], 150 mM NaCl). The aqueous phase after the two extractions was extracted for the final time with 110 μl of fresh cold lysis buffer, and the aqueous phase after this final extraction was kept as the “aqueous phase.”
Western blot analysis.
The Western blot analysis was performed as previously described (15). Briefly, the samples were solubilized in SDS sample buffer, resolved by SDS-PAGE, transferred to nitrocellulose membranes, and blocked with Tris-buffered saline supplemented with 5% nonfat dried milk and 0.05% Tween 20 for 1 h at room temperature. Subsequently, membranes were incubated with the antibodies and analyzed with chemiluminescence reagent (Pierce).
Fluorescence microscopy.
BHK cells grown on coverslips were infected at 0.5 PFU/cell. At 8 hpi, the cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% Saponin (Sigma-Aldrich) for 5 min, blocked with 10% FBS for 60 min, and stained with various antibodies for 1 h and an appropriate secondary antibody for an additional hour. The DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Cells on overslips were imaged with an Olympus IX-81 fluorescence microscope.
RESULTS
A11 coprecipitates with A6.
To gain some insight into the molecular function of A6, we carried out an immunoprecipitation and mass spectrometry study to identify potential binding partners for A6. As a specificity control, we performed in parallel the precipitation of another VACV protein, A46, which is similar to A6 in its expression level and distribution in infected cells (data not shown). We infected HeLa cells with VACV encoding either V5 epitope-tagged A46 or A6 (14). We then used anti-V5-conjugated agarose beads to precipitate proteins from the cells and analyzed the precipitates with mass spectrometry (Fig. 1A). A6 or A46 was the most abundant viral protein in the precipitates as measured by spectrum count, indicating that the target protein was efficiently precipitated. Interestingly, A11 was found in A6 precipitate but not in A46 precipitate, suggesting that A6 specifically interacts with A11. In contrast, several VACV proteins, including F17, A10, I1, and A3, were present in both samples. These proteins were probably nonspecifically precipitated, as they were often found in our precipitations of other unrelated VACV proteins (data not shown).
Fig 1.

(A) Mass spectrometry analysis of proteins coprecipitated with A6. HeLa cells in a 150-mm dish were infected with a VACV that expressed V5-tagged A6 or V5-tagged A46 at an MOI of 5 PFU/cell for 12 h. Proteins were immunoprecipitated from the cells with anti-V5-conjugated agarose beads, digested with trypsin, and analyzed by mass spectrometry. Ion masses were used to search a viral protein sequence database, and VACV proteins identified from A6 precipitation (IP A6) or A46 precipitation (IP A46) are listed along with the spectrum count (the number of tandem mass spectra that are assigned to peptides from the indicated protein) and the percentage of amino acid coverage. (B) The development of anti-A11 MAb 10G11 and the mapping of its epitope. 10G11 was developed from a mouse immunized with an A11-GST fusion protein. Mapping of the 10G11 epitope was done by Western blotting of lysates of Escherichia coli cells overexpressing fragments of A11. The full-length A11 (residues 1 to 287) was fused with GST, while all A11 truncations were fused with MBP. Coomassie staining of the same cell lysates is also shown. Indicated above the lanes are the residue numbers of A11 proteins that were overexpressed. (C) A11 coprecipitates with A6. HeLa cells were infected with an inducible A6 VACV (iA6-GFP) in the presence or absence of IPTG for 12 h and subjected to immunoprecipitation with anti-V5–agarose. The cell lysates (cell), proteins eluted with glycine, pH 1.8 (eluate 1) or with SDS-PAGE sample buffer (eluate 2), and supernatants after V5-agarose binding (sup.) were analyzed by Western blotting, first with anti-A11 MAb 10G11 and subsequently with anti-V5. A11, A6-V5, and V5 antibody heavy chain (HC) are indicated on the SDS-PAGE.
To further study the interaction between A6 and A11, we developed a MAb against A11 by immunizing a mouse with an A11 and GST fusion protein. We mapped the epitope of the antibody (10G11) to within amino acids 184 to 221 of A11 by Western blotting various A11 truncations (Fig. 1B). The MAb was then used to further test whether A11 could be precipitated by A6. Cells were infected with iA6-GFP (13), a recombinant VACV with an IPTG-inducible A6 gene. Immunoprecipitation with the V5 antibody of cells that received no IPTG did not precipitate any A11, while immunoprecipitation of cells that received IPTG precipitated A6 as well as some A11 protein (Fig. 1C). However, the majority of the A11 protein was not precipitated by the V5 antibody, suggesting that A11 interacts with A6 weakly or transiently. Our experiment did not exclude the possibility that A6 precipitates A11 due to an indirect interaction. In the absence of A6, A11 protein was present at a lower level. This is similar to the effect on A11 protein level reported after repression of the expression of L2 (12).
Localization of A11 and D13 to viral factories requires A6 expression.
As A6 is required for the localization of many MV membrane proteins to viral factories (13), we next used immunofluorescence to study whether A6 was also required for proper localization of A11 in infected cells. To allow simultaneous labeling of two viral proteins for immunofluorescence, we infected BHK cells with iA6-GUS (13), an IPTG-inducible A6 VACV that is similar to iA6-GFP but does not express GFP. After the cells were infected in the presence or absence of IPTG for 8 h, the localization of A11 was determined with respect to representative VACV proteins. In the presence of IPTG (+A6), A11 localized to viral factories (Fig. 2), as previously reported for WT VACV (19). Furthermore, A11 colocalized more closely with A13 (an MV membrane protein) than with either A10 (a core protein) or D13 (a nonstructural protein) (Fig. 2, insets). In the absence of IPTG (-A6), A13 localized to secretory compartments outside the viral factories, while A10 accumulated in large inclusions in and around factories, as previously reported (13). Interestingly, A11 also failed to localize to viral factories in the absence of A6, but it demonstrated a diffuse cytoplasmic localization that was distinct from the localization of A13, A10, and D13. D13 accumulated as cytoplasmic inclusions outside the factories in the absence of A6 (Fig. 2C). Additional immunofluorescence studies showed that the D13 inclusions did not overlap the inclusions formed by A10 (data not shown). Some cytoplasmic D13 inclusions were even observed in cells infected with iA6-GUS in the presence of IPTG (data not shown), similar to what has been previously reported for the WT VACV (28).
Fig 2.
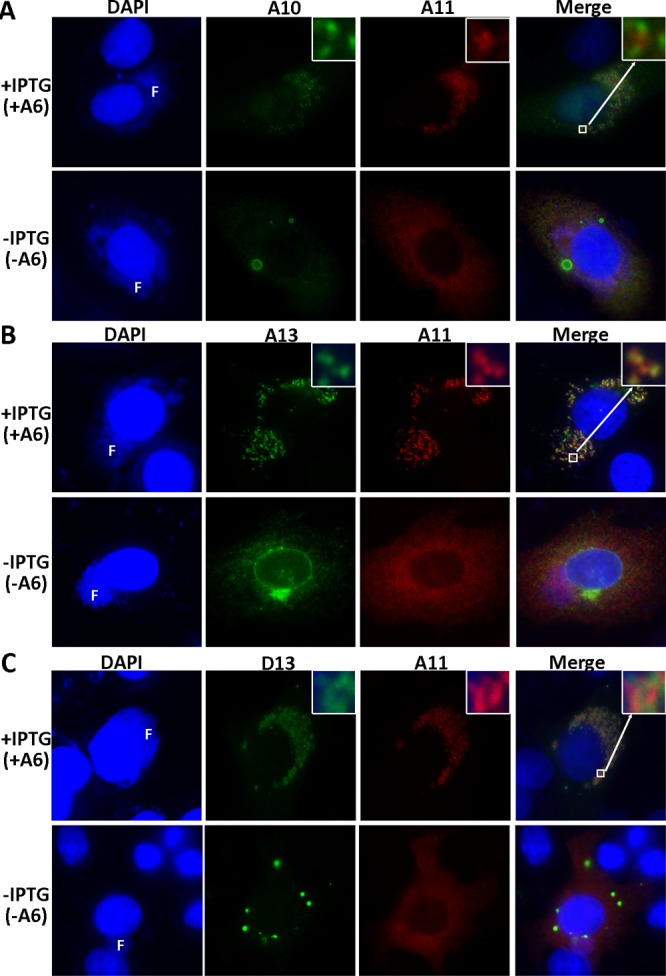
Repression of A6 expression causes a defect in localization of A11 and D13 to viral factories. BHK cells were infected with iA6-GUS at an MOI of 0.5 PFU/cell in the presence (+IPTG) or absence (-IPTG) of 100 μM IPTG. After 8 h, the infected cells were fixed, permeabilized, and stained with anti-A11 MAb 10G11 (IgG2b) and one of the following MAbs: anti-A10 BG3 (IgG2a [A]), anti-A13 11F7 (IgG2a [B]), or anti-D13 HD9 (IgG3 [C]). The cells were further stained with DAPI and isotype-specific secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 594. In some panels, an area of the cell is enlarged (inset, upper right corners). F, viral factory.
To confirm that repression of A6 expression alters the intracellular distribution of A11, we fractionated iA6-GFP-infected HeLa cells with an iodixanol gradient and analyzed the distribution of viral proteins in cell fractions by Western blotting (Fig. 3). The A11 protein from cells that were infected in the presence of IPTG (+A6) primarily sedimented to the parts of the gradient (fractions 10 to 12) that were denser than the protein disulfide isomerase-containing ER fractions (fractions 6 to 10). This suggests that A11 associates with membranes, as proteins alone do not sediment far from the loading zone in this type of velocity sedimentation. D13 was present in two peaks. Some D13 remained near the loading zone (fractions 1 and 2), while the rest of D13 sedimented to fractions 9 and 10. The former probably represents free D13, while the latter represents membrane-bound D13 (2). In contrast, A11 and D13 proteins from cells that were infected in the absence of IPTG (-A6) remained near the loading zone (fractions 1 and 2), suggesting that they did not associate with membranes, consistent with the immunofluorescence results.
Fig 3.

Repression of A6 expression alters the intracellular distribution of A11 and D13. HeLa cells were infected with iA6-GFP at an MOI of 5 PFU/cell in the presence (+IPTG) or absence (-IPTG) of IPTG. At 8 hpi, the cells were harvested, and the postnuclear supernatant was layered on top of a 5 to 25% continuous iodixanol gradient, centrifuged at 200,000 × g for 2.5 h, and fractionated. The fractions were precipitated with TCA, and immunoblotted with antibodies against the indicated proteins. The direction of the density gradient is indicated above the lanes.
A11 localized to viral factories when virion envelope formation was blocked by repression of A14 expression or by rifampin treatment.
A11 localized more closely with A13 than with either D13 or A10 under normal VACV infection conditions (infection by iA6-GUS in the presence of the inducer [Fig. 2] or by WT VACV [data not shown]), suggesting that A11 associates with membrane proteins or viral membranes. However, during normal virion assembly, core proteins, membrane proteins, and D13 localized very closely, making it difficult to rule out a colocalization of A11 with D13 or core proteins. We thus studied the localization of A11 when VACV virion assembly was blocked at specific steps.
The repression of VACV A14 expression was previously shown to result in a block in crescent membrane formation (22, 32). In cells infected by an inducible A14 VACV in the absence of the inducer (-A14), a clear spatial separation between virion core proteins and MV membrane proteins was observed by immunofluorescence (Fig. 4A). While virion core protein A10 and virus-encoded GFP colocalized in large inclusions in the factories, D13 and A13 accumulated in spaces between the GFP inclusions in the factories. A11 also localized to areas between the GFP inclusions in the factories, demonstrating clearly that A11 did not colocalize with core proteins.
Fig 4.
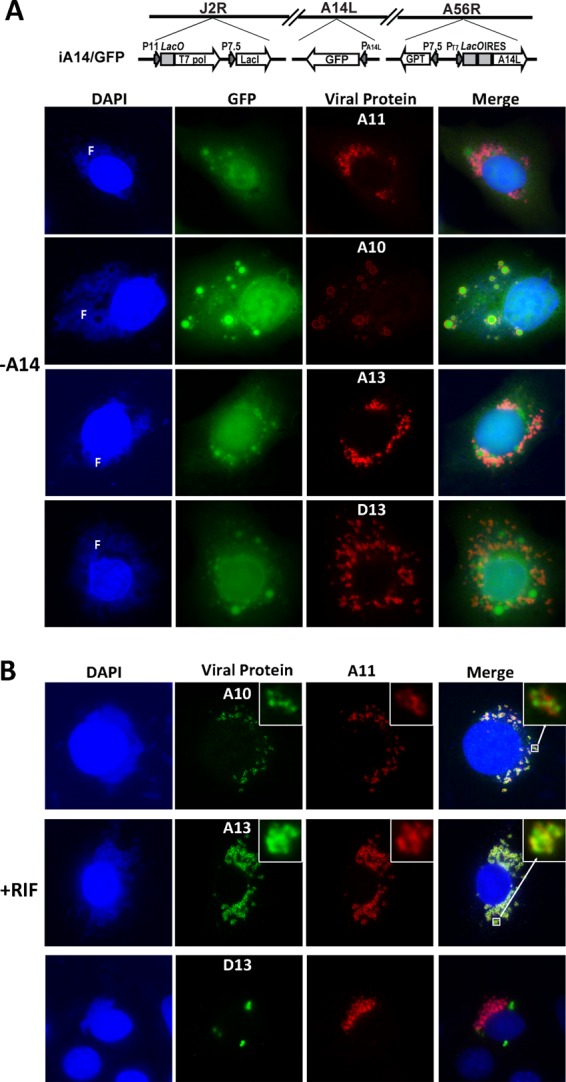
A11 localized to viral factories when virion envelope formation was blocked by repression of A14 expression (A) or by rifampin treatment (B). BHK cells were infected with an inducible A14 VACV (iA14/GFP) in the absence of the inducer (-A14) or with WT VACV in the presence of 100 μg/ml of rifampin (+RIF). After 8 h, immunofluorescence was performed on the cells as described for Fig. 2. An area of the cell in some panels is enlarged (upper right inset). F, viral factory. The genome structure of iA14/GFP is shown schematically in panel A. LacO, lac operator; T7 pol, bacteriophage T7 RNA polymerase; LacI, E. coli lac repressor; GPT, E. coli guanine phosphoribosyltransferase; PT7, T7 promoter.
The antibiotic rifampin prevents the association of D13 with viral membranes and consequently inhibits VACV crescent membrane formation (28). In cells infected by WT VACV in the presence of rifampin, D13 accumulated as cytoplasmic inclusions, but A11, A13, and A10 localized to viral factories (Fig. 4B), demonstrating that the localization of A11 to factories was not due to an association with D13. Again, A11 colocalized more closely with A13 than with A10 (Fig. 4B, insets).
The studies under the conditions of A14 repression and rifampin treatment together showed that A11 colocalized closely with MV membrane proteins but not with either core proteins or D13. They also showed that the cytoplasmic distribution of A11 under A6 repression conditions was not simply due to a block in crescent formation or virion assembly.
A11 associates with membranes.
Similar to results shown in Fig. 3, when HeLa cells infected with WT VACV were fractionated by ultracentrifugation on an iodixanol gradient, A11 also sedimented to the parts of the gradient (fractions 10 to 12) that were denser than the ER fractions. To confirm that A11 associated with membranes during normal VACV replication, we performed three additional biochemical experiments with the A11 fractions derived from WT VACV-infected cells.
To exclude the possibility that A11 might sediment in the gradient due to the formation of large protein aggregates, A11 fractions were treated with buffer containing no or 1% Triton X-100 and then sedimented at 100,000 × g for 2 h. Without detergent treatment, most of the A11, A10, and A13 proteins were in the pellet (Fig. 5A). With detergent treatment, however, all of A11 and A13 were in the supernatant, while A10 remained in the pellet. This suggests that A11 was pelleted due to an association with membranes, which were dissolved by the detergent. A10 was pelleted even after the detergent treatment, presumably because it had been incorporated into the virion core, which was not dissolved by the detergent.
Fig 5.
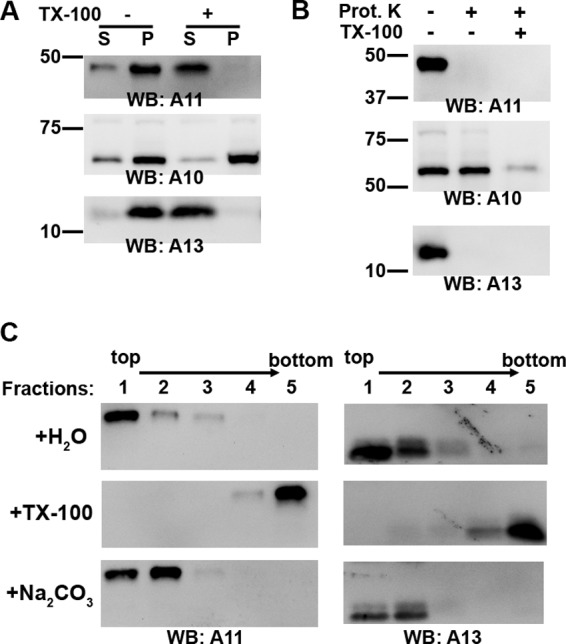
A11 associates with membranes during normal VACV replication. HeLa cells were infected with WT VACV at an MOI of 5 PFU/cell for 8 h, and the cell lysates were fractionated by ultracentrifugation on a 5 to 25% continuous iodixanol gradient as described for Fig. 3. The main fractions containing A11 (fractions 10 to 12) were pooled together and treated as follows. (A) The A11 fraction was mixed with Triton X-100 at a final concentration of 1% (+) or with water (-), layered on top of a 0.5 M sucrose cushion, and centrifuged at 100,000 × g for 2 h. The pellet (P) and TCA-precipitated supernatant (S) were analyzed by Western blotting. The positions and masses (in kilodaltons) of marker proteins are indicated on the left. (B) The A11 fraction was subjected to limited proteinase K digestion in the presence or absence of 1% Triton X-100 and analyzed by Western blotting. (C) The A11 fraction was incubated for 30 min with water (H2O), with Triton X-100 (TX-100) at a final concentration of 1%, or with Na2CO3 (pH 11.5) at a final concentration of 100 mM. The iodixanol concentration of the sample was adjusted to 30%. One milliliter of the sample was placed at the bottom of a centrifuge tube and overlaid with 3 ml of 25% iodixanol and 0.5 ml of 5% iodixanol. Ultracentrifugation was performed at 200,000 × g for 2 h. Fractions of the gradient were precipitated with TCA and analyzed by Western blotting. The direction of the gradient is indicated above the lanes.
To determine whether A11 was external or internal to a membrane structure, A11 fractions were subjected to limited proteinase K digestion. In the absence of detergent, A10 was completely protected from the protease digestion, but both A11 and A13 were completely digested (Fig. 5B), indicating that A11 is not inside a membrane structure.
We also performed a membrane flotation assay (25), which has been widely used to determine whether a protein is membrane associated. A11 fractions were adjusted to buffer containing 30% iodixanol, which has approximately the same density as proteins and is denser than cellular membrane organelles or vesicles. The samples were placed at the bottom of a 5% and 25% iodixanol step gradient. Upon ultracentrifugation, membranes floated out of 30% and 25% iodixanol and into the 5%/25% interface (fraction 1 in Fig. 5C), due to their intrinsic buoyant density, while cytosolic proteins remained at the bottom loading zone (fraction 5 in Fig. 5C). Both A11 and A13 floated to the top of the gradient (fractions 1 and 2), indicating that A11 associated with membranes. In the presence of 1% Triton X-100, both A11 and A13 stayed at the bottom (fraction 5). A11 also floated to the top, even after the samples were treated with sodium carbonate, which has been shown to break up cellular vesicles and strip off peripheral membrane proteins (7). This indicates that A11 associates with membrane more tightly than a peripheral membrane protein.
To determine whether A11 is an integral membrane protein, we performed a Triton X-114 phase separation experiment. Cells infected with WT VACV were solubilized at 4°C with a buffer containing 1% Triton X-114. The detergent and aqueous phases were then separated at 37°C (Fig. 6). While WR148 and D13 partitioned completely in the aqueous phase, A13 and D8 partitioned in the detergent phase, consistent with their characterization as integral membrane proteins. Interestingly, approximately half of the A11 protein partitioned in the detergent phase, with the remaining in the aqueous phase, demonstrating that A11 has some intrinsic affinity to lipids.
Fig 6.
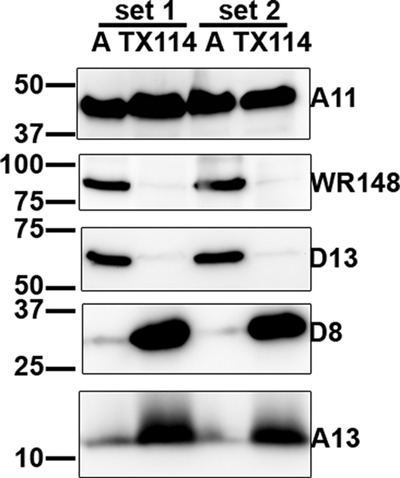
Partial partition of A11 with detergent upon Triton X-114 phase separation. HeLa cells were infected with WT VACV at an MOI of 5 PFU/cell for 20 h and lysed in ice-cold buffer containing 1% Triton X-114. The soluble cell lysate was separated into aqueous (A) and detergent (TX114) phases after a short incubation at 37°C, as described in Materials and Methods. The samples were precipitated with TCA and immunoblotted with antibodies against the indicated viral proteins. The experiment was performed in duplicate, and results from both sets are shown.
A11 does not associate with cellular membranes in the absence of infection or VACV late gene expression.
Although A11 associates with membranes under normal VACV infection conditions, it differs from MV membrane proteins in that it demonstrated cytosolic localization when A6 expression was repressed (Fig. 2 and 3), suggesting that the association of A11 with membranes is regulated during VACV infection. To test this idea further, we determined whether A11 associated with cellular membranes in the absence of infection or VACV late gene expression. To express A11 in the absence of VACV infection, we transfected 293T cells with a mammalian expression plasmid containing a human codon-optimized A11 gene (Fig. 7A). To express A11 during VACV infection but in the absence of VACV late gene expression, we transfected 293T cells with a plasmid containing a bacteriophage T7 promoter-regulated A11 and then infected the cells with a T7 polymerase-expressing VACV (vTF7.3) in the presence of a viral DNA replication inhibitor (Fig. 7B). Under both conditions, A11 localized diffusely in the cytoplasm, as determined by immunofluorescence (data not shown). Furthermore, A11 did not float up to the interface of 5% and 25% iodixanol in the membrane flotation assay, indicating that it did not associate with membranes (Fig. 7). In these experiments, one set of the sample was spiked with cells infected with an inducible A11 virus under A11 repression conditions to provide viral membrane proteins as internal controls for effective flotation of membranes.
Truncation of the C-terminal 31 residues of A11 prevents A11 localization to viral factories.
Residues 288 to 308 and residues 241 to 262 of A11 were identified by a transmembrane domain (TMD) prediction program to be two potential TMDs (Fig. 8A). We performed experiments to determine whether truncation of one or both of these predicted TMDs would affect the localization of A11 to viral factories. BHK cells were infected by an inducible A11 VACV (iA11/GUS) under A11 repression conditions and then transfected with a plasmid encoding full-length or truncated A11 controlled by the native A11 promoter. Immunofluorescence of the cells showed that A11 with truncation of its C-terminal 31 residues (from 288 to 318) or C-terminal 78 residues (from 241 to 318) showed diffuse cytoplasmic staining (Fig. 8B and data not shown). In contrast, full-length A11 or A11 with truncation of its N-terminal 40 residues both localized to viral factories (Fig. 8B). Compared to full-length A11, both the N-terminal and C-terminal truncations of A11 were present at lower level in the cells (Fig. 8C).
Fig 8.

Truncation of the C-terminal predicted transmembrane domain of A11 results in cytoplasmic localization. (A) Kyte-Doolittle hydrophobicity plot of the A11 amino acid sequence. (B) BHK cells were transfected with plasmids with full-length (FL) or truncated A11 (ΔN or ΔC) regulated by the native A11 promoter and infected with an inducible A11 VACV (iA11/GUS) in the absence of the inducer for 12 h. The numbers in parentheses indicate A11 amino acids that were expressed by the plasmid. The plasmids also contained GFP regulated by the VACV p11 late promoter. Immunofluorescence of the cells was performed with anti-A11 MAb 10G11. (C) 293T cells were transfected and infected as described for panel B and analyzed by Western blotting.
Localization of MV membrane proteins to viral factories was defective when A11 expression was repressed.
Both A6 and A11 are required for the appearance of viral membrane precursors in viral factories, but only A6 has been shown to be required for the localization of MV membrane proteins to viral factories. To assess whether A11 is also required for the latter process, we examined the intracellular localization of various VACV proteins under conditions of A11 repression by immunofluorescence (Fig. 9) and cell fractionation analysis (data not shown). Similar to cells infected under A6 repression conditions, cells infected with iA11/GFP in the absence of IPTG had a profound defect in localization of MV membrane proteins to viral factories (Fig. 9). Since this defect was almost identical to the defect previously reported for inducible A6 VACV (13), only images for representative viral proteins are shown in Fig. 9. In the absence of IPTG (-A11), A13 localized to punctate areas outside the viral factories and accumulated in the perinuclear region (Fig. 9A). GFP and the core protein A10 accumulated as large inclusions in and around viral factories (Fig. 9B). D13 accumulated as large cytoplasmic inclusions that were distinct from the core protein-containing GFP inclusions (Fig. 9C). Notably, the protein level (Fig. 9E) or cellular localization of A6 (Fig. 9D) was not affected by repression of A11 expression, in contrast to the effect of A6 on the protein level and cellular localization of A11.
Fig 9.
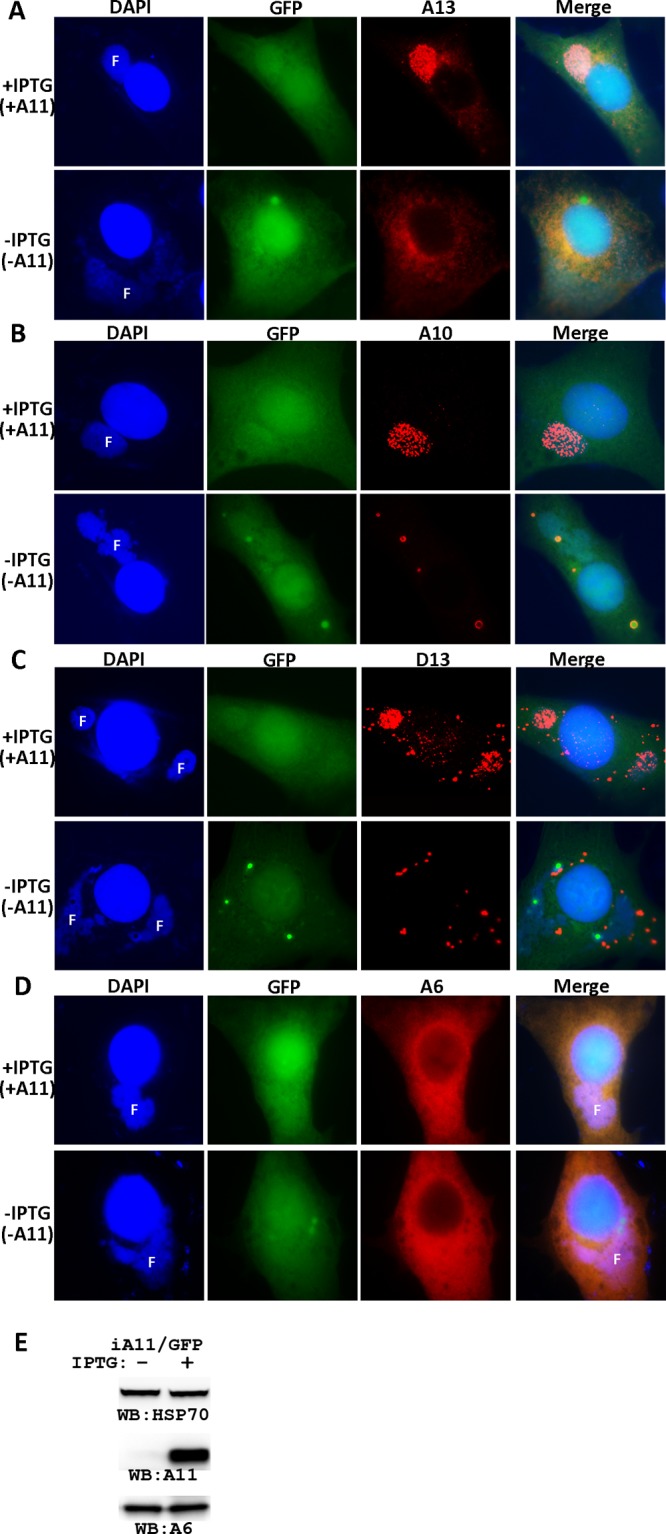
The effect of A11 repression on intracellular localization of VACV proteins. (A to D) BHK cells were infected with a GFP-expressing, inducible A11 VACV (iA11/GFP) at an MOI of 0.5 PFU/cell in the presence or absence of 25 μM IPTG. After 8 h, the infected cells were fixed, permeabilized, and stained with primary antibodies for A13 (A), A10 (B), D13 (C), and A6 (D), followed by staining with DAPI and goat anti-mouse IgG coupled to Cy3. F, viral DNA factory. (E) Repression of A11 expression does not affect the A6 protein level. BS-C-1 cells were infected with iA11/GFP in the presence (+) or absence (−) of 25 μM IPTG. The levels of A11, A6, and cellular heat shock protein 70 (HSP70) were determined by Western blotting.
DISCUSSION
Through analysis of conditional lethal VACV mutants, at least five viral proteins, F10, A11, H7, L2, and A6, have been identified as essential for an early step in virion membrane biogenesis prior to the appearance of viral membrane precursors in viral factories. A6, the newest addition to this group of viral proteins, is also required for localization of MV membrane proteins to factories (13). The mechanisms of action for these proteins, however, are largely unknown. Although their functions are presumably involved in membrane modification or trafficking, with the exception of L2 and F10, they are not known to have any connection with membranes or lipids. Our current study has three main findings that provide a better understanding of the molecular functions of A11 and A6. First, A11 associates with viral membranes during VACV replication, thus providing a physical link between A11 and its purported substrate or site of action. Second, membrane association by A11 requires A6 expression, suggesting that A11 membrane association is regulated by VACV. Finally, A11 is also required for localization of MV membrane proteins to viral factories, further supporting the idea that viral membrane precursors or viral membrane proteins are trafficked to viral factories through an active, virus-mediated process.
A11 was previously considered a nonmembrane protein (19), so we were surprised to find that A11 associates with membranes during normal VACV replication. To confirm this finding, we utilized several biochemical assays that have been widely used for distinguishing cytosolic and membrane-associated proteins. In velocity sedimentation experiments, A11 sedimented like a membrane-associated protein when cell lysates were placed either on the top or at the bottom of a density gradient. With samples layered on top of a cushion or gradient, A11precipitated or sedimented to densities that were similar to cellular organelle membranes (Fig. 3 and 5A). With samples placed at the bottom of a density gradient, A11 floated to the interface of 5%/25% iodixanol (Fig. 5C), similar to the behavior of many membrane proteins in the membrane flotation assay (25). In both experimental settings, dissolving membrane with Triton X-100 prevented A11 from precipitating or floating in the gradient, confirming that the observed sedimentation patterns were due to A11 association with membranes. In addition, immunofluorescence showed that A11 colocalized closely with MV membrane proteins in the factories during normal VACV infection or even when virion envelope formation was blocked by rifampin treatment or repression of A14 expression (Fig. 2 and 4). Based on these biochemical and immunofluorescence data, we conclude that A11 associates with viral membranes in the factories during normal VACV replication. Interestingly, A11 is not incorporated into the MV (19) despite its association with viral membrane. We speculate that A11 may be similar to D13 in that it dissociates from viral membrane before IVs matures into MVs. Alternatively, A11 may be present in MVs at a level that is below the level of detection by conventional methods.
How does A11 associate with membranes? A11 is clearly not internal to an enclosed membrane structure, as A11 was not protected from limited proteinase K digestion (Fig. 5B). Furthermore, membrane flotation experiments showed that A11 associated with membranes even after alkaline treatment (Fig. 5C), which opens up membrane vesicles and strips off peripheral membrane proteins (7). This result also suggested that A11 is not a peripheral membrane protein and led us to consider that A11 might be an integral membrane protein. We have two pieces of data supporting this idea. First, truncations of the C-terminal predicted TMD of A11 resulted in a diffuse cytoplasmic localization of A11 (Fig. 8), suggesting that A11 might associate with membranes through a C-terminal TMD. Second, approximately half of A11 protein partitioned with detergent upon Triton X-114 phase separation (Fig. 6), which is often used to define whether a protein is an integral membrane protein. A similar experiment in a previous report showed that a small, but detectable, amount of A11 protein partitioned with the detergent (19). We are confident that the partition of A11 to the detergent phase in our experiment was not due to insufficient separation of the detergent and aqueous phases, as our negative controls, the cytosolic protein WR148, and peripheral membrane protein D13 did not partition with the detergent. It is obvious that A11 is different from the conventional integral membrane proteins, as A11 was only partially partitioned to the detergent phase, in contrast to the almost-complete partition of A13 and D8 to the detergent phase.
Another key difference between A11 and integral membrane proteins of MV is that A11 membrane association requires VACV infection, and more specifically, the expression of A6. While MV membrane proteins, such as A13 and D8, could be inserted into cellular membranes independent of VACV infection, A11 localized diffusely in the cytoplasm and did not associate with membranes when it was expressed in the absence of either infection or VACV late gene expression (Fig. 7). Furthermore, A11 did not associate with membranes and localized diffusely in the cytoplasm when A6 expression was repressed (Fig. 2 and 3). The effect of A6 on A11 membrane association is quite specific, as preventing crescent formation at a later step by rifampin treatment or by repression of A14 expression did not affect A11 membrane association (Fig. 4). We can envision three possible scenarios in which A6 may affect A11 membrane association. First, A11 may associate with viral membranes indirectly through an interaction with certain viral membrane proteins, the expression of which depends on A6 expression. Among MV membrane proteins examined so far, only those in the VACV entry fusion complex (EFC) are present at reduced levels when A6 expression is repressed (13). However, repressing the expression of EFC proteins did not affect VACV morphogenesis, indicating that A11 functions independently of EFC proteins. Furthermore, A11 does not appear to be a peripheral membrane protein as we discussed above. Second, A11 may associate with lipids on viral membranes, which are absent when A6 expression is repressed. This would, however, imply that lipids on viral membranes are intrinsically different from lipids on cellular membranes, and there is currently no evidence to support this idea. Third, A6 may directly facilitate the insertion of A11 into viral membranes. As we discussed above, A11 has some intrinsic affinity to lipids, which is not sufficient for A11 to associate with membranes in the absence of VACV infection. A6 may help A11 undergo some conformational change that increases the affinity of A11 toward lipids. A direct effect of A6 on A11 is supported by the finding that A6 coprecipitated A11 (Fig. 1). The interaction between A6 and A11, however, is either weak or transient, as only a small amount of A11 coprecipitated with A6, and the cellular localization of A6 and A11 did not overlap extensively. During normal VACV replication, A11 localized to viral factories while A6 localized diffusely in the entire cytoplasm, including the factories (14). It is possible that a weak or transient interaction between A6 and A11 may be sufficient to allow A6 to facilitate A11 membrane association. Regardless of the underlying mechanism, our current study showed clearly that A11 membrane association is regulated by a viral process involving A6. In contrast, the protein level or intracellular distribution of A6 is not affected by the repression of A11 expression (Fig. 9), indicating that A11 does not regulate A6 protein localization or expression.
Among the group of proteins that are involved in virion membrane biogenesis, A6 is the only one that has been previously shown to be required for localization of MV membrane proteins to viral factories. When A6 expression is repressed, many MV membrane proteins no longer localize to viral factories but instead localize to cellular secretory compartments outside the viral factories, suggesting that MV membrane proteins or precursors of MV membranes are actively trafficked to viral factories by a virus-mediated process. Through immunofluorescence and cell fractionation studies, we found that repression of A11 expression also caused a profound defect in localization of MV membrane proteins to viral factories (Fig. 9). In contrast, blocking crescent formation at a later step by rifampin treatment or by repression of A14 expression did not affect localization of MV membrane proteins to viral factories (Fig. 4). These results further support the idea that MV membrane proteins or precursors of MV membranes are actively trafficked to sites of virion assembly through a virus-mediated process, which requires A11 as well as A6.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI079217 (Y.X.) and AI081928 (J.D.).
We thank Steve Hartson for mass spectrometry analyses, performed in the DNA/Protein Resource Facility at Oklahoma State University, using resources supported by the NSF MRI and EPSCoR programs (DBI/0722494).
Footnotes
Published ahead of print 8 August 2012
REFERENCES
- 1. Ansarah-Sobrinho C, Moss B. 2004. Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J. Virol. 78:6335–6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bisht H, Weisberg AS, Szajner P, Moss B. 2009. Assembly and disassembly of the capsid-like external scaffold of immature virions during vaccinia virus morphogenesis. J. Virol. 83:9140–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Byrd CM, Bolken TC, Hruby DE. 2002. The vaccinia virus I7L gene product is the core protein proteinase. J. Virol. 76:8973–8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 66:31–124 [DOI] [PubMed] [Google Scholar]
- 5. Earl PL, Moss B, Wyatt LS, Carroll MW. 2001. Generation of recombinant vaccinia viruses. Curr. Protoc. Mol. Biol. Chapter 16:Unit16.17 [DOI] [PubMed] [Google Scholar]
- 6. Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 83:8122–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. 1982. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93:97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heuser J. 2005. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J. Cell Biol. 169:269–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Husain M, Weisberg AS, Moss B. 2006. Existence of an operative pathway from the endoplasmic reticulum to the immature poxvirus membrane. Proc. Natl. Acad. Sci. U. S. A. 103:19506–19511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keller A, Nesvizhskii AI, Kolker E, Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74:5383–5392 [DOI] [PubMed] [Google Scholar]
- 11. Maruri-Avidal L, Domi A, Weisberg AS, Moss B. 2011. Participation of vaccinia virus l2 protein in the formation of crescent membranes and immature virions. J. Virol. 85:2504–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maruri-Avidal L, Weisberg AS, Moss B. 2011. Vaccinia virus L2 protein associates with the endoplasmic reticulum near the growing edge of crescent precursors of immature virions and stabilizes a subset of viral membrane proteins. J. Virol. 85:12431–12441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meng X, et al. 2012. Vaccinia virus A6 is essential for virion membrane biogenesis and localization of virion membrane proteins to sites of virion assembly. J. Virol. 86:5603–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meng X, Embry A, Sochia D, Xiang Y. 2007. Vaccinia virus A6L encodes a virion core protein required for formation of mature virion. J. Virol. 81:1433–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meng X, Xiang Y. 2006. Vaccinia virus K1L protein supports viral replication in human and rabbit cells through a cell-type-specific set of its ankyrin repeat residues that are distinct from its binding site for ACAP2. Virology 353:220–233 [DOI] [PubMed] [Google Scholar]
- 16. Meng X, et al. 2011. Generation and characterization of a large panel of murine monoclonal antibodies against vaccinia virus. Virology 409:271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moss B. 2007. Poxviridae: the viruses and their replication, p 2905–2946 In Knipe DM, Howley PM. (ed), Fields virology, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 18. Punjabi A, Traktman P. 2005. Cell biological and functional characterization of the vaccinia virus F10 kinase: implications for the mechanism of virion morphogenesis. J. Virol. 79:2171–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Resch W, Weisberg AS, Moss B. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J. Virol. 79:6598–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Risco C, et al. 2002. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 76:1839–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez JR, Risco C, Carrascosa JL, Esteban M, Rodriguez D. 1997. Characterization of early stages in vaccinia virus membrane biogenesis: implications of the 21-kilodalton protein and a newly identified 15-kilodalton envelope protein. J. Virol. 71:1821–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rodriguez JR, Risco C, Carrascosa JL, Esteban M, Rodriguez D. 1998. Vaccinia virus 15-kilodalton (A14L) protein is essential for assembly and attachment of viral crescents to virosomes. J. Virol. 72:1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salmons T, et al. 1997. Vaccinia virus membrane proteins p8 and p16 are cotranslationally inserted into the rough endoplasmic reticulum and retained in the intermediate compartment. J. Virol. 71:7404–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Satheshkumar PS, Weisberg A, Moss B. 2009. Vaccinia virus H7 protein contributes to the formation of crescent membrane precursors of immature virions. J. Virol. 83:8439–8450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scheiffele P, et al. 1998. Caveolin-1 and -2 in the exocytic pathway of MDCK cells. J. Cell Biol. 140:795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith GL, Vanderplasschen A, Law M. 2002. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 83:2915–2931 [DOI] [PubMed] [Google Scholar]
- 27. Sodeik B, et al. 1993. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J. Cell Biol. 121:521–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Szajner P, Weisberg AS, Lebowitz J, Heuser J, Moss B. 2005. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J. Cell Biol. 170:971–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Szajner P, Weisberg AS, Moss B. 2004. Evidence for an essential catalytic role of the F10 protein kinase in vaccinia virus morphogenesis. J. Virol. 78:257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Traktman P, Caligiuri A, Jesty SA, Liu K, Sankar U. 1995. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of virion morphogenesis. J. Virol. 69:6581–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Traktman P, et al. 2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74:3682–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ward G, Stover C, Moss B, Fuerst T. 1995. Stringent chemical and thermal regulation of recombinant gene expression by vaccinia virus vectors in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 92:6773–6777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wolffe EJ, Moore DM, Peters PJ, Moss B. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu C, et al. 2011. An epitope conserved in orthopoxvirus A13 envelope protein is the target of neutralizing and protective antibodies. Virology 418:67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]