

Abstract
Bats are natural hosts for a large variety of zoonotic viruses. This study aimed to describe the range of bat viromes, including viruses from mammals, insects, fungi, plants, and phages, in 11 insectivorous bat species (216 bats in total) common in six provinces of China. To analyze viromes, we used sequence-independent PCR amplification and next-generation sequencing technology (Solexa Genome Analyzer II; Illumina). The viromes were identified by sequence similarity comparisons to known viruses. The mammalian viruses included those of the Adenoviridae, Herpesviridae, Papillomaviridae, Retroviridae, Circoviridae, Rhabdoviridae, Astroviridae, Flaviridae, Coronaviridae, Picornaviridae, and Parvovirinae; insect viruses included those of the Baculoviridae, Iflaviridae, Dicistroviridae, Tetraviridae, and Densovirinae; fungal viruses included those of the Chrysoviridae, Hypoviridae, Partitiviridae, and Totiviridae; and phages included those of the Caudovirales, Inoviridae, and Microviridae and unclassified phages. In addition to the viruses and phages associated with the insects, plants, and bacterial flora related to the diet and habitation of bats, we identified the complete or partial genome sequences of 13 novel mammalian viruses. These included herpesviruses, papillomaviruses, a circovirus, a bocavirus, picornaviruses, a pestivirus, and a foamy virus. Pairwise alignments and phylogenetic analyses indicated that these novel viruses showed little genetic similarity with previously reported viruses. This study also revealed a high prevalence and diversity of bat astroviruses and coronaviruses in some provinces. These findings have expanded our understanding of the viromes of bats in China and hinted at the presence of a large variety of unknown mammalian viruses in many common bat species of mainland China.
INTRODUCTION
The order Chiroptera comprises two suborders, frugivorous Megachiroptera and insectivorous Microchiroptera. Chiroptera is the second largest group of mammalian species, accounting for about 25% of all mammalian species. With the exception of the Primates, Chiroptera has the broadest geographical distribution among mammalian species (64). There are 1,240 bat species widely distributed all over the world, except at the South and North Poles. Mainland China harbors 120 bat species that belong to 30 genera and 7 families (60).
Bats are considered natural carriers of a large variety of viruses. Many studies have successfully identified novel bat viruses on the basis of consensus primers or sequence-independent PCR amplification (27, 34, 63). Over 80 virus species have been detected in bats, including several emergent human pathogens, like severe acute respiratory syndrome coronaviruses (SARS-CoVs), lyssaviruses, henipaviruses, Marburg virus, and Ebola viruses (5, 8, 16, 27–29, 34, 48, 51, 59, 61). Because the distribution and habitats of bats are closely related to those of humans (32, 53), zoonotic diseases may be transmitted from bats to humans.
The recent advent of next-generation sequencing (NGS) technology, including pyrosequencing (454; Roche) and sequencing by synthesis (Solexa genome analyzer; Illumina) (36), has facilitated the use of metagenomic analyses to characterize viruses in many types of samples. NGS-based surveys of the range of viruses present in natural hosts have become important research tools in basic virology, diagnostic virology, and disease prevention and control. In previous studies, this approach was successful for analyzing the viromes and identifying viruses in wild rodent feces, bat feces, human nasopharyngeal aspirates, and human feces (11, 39, 57, 67). The application of NGS to bats is expected to provide more information for identifying novel viruses and characterizing the range of viruses present in different samples.
In this study, we collected pharyngeal and anal swab samples from 11 insectivorous bat species from six provinces in China for metagenomic analyses. On the basis of sequence-independent PCR amplification, NGS (Solexa Genome Analyzer II [GA II]; Illumina), and sequence similarity comparisons, we characterized the viromes of these 11 insectivorous bat species. This revealed the complete or partial genome sequences of 13 novel mammalian viruses that were closely related to human viruses, including herpesviruses (HVs), papillomaviruses (PVs), circovirus (CV), bocavirus (BoV), pestivirus (PestV), foamy virus (FV), and three new members of the Picornaviridae. We also confirmed and analyzed other viruses that had previously been reported in different bat species from other regions, including coronaviruses (CoVs), astroviruses (AstVs), adenoviruses (AdVs), and adeno-associated viruses (AAVs).
MATERIALS AND METHODS
Ethics statement.
Bats were treated according to the guidelines of Regulations for the Administration of Laboratory Animals (Decree No. 2 of the State Science and Technology Commission of the People's Republic of China, 1988). The sampling was approved by the Ethics Committee of the Institute of Pathogen Biology, Chinese Academy of Medical Sciences & Beijing Union Medical College.
Collection of samples.
Pharyngeal swab and anal swab samples from captured bats were immersed into maintenance medium in a virus sampling tube (Yocon, China) and temporarily stored at −20°C. After the sampling was finished, samples were transported to the laboratory and stored at −80°C.
Purification of viral nucleic acids.
Tubes with either the pharyngeal swab or anal swab sample in maintenance medium were vigorously vortexed to resuspend the samples completely into solution. Samples of each species were then pooled by adding 1 ml from each maintenance medium sample into one fresh sample tube. The 11 pooled samples, classified by species, were then separately filtered through a 0.45-μm-pore-size polyvinylidene difluoride filter (Millipore) to remove eukaryotic cell- and bacterium-sized particles. The filtered samples were then centrifuged at 150,000 × g for 3 h at 4°C. The pellets from ultracentrifugation were resuspended in 100 μl of Hanks' balanced salt solution. To remove the naked DNA and RNA, 100 μl of the resuspended pellet from each pooled sample was digested in a cocktail of DNase and RNase enzymes consisting of 14 U of Turbo DNase (Ambion), 20 U of benzonase (Novagen), and 20 U of RNase One (Promega) at 37°C for 2 h in 1× DNase buffer (Ambion). The viral DNA and RNA were simultaneously isolated and eluted from the enzyme-digested solution into 60 μl AVE buffer with a QIAamp viral RNA minikit (Qiagen).
Reverse transcription (RT) and sequence-independent PCR amplification of viral nucleic acids.
Viral first-strand cDNA was synthesized in a 20-μl reaction mixture with 2 μl of viral nucleic acids from each pooled sample and 100 pmol of primer K-8N (GACCATCTAGCGACCTCCACNNNNNNNN), as previously described (10, 56). To convert the first-strand cDNA into double-stranded cDNA, 20 μl of the first-strand cDNA was incubated at 37°C for 1 h and then at 75°C for 10 min in the presence of 5 U of Klenow fragment (NEB) in 1× NEB buffer 2 (final volume, 25 μl).
Sequence-independent PCR amplification was conducted with 5 μl of the double-stranded cDNA template in a final reaction volume of 50 μl, which contained 1× Phusion HF buffer, 200 μM deoxynucleoside triphosphate (dNTP), 1 μM primer K (GACCATCTAGCGACCTCCAC), and 0.5 U Phusion DNA polymerase (NEB). The PCR cycling was performed as follows: 98°C for 30 s, followed by 35 cycles of 98°C for 10 s, 55°C for 30 s, and 72°C for 1 min, with a final extension at 72°C for 10 min. A DNA smear larger than 500 bp was excised and extracted with a MinElute gel extraction kit (Qiagen).
Next-generation sequencing by GA II.
As described in our previous study (67), DNA libraries based on the PCR products described above were constructed according to the manufacturer's instructions (Illumina). Each library was sequenced with a GA II sequencer for a single read of 81 bp in length. To maximize the available length and the total output of raw data, each library of a pooled sample was sequenced in an individual lane without indexing (11 lanes in total). Initial image analysis and base calling were performed with the GAPipeline program and the default parameters. A series of in-house Perl scripts was then employed for further quality control, and reads were culled according to the following criteria: (i) reads filtered with Illumina's Consensus Assessment of Sequence and Variation (CASAVA) software, (ii) reads with no call sites, (iii) reads with similarity to the sequencing adaptor and the primer K sequence, and (iv) duplicate reads and low-complexity reads. Only reads that passed the quality control were considered valid sequences.
Taxonomic assignment.
Sequence similarity-based taxonomic assignments were conducted as described in our previous study (67). The valid sequence reads were aligned to sequences in the NCBI nonredundant nucleotide database (NT) and the nonredundant protein database (NR) (downloaded from the NCBI FTP server in October 2011) using BLASTn and BLASTx, respectively. The taxonomies of the aligned reads with the best BLAST scores (E scores < 10−5) from all 11 lanes were parsed and exported with the MEGAN (version 4) MetaGenome Analyzer (MEGAN 4) (Fig. 1).
Fig 1.

Taxonomic classification based on bat virome alignments. Aligned reads with the best BLAST scores (E < 10−5) from 11 viromes (lanes 1 to 11) were exported from MEGAN 4. Reads from different viromes (lanes 1 to 11) are labeled with different colors. The number of reads in each family is given after the family name. dsDNA, double-stranded DNA; dsRNA, double-stranded RNA; ssDNA, single-stranded DNA; ssRNA, single-stranded RNA.
Partial genomes of novel viruses sequenced by reads-based PCR.
Sequence reads classified into the same virus genus or species found in MEGAN 4 were extracted. The extracted reads were then sorted and assembled with the SeqMan program (Lasergene). The accurate locations of the extracted reads and the relative distances between reads of the same virus were determined on the basis of the alignment results exported with MEGAN 4. Specific primers were designed from the located sequence reads. Fragments between reads were amplified (PCR for DNA viruses and RT-PCR for RNA viruses) with nested specific primers and then sequenced. Partial genome sequences were obtained by assembling amplified fragments and sequence reads. The primers used for amplifying fragments of each virus are available on request.
Complete genome sequencing and structure prediction of novel viruses.
On the basis of the partial genome sequences of viruses, the remaining genome sequences were determined with inverse PCR, genome walking, and 5′ and 3′ rapid amplification of cDNA ends (RACE). For bat HVs, the complete open reading frames (ORFs) of glycoprotein B (gB) and DNA polymerase (DPOL) were obtained with the genome walking kit (TaKaRa), based on fragments located in these two genes. For bat PVs and the CV, inverse PCR was used to determine the complete circular genome of each virus on the basis of the fragments obtained in the reads-based PCR. For bat BoV, based on the sequenced intervening fragment, the 5′ and 3′ ends of the genome were amplified and sequenced with inverse linear PCR and a 5′ RACE kit (Invitrogen), as described previously (23). For bat picornaviruses, the 5′ and 3′ ends of the genomes were completed with a 5′ RACE kit (Invitrogen) and a 3′ full RACE core set, version 2.0 (TaKaRa). All identified viruses were reconfirmed by long-distance nested PCR with specific primers derived from the completed viral genomes (the primer sequences are available on request).
The ORFs of complete or partial genomes of the sequenced viruses were predicted with Vector NTI software (Invitrogen) or with the ORF Finder tool of NCBI (http://www.ncbi.nlm.nih.gov/gorf/gorf.html).
Molecular epidemiology of bat astroviruses.
Random hexamer-generated cDNAs of the 11 pooled samples were synthesized separately with a Superscript III first-strand synthesis system (Invitrogen). Previously described heminested PCR primers were used to amplify the conserved regions (387 nucleotides [nt]) of the RNA-dependent RNA polymerase (RdRp) gene of AstVs (8, 66). The expected products were then gel extracted and subcloned into the pGEM-T Easy vector (Promega) for sequencing.
Phylogenetic analysis.
Reference viral sequences were downloaded from GenBank. We used the MEGA (version 5.0) program (52) to align nucleotide sequences and deduced amino acid sequences with the MUSCLE package and default parameters. The best substitution model was then evaluated with the Model Selection package; finally, we constructed a maximum likelihood method with an appropriate model to process the phylogenetic analyses and create phylogenetic trees with 1,000 bootstrap replicates.
Nucleotide sequence accession numbers.
All complete and partial genome sequences have been submitted to GenBank. The accession numbers for gB and DPOL genes of bat HVs are JQ814845, JQ814846, JN692429, and JN692430. The accession numbers for bat PVs are JQ814847 and JQ814848. The accession number for bat CV is JQ814849. The accession number for bat BoV is JQ814850. The accession numbers for bat picornaviruses are JQ814851, JQ814852, and JQ814853. The accession number for bat PestV is JQ814854. The accession number for bat FV is JQ814855. The accession numbers for bat AstVs are JQ814856 to JQ814871. The GA II sequence data have been deposited in the NCBI sequence reads archive (SRA) under accession number SRA051252.
RESULTS
Bat sampling.
Bat samples were collected from October 2010 through February 2011 in six provinces (Beijing, Hunan, Jiangxi, Yunnan, Guizhou, and Hainan) in China. We sampled a total of 216 insectivorous bats (11 species) with both pharyngeal and anal swabs (Table 1). All bat roosts in the six provinces were in or close to human gathering places.
Table 1.
Samples of the 11 bat species used in this study and the provinces and dates of collection
| Bat species | No. of samplesa collected in the following province (collection date [yr.mo]): |
Lane for Illumina GA II | |||||
|---|---|---|---|---|---|---|---|
| Beijing (2011.2) | Hunan (2010.10) | Jiangxi (2010.10) | Yunnan (2010.12) | Guizhou (2010.10) | Hainan (2010.12) | ||
| Myotis ricketti | 15 | 14 | 1 | ||||
| Rhinolophus ferrumequinum | 15 | 2 | |||||
| Myotis myotis | 16 | 12 | 3 | ||||
| Rhinolophus sinicus | 13 | 6 | 4 | ||||
| Hipposideros armiger | 15 | 5 | |||||
| Hipposideros pomona | 23 | 6 | |||||
| Hipposideros cineraceus | 20 | 7 | |||||
| Ia io | 11 | 8 | |||||
| Tylonycteris robustula | 10 | 9 | |||||
| Rhinolophus affinis | 19 | 10 | |||||
| Miniopterus schreibersii | 27 | 11 | |||||
Each bat sample contained a copy from the pharyngeal swab sample and a copy from the anal swab sample obtained from the same bat.
Metagenomic analysis of NGS results.
All pharyngeal swab and anal swab samples were processed with a viral particle-protected, nucleic acid purification method. The extracted RNA and DNA were amplified by sequence-independent RT-PCR. The amplified viral nucleic acid libraries of the 11 bat species were then sequenced with an Illumina GA II (one lane per species; Table 1). A total of 29,272,685 sequence reads of 81 bp in length were obtained from the 11 bat species (Fig. 1; the number of sequences in each family is shown after the family name). A total of 9,103,279 sequence reads were classified as cellular organisms (∼31.1% of total sequence reads, including bacteria, archaea, and eukaryota). A total of 353,973 sequences were best matched with viral proteins (∼1.2% of total sequence reads). The remaining 19,584,097 sequence reads had no significant similarity to any amino acid sequence in NR (∼66.9% of the total number of sequence reads, including unclassified sequences, other sequences, unassigned sequences, and no hits). The complete data on the virome identified for the 11 bat species were released to the NCBI SRA database with accession number SRA051252.
In agreement with previous studies, the total sequence reads related to viruses included mostly those of phages (order Caudovirales, families Inoviridae and Microviridae, and unclassified phages), insect viruses (families Baculoviridae, Iflaviridae, Dicistroviridae, and Tetraviridae; subfamily Densovirinae), fungal viruses (families Chrysoviridae, Hypoviridae, Partitiviridae, and Totiviridae), and a small quantity of plant viruses. Viral sequences related to mammalian viruses (31,744 sequence reads) mainly included those of the families Adenoviridae, Herpesvirdae, Papillomaviridae, Retroviridae, Circoviridae, Rhabdoviridae, Astroviridae, Flaviridae, Coronaviridae, and Picornaviridae and the subfamily Parvovirinae (labeled with red blocks in Fig. 1). These comprised ∼9% of the total viral hits. Most of the sequence reads related to mammalian viruses showed low percentages of nucleotide and amino acid sequence identities with known viruses.
Bat HVs.
HVs are large, enveloped, double-stranded DNA viruses that infect the skin, mucous membranes, and nervous systems of a wide variety of vertebrate hosts, including humans (13, 63). Over 80 HV species are currently registered by the International Committee on Taxonomy of Viruses (ICTV). These HV species are classified into three subfamilies (Alpha-, Beta-, and Gammaherpesvirinae) within the family Herpesviridae.
We identified four new HVs in this study. Two betaherpesviruses (BHVs) were detected in Rhinolophus ferrumequinum of Beijing (RfBHV-1) and Tylonycteris robustula of Hainan (TrBHV-1); two gammaherpesviruses (GHVs) were detected in Myotis ricketti of Beijing (MrGHV-1 and MrGHV-2). On the basis of the NGS reads, we established the complete sequences of the gB and DPOL genes of the four bat HVs (RfBHV-1, 5,534 bp; TrBHV-1, 5,912 bp; MrGHV-1, 5,874 bp; MrGHV-2, 5,983 bp). Pairwise similarity analysis of concatenated gB and DPOL genes indicated 52.1% nucleotide sequence identity and 44.2% amino acid sequence identity (1,843 versus 1,969 amino acids [aa]) between the two bat betaherpesviruses (RfBHV-1 and TrBHV-1) and 58% nucleotide sequence identity and 56% amino acid sequence identity (1,862 versus 1,873 aa) between the two bat gammaherpesviruses (MrGHV-1 and MrGHV-2). Furthermore, among bats, both RfBHV-1 and TrBHV-1 showed the highest similarity with bat BHV-2 (52.1% and 57.2% nucleotide sequence identities, respectively; 45.3% and 54.8% amino acid sequence identities, respectively); among betaherpesviruses, RfBHV-1 showed high similarity to human HV 6 (HHV-6; 57.8% nucleotide sequence identity and 54.3% amino acid sequence identity) and TrBHV-1 showed high similarity to cercopithecine HV 8 (CeHV-8; 56.4% nucleotide sequence identity and 51.6% amino acid sequence identity). A BLAST search indicated that among the bat gammaherpesviruses, MrGHV-1 and MrGHV-2 showed the highest similarity to bat GHV4 (59.4% amino acid sequence identity); among gammaherpesviruses, MrGHV-1 was most similar to saimiriine HV 2 (SaHV2; 63.7% nucleotide sequence identity and 57% amino sequence acid sequence identity) and MrGHV-2 was most similar to equid HV 2 (EHV2; 63.8% nucleotide sequence identity and 66.1% amino acid sequence identity). From the phylogenetic tree based on the concatenated gB and DPOL proteins (Fig. 2), RfBHV-1, TrBHV-1, and MrGHV1 could not be assigned to any known genus; however, MrGHV2 might be assigned to the genus Percavirus (the same root was shared with Badger herpesvirus and EHV2 in the genus Percavirus with 100% bootstrap support). Further characterization of these four bat HVs is needed to determine the exact taxonomy.
Fig 2.

Phylogenetic analysis of concatenated gB and DPOL proteins from the four bat HVs and other representative HV species. The phylogenetic tree was based on deduced amino acid sequences of concatenated gB and DPOL ORFs. Two novel bat gammaherpesviruses are labeled with black circles, and two novel bat betaherpesviruses are marked with black triangles. RRV, Macaca mulatta rhadinovirus; HDHV1, Hipposideros diadema HV1; AHV1, alcelaphine HV1; PLHV1, porcine lymphotropic HV1; EBV, Epstein-Barr virus; CavHV2, caviid HV2; HCMV, human cytomegalovirus.
Bat PVs.
The large Papillomaviridae family includes 30 genera (ICTV) (2) of small nonenveloped, double-stranded, circular DNAs (∼8 kb in length). PVs infect skin and mucosa and cause a wide variety of benign and malignant epithelial tumors in higher vertebrate species (2, 42–44); human PV has a special relationship with female cervical carcinoma (19). The majority of PVs were isolated from humans. Recently, many groups have successfully identified PVs with high host specificity in other nonhuman species, including manatee, raccoon, and wild rodents (39, 42–44).
In this study, we identified two full-length PV genomes in two insectivorous bats: one in Myotis ricketti of Beijing (MrPV-1) and the other in Miniopterus schreibersii of Hainan (MsPV-1). The complete circular genomes of the two bat PVs were 7,339 bp (MrPV-1) and 7,632 bp (MsPV-1).
MrPV-1 contained five clear ORFs that encoded early proteins E6 (184 aa), E1 (698 aa), and E2 (357 aa) and late proteins L2 (435 aa) and L1 (500 aa) (Fig. 3A). The E6 protein contained two characteristic zinc binding domains (C-X2-C-X29-C-X2-C) separated by 36 aa. The E1 protein contained a C-terminal conserved ATP-binding site specific for the ATP-dependent helicase (GPPNTGKS). There was a noncoding region (NCR; 447 bp) between the end of L1 and the start of E6 (nt 6,893 to 7,339). The NCR contained a typical E2-binding site (ACC-X6-GGT) at nucleotide position 7,217, a modified E2-binding site (AAC-X6-GGT) at position 7,303, a polyadenylation site (AATAAA) for L1 and L2 mRNA at position 6,985, and a TATA box for the E6 promoter at position 7,168. An E7 gene, presents in most PV species with a zinc binding domain and a retinoblastoma tumor suppressor binding motif, could not be found in MrPV-1.
Fig 3.

Two bat papillomaviruses. (A) Genomic organizations of MrPV-1 and MsPV-1; (B) phylogenetic tree based on concatenated L2 and L1 proteins from MrPV-1, MsPV-1, and other species of 30 genera. The two novel bat papillomaviruses are labeled with black triangles. ChPV, Capra hircus PV; RAPV, Rousettus aegyptiacus PV; PsPV, Phocoena spinipinnis PV; TtPV, Tursiops truncatus PV; HPV-4, human PV type 4; MmPV, Mus musculus PV; EePV, Erinaceus europaeus PV; TmPV, Trichechus manatus PV; MsPV, Miniopterus schreibersii PV; SsPV, Sus scrofa PV; RhPV, rhesus monkey PV; MnPV, Mastomys natalensis PV; CrPV, cottontail rabbit PV; CoPV, canine oral PV; EdPV, Erethizon dorsatum PV; EcPV, Equus caballus PV; CcPV, Caretta caretta PV; FIPV, Francolinus leucoscepus PV; FcPV, Fringilla coelebs PV; PePV, Psittacus erithacus PV.
MsPV-1 contained six clear ORFs, including early genes E6 (135 aa), E7 (98 aa), E1 (623 aa), and E2 (467 aa) and late genes, L2 (482 aa) and L1 (565 aa) (Fig. 3A). The E7 protein contained a zinc binding domain and a conserved retinoblastoma tumor suppressor binding domain (with the L-X-C-X-E motif). The ATP-binding site of E1 was GPANTGKS. The NCR contained five typical E2-binding sites. Other characteristics of MsPV-1 were similar to those of MrPV-1.
The phylogenetic tree based on the concatenated L2 and L1 proteins (Fig. 3B) showed that MrPV-1 shared the same root with Ursus maritimus PV 1 (UmPV-1; 92% bootstrap support) in the genus Omegapapillomavirus, A pairwise alignment indicated that the L1 gene of MrPV-1 shared 65.2% nucleotide sequence identity and 68.5% amino acid sequence identity with the L1 of UmPV-1. According to the general rules (not absolute criteria) of ICTV (2), all PV species in the same genus share 60% to 70% nucleotide sequence homology in the L1 gene; thus, MrPV-1 could be considered a new species of the genus Omegapapillomavirus. In addition, the absence of an E7 ORF in MrPV-1 and UmPV-1 was a common feature among species in the genus Omegapapillomavirus (49). The pairwise alignment analysis indicated that the L1 gene of MsPV-1 shared 57.3% to 58.5% nucleotide sequence identities and 55.6% to 59.9% amino acid sequence identities with the L1 gene of closely related PVs. Thus, the phylogenetic tree indicated that MsPV-1 was located in a separate branch from MrPV-1. Consequently, MsPV-1 might be regarded as the prototype of a new genus. The two bat PVs shared only 58.7% nucleotide sequence identity and 59.0% amino acid sequence identity. It is worth mentioning that the 6 new genera (Dyoeta-, Dyoiota-, Dyotheta-, Dyodelta-, Dyozeta-, and Dyoepsilonpapillomavirus) were not considered in the taxonomic classification process of the two bat PVs, because these genera failed to comply with the general ICTV taxonomy rules.
Bat CV.
The genus Circovirus, in the family Circoviridae, is a group of viruses with small, nonenveloped, circular single-stranded (ssDNAs; 1.7 to 2 kb in length) (14). CVs were previously found to infect pig, bat, mouse, and a wide variety of bird species (6, 17, 31, 54). Porcine CV 1 (PCV-1) and PCV-2 are widespread in the swine population. PCV-2 is the main pathogen associated with postweaning multisystemic wasting syndrome (PMWS) and porcine dermatitis nephropathy syndrome (PDNS) (6, 15, 45).
We characterized one CV from Rhinolophus ferrumequinum of Beijing, and the full length was sequenced. The complete circular genome of Rhinolophus ferrumequinum circovirus 1 (RfCV-1) comprised 1,760 nt. Two ORFs that encoded the Rep protein (294 aa) and the Cap protein (218 aa) were separated by a 3′ intergenic region (128 nt) between the two stop codons and a 5′ intergenic region (90 nt) between the two start codons (Fig. 4). The Rep protein sequence included three conserved motifs (22) involved in rolling-circle replication and a putative dNTP-binding box (GKS). The 5′ intergenic region included a potential stem-loop structure (loop motif, ATAGTATTA; inverted repeat, GTGCCGGGG).
Fig 4.

Bat circovirus. Genomic organization of RfCV-1 and phylogenetic tree based on Rep proteins of circoviruses. RfCV-1 is labeled with a black triangle.
The pairwise alignment of Rep showed 37.2% to 57.3% nucleotide sequence identities and 27.6% to 47.6% amino acid sequence identities with Reps of other known CVs. The capsid protein sequence of RfCV-1 showed less than 33% amino acid sequence identity with other known CVs. In accordance with the criteria of ICTV, RfCV-1 could be considered a new species of the genus Circovirus. From the phylogenetic analysis based on the full-length Rep proteins (Fig. 4), we found that RfCV-1 could not be sorted into any other CV clade and not even the reported bat CV clade.
Bat BoV.
The genus Bocavirus, a group of small, nonenveloped, linear ssDNA viruses with ∼5-kb genomes, is one of five genera (Amdovirus, Bocavirus, Dependovirus, Erythrovirus, and Parvovirus) in the subfamily Parvovirinae of the family Parvoviridae. To date, BoVs have been identified in bovines (bovine parvovirus), canines (canine minute virus), swine, gorillas, sea lions, and humans (human bocavirus [HBoV] 1 to 4) (1, 20, 24, 31, 40, 46, 50). Bovine parvovirus is associated with diarrhea in neonatal calves and respiratory and reproductive disease in adult cattle. Canine minute virus causes severe diarrhea, difficulty breathing, myocarditis, anorexia, and even death in dogs. HBoVs are associated with lower respiratory tract infections and gastroenteritis. Recently, BoVs have been associated with gorilla diarrhea and porcine PMWS (4, 25).
We identified a novel BoV in Myotis myotis of Hainan. The complete genome of Myotis myotis BoV (MmBoV-1) comprised 5,038 nt. It harbored three distinct ORFs that encoded the nonstructural proteins NS1 (720 aa) and NP1 (167 aa) and the structural protein VP1/VP2 (659/535 aa) (Fig. 5A). The N terminus of VP1 encoded a putative secretory phospholipase A2 (sPLA2) motif with a Ca2+ binding loop (Y-X-G-X-F) and a conserved motif (HD-X2-Y) in the catalytic center. Multiple-sequence alignments revealed that NS1 of MmBoV-1 shared 43.8% to 54.5% nucleotide sequence identities and 32.6% to 44.7% amino acid sequence identities with other members of the genus Bocavirus. Moreover, VP1 shared 49.0% to 53.8% nucleotide sequence identities and 44.2% to 46.4% amino acid sequence identities with other BoVs. In accordance with the ICTV (25), species of BoVs with less than 95% similarity in nonstructural genes are defined as new species. Therefore, MmBoV-1 was defined as a new species. Two separate phylogenetic trees based on the complete NS1 (Fig. 5B) and VP1 (Fig. 5C) proteins were used to described the evolutionary relationships between MmBoV-1 and other BoVs. The clade of MmBoV-1 was distant from other clusters.
Fig 5.

Bat bocavirus. (A) Genomic organization of MmBoV-1. Phylogenetic trees based on the NS1 protein (B) and the VP1 protein (C) of bocaviruses. MmBoV-1 is labeled with a black triangle. Csl, California sea lion; PBoV, porcine BoV.
Bat picornaviruses.
The family Picornaviridae includes 12 genera (Aphthovirus, Avihepatovirus, Cardiovirus, Enterovirus, Erbovirus, Hepatovirus, Kobuvirus, Parechovirus, Sapelovirus, Senecavirus, Teschovirus, and Tremovirus). It is a group of small, nonenveloped, positive single-stranded RNA viruses with a genome of 7 to 9 kb and a single ORF that encodes a polyprotein. The members of the family Picornaviridae can cause mucocutaneous, encephalic, cardiac, hepatic, neurological, and respiratory diseases in a wide variety of vertebrate hosts, including duck, chicken, bovine, swine, horse, chimpanzee, and human (55). We identified three new members of the family Picornaviridae in bats.
First, we identified a virus in Miniopterus schreibersii of Hainan, which we named Miniopterus schreibersii picornavirus 1. The complete genome of this virus (8,468 nt) contained a single ORF that encoded a large polyprotein (2,280 aa) flanked by a 5′ untranslated region (UTR) (1,407 nt) and a 3′ UTR (215 nt) (Fig. 6A). An alignment analysis of Miniopterus schreibersii picornavirus 1 and other known picornaviruses showed the characteristic gene order of 5′-L (92 aa)-P1 (810 aa; VP4, VP2, VP3, and VP1)-P2 (575 aa; 2A, 2B, and 2C)-P3 (804 aa; 3A, 3B, 3C, and 3D)-3′. It also showed the theorized cleavage sites in the polyprotein. The P1, P2, and P3 polypeptides were hypothetically cleaved at VP4/VP2 (M/D), VP2/VP3 (Q/G), VP3/VP1 (Q/G), 2A/2B (G/P), 2B/2C (A/G), 3A/3B (Q/G), 3B/3C (Q/G), and 3C/3D (E/G). The conserved cleavage motif (NPG/P) for 2A/2B of the genus Cardiovirus was also found in this virus. Similar to other picornaviruses, the 2C protein contained a conserved NTP-binding motif (G-X2-G-X-GKS) and a helicase motif (DDL-X-Q); the 3C protein contained a conserved active protease site (G-X-CG) and a G-X-H motif; and the 3D protein contained conserved KDEIR, GGLPSG, YGDD, and FLKR motifs. The BLASTp results showed that the P1, P2, and P3 regions of this virus had 40.3% to 41.7%, 32.2% to 35.4%, and 42.4% to 45.7% amino acid sequence identities with members of the genus Cardiovirus, respectively. According to the ICTV criteria, members of a genus of the family Picornaviridae should share >40%, >40%, and > 50% amino acid sequence identity with phylogenetically related P1, P2, and P3 genomic regions, respectively (Picornaviridae Study Group, ICTV). Thus, Miniopterus schreibersii picornavirus 1 may be defined as the prototype of a new genus closely related to the genus Cardiovirus.
Fig 6.

Three novel bat picornaviruses. (A) Genomic organizations of Miniopterus schreibersii picornavirus 1 and Ia io picornavirus 1; (B) phylogenetic tree based on the complete 3D protein sequence of picornaviruses; (C) partial genomic organization of Rhinolophus affinis picornavirus 1; (D) phylogenetic tree based on the complete or partial P1 protein sequence of picornaviruses. The three bat picornaviruses are labeled with black triangles. HuTLCV, muman TMEV-like cardiovirus; ThV, theilovirus; EMCV, encephalomyocarditis virus; SVV, Seneca valley virus; ERAV, equine rhinitis A virus; HCoSV-A, human cosavirus A; FMDV-O, foot-and-mouth disease virus O; ERBV, equine rhinitis B virus; PTV, porcine teschovirus; TurdiV-2, turdivirus 2; AiV, Aichi virus; DPTV, avian sapelovirus; PEVA, porcine sapelovirus; SpV-1, simian sapelovirus 1; BEV, bovine enterovirus; HEV71,human enterovirus 71; AEV, avian encephalomyelitis virus; HAV, hepatitis A virus; DHAV-1, duck hepatitis A virus 1; HPeV-2, human parechovirus 2; LV, Ljungan virus.
We found the second virus in Ia io of Guizhou, which we named Ia io picornavirus 1. The nearly complete genome sequence of this virus was established (7,543 nt lacking the 5′ terminus of 5′ UTR). We identified a single ORF that encoded a large polyprotein (2,371 aa). As shown in Fig. 6A, the characteristic gene order of the polyprotein was 5′-L (70 aa)-P1 (841 aa; VP4, VP2, VP3, and VP1)-P2 (667 aa; 2A, 2B, and 2C)-P3 (793 aa; 3A, 3B, 3C, and 3D)-3′, with potential polyprotein cleavage sites. The hypothetical cleavage sites between proteins were VP4/VP2 (A/S), VP2/VP3 (Q/G), VP3/VP1 (Q/G), 2A/2B (Q/G), 2B/2C (Q/G), 3A/3B (Q/G), 3B/3C (Q/G), and 3C/3D (Q/G). Similar to the previously reported bat picornaviruses, enteroviruses, and sapeloviruses, the VP1 protein of Ia io picornavirus 1 had a PAL-X-A-X-ETG motif. Two potential cleavage sites (NFQ/GPEEQ/GIK) were found at the 2A/2B junction. Like the 2A protein of enterovirus and rhinovirus, the 2A protein of Ia io picornavirus 1 contained two conserved G-X-CG motifs that are targeted by a chymotrypsin-like protease. Similar to Miniopterus schreibersii picornavirus 1, the 2C protein of Ia io picornavirus 1 also contained a G-X2-G-X-GKS motif and a DDL-X-Q motif. The 3C protein contained a G-X-CG motif and a conserved RNA-binding motif (DFRDI), and the 3D protein contained the conserved KDELR, GGMPSG, YGDD, and FLKR motifs. Pairwise alignment results revealed that the P1, P2, and P3 regions shared 53.5% to 56.5%, 46.7% to 56.3%, and 61.5% to 67.6% amino acid sequence similarities, respectively, with members of the previously described bat picornavirus group (bat picornavirus 1 from Miniopterus pusillus, bat picornavirus 2 from Miniopterus magnate, and bat picornavirus 3 from Rhinolophus sinicus [27]). Therefore, consistent with that previous study, we could establish a new genus that consisted of these four bat picornaviruses.
The phylogenetic tree based on the complete 3D proteins (Fig. 6B) showed that the clade of Miniopterus schreibersii picornavirus 1 was closely related to the genera Cardiovirus and Senecavirus and Ia io picornavirus 1 was clustered with the three previously reported bat picornaviruses.
We found the third virus in Rhinolophus affinis of Hainan; we named it Rhinolophus affinis picornavirus 1. Its partial sequence (2,317 nt), which covered nearly the complete region of P1 (VP3, 243 aa; VP1, 272 aa; and part of VP0, 214 aa) and 2A (43 aa), was identified (Fig. 6C). The partial P1 region of Rhinolophus affinis picornavirus 1 shared less than 25% amino acid sequence identity with other picornaviruses. The most closely related virus was Ljungan virus, which shared 25.0% amino acid sequence identity with the P1 region of Rhinolophus affinis picornavirus 1. A P1-based phylogenetic analysis (Fig. 6D) indicated that Rhinolophus affinis picornavirus 1 harbored a single phylogenetic root close to the genera Avihepatovirus and Parechovirus, which may represent a new genus in the family Picornaviridae.
Bat PestV.
Pestivirus is a genus in the family Flaviviridae. Pestivirus comprises positive single-stranded RNA viruses with a genome size of ∼12 kb. PestVs have a single ORF that encodes a polyprotein which is flanked by 5′ and 3′ UTRs. PestVs of four species (Bovine viral diarrhea virus 1 [BVDV-1] and BVDV-2, Classical swine fever virus [CSFV], and Border disease virus [BDV]) and other potential species can cross species barriers and infect a wide variety of artiodactylous hosts (hoofed mammals in the order Artiodactyla having an even number of toes, including swine and ruminants) (14, 26, 58). BVDV and CSFV are severely pathogenic in cattle and swine (47, 65).
All previous PestVs were found in animals in the order Artiodactyla, but in this study, one PestV was found in animals in the order Chiroptera. This PestV was identified in Rhinolophus affinis of Hainan (RaPestV-1). We identified a partial genome sequence (5,130 nt) of RaPestV-1 that covered a partial coding sequence of the nonstructural protein NS4B and the complete coding sequences of the structural protein E2 and the nonstructural proteins p7, NS2-3, and NS4A (Fig. 7A). The confirmed partial sequence of RaPestV-1 showed 46.5% to 47.3% nucleotide sequence identities and 32.4% to 32.9% amino acid sequence identities to other PestVs. Multiple-sequence alignment analyses of the E2 region of RaPestV-1 showed lower amino acid sequence identities (∼28%) than alignments with the whole partial sequence. We constructed a phylogenetic tree based on the confirmed partial protein sequence of RaPestV-1 and the corresponding positions of other members of the genus Pestivirus (Fig. 7B). Japanese encephalitis virus (JEV), in the genus Flavivirus, served as an outgroup. The RaPestV-1 clade was distant from the other pestivirus clades. Thus, the RaPestV-1 represented another novel species that occurred evolutionarily distant from the known PestVs in the genus Pestivirus.
Fig 7.

Bat pestivirus. (A) Partial genomic segment of RaPestV-1; (B) phylogenetic analysis based on amino acid sequences of partial regions in RaPestV-1 and the corresponding positions in other pestiviruses. RaPestV-1 is labeled with a black triangle.
Bat FV.
The FVs, also called spumaviruses, are in the family Retroviridae and are complex retroviruses with 12- to 13-kb DNA genomes. They infect cattle, cats, horses, gorillas, monkeys, and humans. In this study, we found an FV in Rhinolophus affinis of Hainan, and we named it RaFV-1. We identified a partial genomic sequence (2,856 nt) that encoded the C terminus of the pol gene and the N terminus of the env gene (Fig. 8A). The partial pol gene of RaFV-1 showed 52% to 59% amino acid sequence similarity with the pol genes of other FVs, and the partial env gene of RaFV-1 showed 35% to 38% amino acid sequence identity with the env genes of other FVs. We constructed a phylogenetic tree based on the concatenated Pol and Env protein sequences of RaFV-1 and other FVs. We used two members of the subfamily Orthoretrovirinae, the avian leukosis virus (ALV) and human immunodeficiency virus type 1 (HIV-1), as outgroups (Fig. 8B). RaFV-1 could be clustered with simian FV (SFV), human FV (HFV), bovine FV (BFV), equid FV (EFV), and feline FV (FFV) in the same genus Spumavirus with strong bootstrap support. This finding indicated that FVs could infect bats, and they showed low genomic similarity with FVs that infect other hosts.
Fig 8.

Bat foamy virus. (A) Partial genomic segment of RaFV-1; (B) phylogenetic tree generated with concatenated partial sequences of the Pol and Env proteins of RaFV-1 and other members of the genus Spumavirus. RaFV-1 is labeled with a black triangle.
Prevalence and genetic diversity of bat AstVs.
Members of the genus Mamastrovirus in the family Astroviridae infect many mammals, including humans, and cause gastroenteritis. In this study, several NGS sequence reads of AstVs were found in Myotis rickeeti of Jiangxi, Rhinolophus sinicus of Hunan, and Miniopterus schreibersii and Tylonycteris robustula of Hainan. Therefore, we used a set of nested PCR primers described previously to detect the conserved regions of the RdRp gene of AstVs in each bat species (8, 66). We identified 16 bat AstVs, including 1 Myotis rickeeti AstV (MrAstV-1), 1 Rhinolophus sinicus AstV (RsAstV-1), 13 Miniopterus schreibersii AstVs (MsAstV-1 to MsAstV-13), and 1 Tylonycteris robustula AstV (TrAstV-1). These 16 bat AstVs showed 63.0% to 100% nucleotide sequence identities to each other and 66.1% to 99.7% nucleotide sequence identities with bat AstVs reported previously in Hong Kong. In particular, two of the bat AstVs that we found in Hainan, MsAstV-5 and MsAstV-13, showed 99.7% and 99.0% nucleotide sequence identities, respectively, with bat AstVs AFCD82 and AFCD269 previously found in Miniopterus spp. of Hong Kong; in addition, three of the bat AstVs, MrAstV-1, MsAstV-1, and TrAstV-1, that we found in Myotis rickeeti, Miniopterus schreibersii, and Tylonycteris robustula, respectively, from Jiangxi and Hainan shared more than 99% nucleotide sequence identities. We constructed a phylogenetic tree based on partial sequences (387 nt) of the RdRp gene to show the evolutionary relationship between these 16 bat AstVs and other AstVs. All bat AstVs were clustered in a single clade, and a large branch of bat AstVs was distinct from other branches of human or ovine AstVs. Two members of the genus Avastrovirus, chicken and turkey astroviruses, were introduced as outgroups (Fig. 9).
Fig 9.
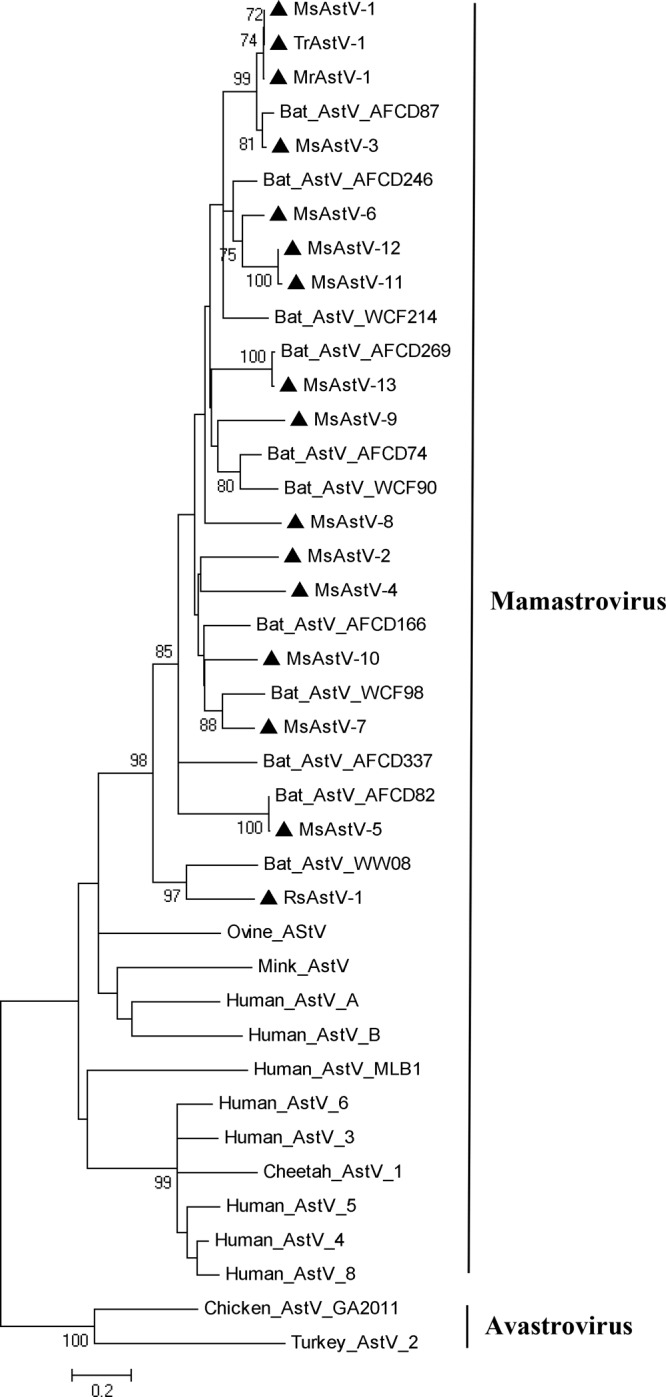
Bat astroviruses. The phylogenetic tree was constructed based on 387 nucleotides of the partial RdRp gene. Novel bat astroviruses are labeled with black triangles.
Bat coronaviruses CoVs.
Bats were considered to be the natural carriers of CoV (BtCoV) in previous studies, because a large number of BtCoVs were identified in a variety of bat species (7, 9, 11, 28, 59). In this study, we found 227 reads of BtCoV in Rhinolophus affinis of Hainan and 210 reads of BtCoV in Miniopterus schreibersii of Hainan. The NR alignment results showed that all these reads (81 nucleotides long) showed 94% to 100% amino acid sequence identities with either BtCoV 1A (GenBank accession number EU420138) or BtCoV 1B (GenBank accession number EU420137), which were previously identified in Miniopterus magnater and Miniopterus pusillus of Hong Kong, respectively. This finding suggested that BtCoVs in Hainan shared a very close genetic relationship with the BtCoV 1A and BtCoV 1B in Hong Kong.
DISCUSSION
This study detailed the viromes found in respiratory fluids and feces of bats in China. We chose 11 insectivorous bat species from six provinces for metagenomic analysis. To facilitate the identification and tracking of viral communities near humans, all the bat habitats in this study were in or close to suburban villages; thus, the bats shared the same living environment and had close contact with human beings.
As described in previous metagenomic analyses (10, 11, 32, 56, 57), we detected known or unknown viruses in this study by analyzing purified, viral particle-protected nucleic acid that had been amplified with sequence-independent PCR amplification. Initially, we de novo assembled the sequence reads from each lane into contigs and then aligned the contigs against NT and NR. Because the sequence reads of NGS were randomly distributed throughout the whole genome of viruses, many of them could not be assembled into longer contiguous segments. Thus, this type of pretreatment of the NGS data incurred losses of valuable read information for NT and NR alignments (data not shown). Considering that the length and accuracy of our valid sequence reads (81 nt) were sufficient for initial taxonomic assignment, we conducted the alignments with the valid reads directly. This change in data processing provided an unprecedented quantity of taxonomic information. The assembled contigs were used in the subsequent genome sequencing to facilitate the reads-based PCR.
Despite the background sequence reads related to cellular organisms (which may be the result of incomplete DNase and RNase enzyme digestion) and the reads with no hits (which may be the result of unknown host and virus sequences), the similarity comparisons showed that a large number of reads could be classified into families of phages, insect viruses, fungal viruses, and plant viruses. The abundance of insect viruses, fungal viruses, and plant viruses was associated with the insectivorous, cavernicolous, and social habits of bats (it is worth mentioning that a large number of reads related to Chrysoviridae, Partitiviridae, and Totiviridae were found in Tylonycteris robustula; this may indicate fungal infections in the bat species that we collected); thus, these were not initially harbored in bats. The presence of phages reflected the bacterial flora harbored in bats of different species. These viruses were described in several previous studies, and they have little relationship with mammalian infectious diseases (11, 32, 37); thus, we described them only in the section that described the metagenomic analysis of NGS results, and we did not perform further verifications for these viruses. The sequence reads related to mammalian viruses were the emphasis of the present study.
Previous studies have reported HVs from the subfamilies Alpha-, Beta-, to Gammaherpesviruses in many bat species from other regions (41, 61–63). In this study, the reads classified in the family Herpesviridae comprised the second largest proportion of mammalian viruses (24.8%). This indicated that HVs represent a large class of viruses associated with bat species common in mainland China. This conclusion was confirmed by our prevalence survey of individual samples, where we used nested PCR to target a partial region of the DPOL gene for RfBHV-1, TrBHV-1, MrGHV-1, and MrGHV-2 (data not shown). The phylogenetic analysis revealed the genetic relationships among these bat HVs and showed that HVs from different bat species were located in different phylogenetic positions at far distances from each other.
For bat PVs, this report was the first to identify PVs in the suborder Microchiroptera (insectivorous bats). Although Rousettus aegyptiacus papillomavirus type 1 (RaPV-1) was previously described in a basosquamous carcinoma of the Egyptian fruit bat in the suborder Megachiroptera (43), PVs found in the two suborders showed only ∼50% nucleotide and amino acid sequence identities, and these three bat PVs were located in distinct clusters on different phylogenetic branches. PVs have high species-specific and low cross-species transmission characteristics (42–44, 49). The broad genetic diversity and strict host specificity exhibited in the phylogenetic analysis confirmed the hypothesis that PVs are ancient viruses that coevolved with the evolution of their vertebrate host species. The symptoms of PV infections in insectivorous bats need to be characterized further; for example, PV-associated carcinomatosis was documented in fruit bats (43).
Previous studies have described broad genetic diversity in bat CVs (17, 32). We identified only one new species of circovirus, RfCV-1, in our bat samples. However, RfCV-1 showed little sequence similarity and a distant phylogenetic relationship with other CVs, particularly with the two bat CVs (bat CV YN-BtCV-1 and TM-6c). Most avian CVs are pathogenic, and PCV-2 has been reported to be pathogenic. Given the potential cross-species transmission and highly variable characteristics of CVs (30, 35), it is important to analyze the diversity of CVs among different hosts.
We simultaneously identified three new bat picornaviruses in this study. These picornaviruses shared many common characteristics but few amino acid sequence similarities with members of other genera in the family Picornaviridae. According to the ICTV and with the support of P1- and 3D-based phylogenetic analyses, two of the three bat picornaviruses may represent two distinct new genera. Members of the genus Cardiovirus are associated with severe mammalian diseases. For example, Saffold viruses (SaffVs) are associated with respiratory or gastrointestinal symptoms and nonpolio acute flaccid paralysis in children (38). Additionally, encephalomyocarditis virus and Theiler's murine encephalomyelitis virus can cause encephalitis and myocarditis mainly in rodents, and they can be transmitted from rodents to other hosts, including mammals and birds (21). Our results showed that Miniopterus schreibersii picornavirus 1 was a close relative of cardioviruses, but further studies are required to determine whether it can cause diseases in bat species. The close genetic relationship between Ia io picornavirus 1 and three previously documented bat picornaviruses from other bat species (Miniopterus pusillus, Miniopterus magnate, and Rhinolophus sinicus) in China indicated that the hypothetical genus may consist of bat picornaviruses that are widely distributed in Chinese bat species. Considering that most picornaviruses can cause many distinct diseases in their hosts (55), the pathogenicity of the three bat picornaviruses should be further investigated.
The present study was the first to show that bats carried three mammalian viruses, BoV, PestV, and FV. This finding showed that members of the genera Bocavirus and Pestivirus and the family Retroviridae could infect more mammalian hosts than previously thought. Bats could be considered potential carriers and disseminators of viruses in this family and these genera. Previous studies have documented that cross-species transmission of some viruses, like simian FV and BoV, represent potential risks to humans (3, 25). Thus, closer monitoring of common bat species that are widely distributed in or around human gathering places is required.
This study revealed very close genetic relationships among bat AstVs in different species (Myotis rickeeti, Tylonycteris robustula, and three members of the genus Miniopterus) from distant locations (Jiangxi, Hainan, and Hong Kong). These viruses may arise from the same prototype AstV strain. In this study, reads of highly homologous BtCoV 1A and BtCoV 1B, validated previously in Miniopterus magnater and Miniopterus pusillus of Hong Kong (7), were found in Rhinolophus affinis and Miniopterus schreibersii in Hainan. This finding also supported the previously reported hypothesis that the distribution and cross-species transmission of BtCoVs were closely associated with the migration and coroosting of bat species (9).
The bat virome identified in this study was compared with three previously reported bat viromes (11, 18, 32). All four studies showed that sequences related to mammalian viruses comprised less than 10% of the total sequences related to viruses; the largest proportions of the sequence reads were related to phages, insect viruses, and fungal viruses. Two bat virome analyses conducted by Li et al. (32) and Donaldson et al. (11) with the Roche 454 method described the presence of CoVs, HVs, picornaviruses, CVs, AdVs, AAVs, and AstVs in some bat species of North America. One bat virome analysis conducted by Ge et al. (18) with the Illumina Solexa GA method for a single read of 35 bp mainly described the insect viruses in some bat species of China. In this study, the Illumina Solexa GA II method was used for a single read of 81 bp, and 11 species were separately sequenced. We also found CoVs, HVs, picornaviruses, CV, AdVs, AAVs, and AstVs, but they shared little similarity with viruses in the two previously reported bat viromes. In addition, this study was the first to characterize PVs, BoVs, PestV, and FV in bat species. The sequence reads of AdVs and AAVs in our bat species showed high similarities with previously reported AdVs and AAVs in different bat species of different Chinese provinces (85% to 100% amino acid sequence identities; data not shown) (33, 34). Based on several sequence reads related to the family Paramyxoviridae, we obtained a partial region (330 nt) of matrix gene in Miniopterus schreibersii of Hainan. This sequence showed low nucleotide and amino acid sequence similarity with known paramyxoviruses (data not shown). Considering the result of a previous study that indicated that RT-PCR assays were more sensitive than NGS in detecting paramyxoviruses (12), further work is needed to detect bat paramyxoviruses in China.
This description of bat viromes in China provides a more comprehensive understanding of the virus communities present in common bat species found in human habitats. The NGS technology should facilitate further metagenomic analyses to investigate more bat species in other regions and other natural virus hosts in human environments.
ACKNOWLEDGMENTS
This work was supported by the National S&T Major Project China Mega-Project for Infectious Disease (grant no. 2011ZX10004-001) from the People's Republic of China and Basic Research and Operating Expenses (grant no. 2010IPB111) from the Institute of Pathogen Biology, Chinese Academy of Medical Sciences & Beijing Union Medical College.
Footnotes
Published ahead of print 1 August 2012
REFERENCES
- 1. Allander T, et al. 2005. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. U. S. A. 102:12891–12896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bernard HU, et al. 2010. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401:70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Betsem E, Rua R, Tortevoye P, Froment A, Gessain A. 2011. Frequent and recent human acquisition of simian foamy viruses through apes' bites in central Africa. PLoS Pathog. 7:e1002306 doi:10.1371/journal.ppat.1002306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blomstrom AL, et al. 2009. Detection of a novel porcine boca-like virus in the background of porcine circovirus type 2 induced postweaning multisystemic wasting syndrome. Virus Res. 146:125–129 [DOI] [PubMed] [Google Scholar]
- 5. Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. 2006. Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 19:531–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chae C. 2005. A review of porcine circovirus 2-associated syndromes and diseases. Vet. J. 169:326–336 [DOI] [PubMed] [Google Scholar]
- 7. Chu DK, Peiris JS, Chen H, Guan Y, Poon LL. 2008. Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co-infections in Miniopterus bats. J. Gen. Virol. 89:1282–1287 [DOI] [PubMed] [Google Scholar]
- 8. Chu DK, Poon LL, Guan Y, Peiris JS. 2008. Novel astroviruses in insectivorous bats. J. Virol. 82:9107–9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cui J, et al. 2007. Evolutionary relationships between bat coronaviruses and their hosts. Emerg. Infect. Dis. 13:1526–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Djikeng A, et al. 2008. Viral genome sequencing by random priming methods. BMC Genomics 9:5 doi:10.1186/1471-2164-9-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donaldson EF, et al. 2010. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J. Virol. 84:13004–13018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Drexler JF, et al. 2012. Bats host major mammalian paramyxoviruses. Nat. Commun. 3:796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ehlers B, et al. 1999. Detection of new DNA polymerase genes of known and potentially novel herpesviruses by PCR with degenerate and deoxyinosine-substituted primers. Virus Genes 18:211–220 [DOI] [PubMed] [Google Scholar]
- 14. Fauquet CM, Fargette D. 2005. International Committee on Taxonomy of Viruses and the 3,142 unassigned species. Virol. J. 2:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Finsterbusch T, Mankertz A. 2009. Porcine circoviruses—small but powerful. Virus Res. 143:177–183 [DOI] [PubMed] [Google Scholar]
- 16. Freuling CM, et al. 2011. Novel lyssavirus in Natterer's bat, Germany. Emerg. Infect. Dis. 17:1519–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ge X, et al. 2011. Genetic diversity of novel circular ssDNA viruses in bats in China. J. Gen. Virol. 92:2646–2653 [DOI] [PubMed] [Google Scholar]
- 18. Ge X, et al. 2012. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 86:4620–4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gravitt PE. 2011. The known unknowns of HPV natural history. J. Clin. Invest. 121:4593–4599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo L, et al. 2011. Bocavirus in children with respiratory tract infections. Emerg. Infect. Dis. 17:1775–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Himeda T, Ohara Y. 2012. Saffold virus, a novel human cardiovirus with unknown pathogenicity. J. Virol. 86:1292–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ilyina TV, Koonin EV. 1992. Conserved sequence motifs in the initiator proteins for rolling circle DNA replication encoded by diverse replicons from eubacteria, eucaryotes and archaebacteria. Nucleic Acids Res. 20:3279–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jones MS, et al. 2005. New DNA viruses identified in patients with acute viral infection syndrome. J. Virol. 79:8230–8236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kantola K, et al. 2011. Seroepidemiology of human bocaviruses 1-4. J. Infect. Dis. 204:1403–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kapoor A, et al. 2010. Identification and characterization of a new bocavirus species in gorillas. PLoS One 5:e11948 doi:10.1371/journal.pone.0011948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kirkland PD, et al. 2007. Identification of a novel virus in pigs—Bungowannah virus: a possible new species of pestivirus. Virus Res. 129:26–34 [DOI] [PubMed] [Google Scholar]
- 27. Lau SK, et al. 2011. Complete genome analysis of three novel picornaviruses from diverse bat species. J. Virol. 85:8819–8828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lau SK, et al. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102:14040–14045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leroy EM, et al. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438:575–576 [DOI] [PubMed] [Google Scholar]
- 30. Li L, et al. 2010. Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J. Virol. 84:1674–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li L, et al. 2011. The fecal viral flora of California sea lions. J. Virol. 85:9909–9917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li L, et al. 2010. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 84:6955–6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li Y, et al. 2010. Prevalence and genetic diversity of adeno-associated viruses in bats from China. J. Gen. Virol. 91:2601–2609 [DOI] [PubMed] [Google Scholar]
- 34. Li Y, et al. 2010. Host range, prevalence, and genetic diversity of adenoviruses in bats. J. Virol. 84:3889–3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lorincz M, et al. 2010. Detection of porcine circovirus in rodents—short communication. Acta Vet. Hung. 58:265–268 [DOI] [PubMed] [Google Scholar]
- 36. Metzker ML. 2010. Sequencing technologies—the next generation. Nat. Rev. Genet. 11:31–46 [DOI] [PubMed] [Google Scholar]
- 37. Mouinga-Ondeme A, et al. 2012. Cross-species transmission of simian foamy virus to humans in rural Gabon, Central Africa. J. Virol. 86:1255–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nielsen AC, Bottiger B, Banner J, Hoffmann T, Nielsen LP. 2012. Serious invasive Saffold virus infections in children, 2009. Emerg. Infect. Dis. 18:7–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Phan TG, et al. 2011. The fecal viral flora of wild rodents. PLoS Pathog. 7:e1002218 doi:10.1371/journal.ppat.1002218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qiu J, Cheng F, Johnson FB, Pintel D. 2007. The transcription profile of the bocavirus bovine parvovirus is unlike those of previously characterized parvoviruses. J. Virol. 81:12080–12085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Razafindratsimandresy R, et al. 2009. Partial molecular characterization of alphaherpesviruses isolated from tropical bats. J. Gen. Virol. 90:44–47 [DOI] [PubMed] [Google Scholar]
- 42. Rector A, et al. 2004. Characterization of a novel close-to-root papillomavirus from a Florida manatee by using multiply primed rolling-circle amplification: Trichechus manatus latirostris papillomavirus type 1. J. Virol. 78:12698–12702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rector A, et al. 2006. Genetic characterization of the first chiropteran papillomavirus, isolated from a basosquamous carcinoma in an Egyptian fruit bat: the Rousettus aegyptiacus papillomavirus type 1. Vet. Microbiol. 117:267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rector A, et al. 2005. Isolation and cloning of the raccoon (Procyon lotor) papillomavirus type 1 by using degenerate papillomavirus-specific primers. J. Gen. Virol. 86:2029–2033 [DOI] [PubMed] [Google Scholar]
- 45. Rosell C, et al. 2000. Identification of porcine circovirus in tissues of pigs with porcine dermatitis and nephropathy syndrome. Vet. Rec. 146:40–43 [DOI] [PubMed] [Google Scholar]
- 46. Shan T, et al. 2011. Genomic characterization and high prevalence of bocaviruses in swine. PLoS One 6:e17292 doi:10.1371/journal.pone.0017292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shen H, et al. 2011. Genetic diversity and positive selection analysis of classical swine fever virus isolates in south China. Virus Genes 43:234–242 [DOI] [PubMed] [Google Scholar]
- 48. Smith I, et al. 2011. Identifying Hendra virus diversity in pteropid bats. PLoS One 6:e25275 doi:10.1371/journal.pone.0025275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stevens H, Rector A, Bertelsen MF, Leifsson PS, Van Ranst M. 2008. Novel papillomavirus isolated from the oral mucosa of a polar bear does not cluster with other papillomaviruses of carnivores. Vet. Microbiol. 129:108–116 [DOI] [PubMed] [Google Scholar]
- 50. Sun Y, et al. 2009. Molecular characterization of infectious clones of the minute virus of canines reveals unique features of bocaviruses. J. Virol. 83:3956–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Swanepoel R, et al. 2007. Studies of reservoir hosts for Marburg virus. Emerg. Infect. Dis. 13:1847–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Teeling EC, et al. 2005. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science 307:580–584 [DOI] [PubMed] [Google Scholar]
- 54. Todd D, Weston JH, Soike D, Smyth JA. 2001. Genome sequence determinations and analyses of novel circoviruses from goose and pigeon. Virology 286:354–362 [DOI] [PubMed] [Google Scholar]
- 55. Tracy S, Chapman NM, Drescher KM, Kono K, Tapprich W. 2006. Evolution of virulence in picornaviruses. Curr. Top. Microbiol. Immunol. 299:193–209 [DOI] [PubMed] [Google Scholar]
- 56. Victoria JG, Kapoor A, Dupuis K, Schnurr DP, Delwart EL. 2008. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog. 4:e1000163 doi:10.1371/journal.ppat.1000163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Victoria JG, et al. 2009. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J. Virol. 83:4642–4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vilcek S, Nettleton PF. 2006. Pestiviruses in wild animals. Vet. Microbiol. 116:1–12 [DOI] [PubMed] [Google Scholar]
- 59. Wang LF, et al. 2006. Review of bats and SARS. Emerg. Infect. Dis. 12:1834–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang YX. (ed). 2003. A complete checklist of mammal species and subspecies in China: a taxonomic and geographic reference. China Forestry Publishing House, Beijing. China [Google Scholar]
- 61. Watanabe S, et al. 2010. Novel betaherpesvirus in bats. Emerg. Infect. Dis. 16:986–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Watanabe S, et al. 2009. Detection of a new bat gammaherpesvirus in the Philippines. Virus Genes 39:90–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wibbelt G, et al. 2007. Discovery of herpesviruses in bats. J. Gen. Virol. 88:2651–2655 [DOI] [PubMed] [Google Scholar]
- 64. Wilson DE, Reeder DM. (ed). 2005. Mammal species of the world. A taxonomic and geographic reference, 3rd ed Johns Hopkins University Press, Baltimore, MD [Google Scholar]
- 65. Xia H, Vijayaraghavan B, Belak S, Liu L. 2011. Detection and identification of the atypical bovine pestiviruses in commercial foetal bovine serum batches. PLoS One 6:e28553 doi:10.1371/journal.pone.0028553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xiao J, et al. 2011. Isolation and phylogenetic characterization of bat astroviruses in southern China. Arch. Virol. 156:1415–1423 [DOI] [PubMed] [Google Scholar]
- 67. Yang J, et al. 2011. Unbiased parallel detection of viral pathogens in clinical samples by use of a metagenomic approach. J. Clin. Microbiol. 49:3463–3469 [DOI] [PMC free article] [PubMed] [Google Scholar]