

Abstract
Pseudomonas aeruginosa secretes an epoxide hydrolase virulence factor that reduces the apical membrane expression of ABC transporters such as the cystic fibrosis transmembrane conductance regulator (CFTR). This virulence factor, named CFTR inhibitory factor (Cif), is regulated by a TetR-family, epoxide-responsive repressor known as CifR via direct binding and repression. We identified two sites of CifR binding in the intergenic space between cifR and morB, the first gene in the operon containing the cif gene. We have mapped these binding sites and found they are 27 bp in length, and they overlap the −10 and +1 sites of both the cifR and morB regulatory region and the start of transcription, respectively. In addition, we found that CifR binds to each repression site with differing affinity. Mutagenesis of these binding sites resulted in a loss of DNA binding in vitro, and mutation of one of these sites in vivo resulted in an increase in transcription of both the cif and cifR genes. We characterized cif and cifR gene expression in sputum and found that, whereas cif gene expression varied relative to an in vitro coculture control, cifR gene expression was consistently higher. Analysis of a longitudinal sample of CF isolates from nine patients revealed that Cif protein was expressed over time, although variably, and these changes could not be linked to mutations in the cifR gene or the promoters of these genes. Finally, we tested CifR responsiveness to other epoxides and showed that CifR can respond to multiple epoxides to various degrees.
INTRODUCTION
Pseudomonas aeruginosa is a highly adaptable, opportunistic organism that can infect a variety of plant and animal hosts (32, 43). This versatility allows P. aeruginosa to infect immunocompromised humans with great efficiency. In humans, P. aeruginosa can colonize the skin, urinary tract, eyes, ears, and lungs (15, 19, 29, 30, 41), causing debilitating, even fatal, disease. The hyperadaptability exhibited by P. aeruginosa is due, in large part, to the numerous virulence factors it possesses. These myriad factors include phosphatases, phospholipases, phenazines, a type 3 secretion system, elastase, and β-lactamases (18, 22, 36, 44), which act in concert to establish P. aeruginosa in its many niches.
P. aeruginosa infection is particularly detrimental in the context of cystic fibrosis (CF) lung infection. CF is an inherited disorder in which the individual harbors mutations in both copies of the CF transmembrane conductance regulator (CFTR). CFTR is an ABC transporter family protein that moves chloride across the apical membrane of the epithelia (20). In the lung, chloride secretion is important for maintenance of the airway surface liquid, which in turn, is required for mucociliary clearance (9). In CF, the resulting ciliostasis causes a thick mucus layer to form in which a polymicrobial infection develops (14). P. aeruginosa outcompetes other bacteria and often becomes the most common, persistent organism in the CF airway and frequently the cause of mortality (6, 14). Over the course of this chronic infection, P. aeruginosa will often decrease or lose expression of several virulence factors (28, 41). This loss of expression can be due to mutations in the gene coding for the virulence factors themselves or, more often, mutations in the vast regulatory network that controls expression of these virulence factors (17, 34).
Previously, we characterized a virulence factor, Cif, which is capable of reducing apical membrane expression of ABC transporters, including CFTR (26, 42). Cif, delivered to epithelial cells either by direct secretion from the bacterial cell to the extracellular milieu or via outer membrane vesicles (7), decreases CFTR by stabilizing the inhibitory interaction between G3BP1 and a deubiquitinating enzyme (USP10), resulting in the polyubiquitination of CFTR and its lysosomal degradation (8). Structural and biochemical characterization of Cif have revealed that is it an epoxide hydrolase (1, 2, 3). Recently, we have shown that the transcriptional regulator CifR mediates cif gene expression (25). The cifR gene is adjacent to and divergently transcribed from the three-gene operon that contains the cif gene. This TetR-family regulator represses the transcription of the polycistronic transcript encoding the Cif protein by binding to the intergenic region between this cif-encoding operon and the cifR gene. We previously showed that in the presence of epibromohydrin (EBH), an inducing epoxide, CifR releases from the DNA to allow transcription to occur resulting in increased Cif production (25). We have shown that the cif gene is expressed in the lung and that there is variability in cif gene transcription in clinical isolates (26). However, a better understanding of CifR regulation of cif gene expression will assist in the identification of epoxides and mutations that lead to changes in this virulence factor's expression during chronic lung infection.
The goal of the present study was to better characterize the regulation of the cif gene by CifR. Here, we identify the binding sites of CifR and place them in the context of transcription of the cif and cifR genes by characterizing the promoters and transcriptional start sites of these loci. Furthermore, we identify two putative CifR recognition sites within these binding regions and demonstrate that mutation of one of these sites results in an increase in transcription of both the cif and the cifR genes in vivo, as well as the loss of CifR-DNA binding in vitro. We characterize cif and cifR gene expression in sputum samples from CF patients and show differences in transcript level of the message encoded by these genes. Next, with a clearer picture of CifR-mediated repression of the cif gene, we characterize Cif expression in multiple clinical Pseudomonas isolates from CF patients isolated over time. These data support our hypothesis that CifR-mediated regulation of Cif is important in the context of lung infection. Toward our endeavor of identifying an epoxide inducer of the Cif protein in the lung, we also characterize cif gene expression in the presence of several epoxides.
MATERIALS AND METHODS
Strains, plasmids, primers, and culture conditions.
The bacterial strains, plasmids and primers used in the present study are listed in Table S1 in the supplemental material. Cultures were grown in lysogeny broth (LB) at 37°C. The antibiotics gentamicin (10 μg/ml for Escherichia coli and 100 μg/ml for P. aeruginosa strain UCBPP-PA14 [PA14]), nalidixic acid (20 μg/ml for P. aeruginosa), and ampicillin (150 μg/ml for E. coli) were added to the growth medium for maintenance of plasmids. Yeast was grown either in yeast extract-peptone-dextrose medium or SD-URA minimal medium (Sunrise Science Products) at 30°C. Cloning was performed using restriction enzymes (NEB) and standard molecular techniques (27) or Saccharomyces cerevisiae-based recombineering (38).
Mutagenesis of a CifR binding site in P. aeruginosa.
To mutate the morB proximal CifR binding site in P. aeruginosa, two ∼1-kb regions surrounding the site were amplified with Phusion polymerase (Finnzymes) using primers containing the mutated CifR consensus sequence (30/5′ for Mut, MorB proximal 6mix F, MorB proximal 6mix R, and 3′ rev Mut/30), these amplicons were cloned into the suicide vector (pMQ30) by yeast-based homologous recombination (38) and transformed into E. coli S17-1. Conjugation and counterselection for recombinant P. aeruginosa were performed as described previously (21, 38) with integrants grown on medium with gentamicin and nalidixic acid, and mutants selected on medium containing sucrose. Mutations were verified by sequencing.
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed as described previously [the buffer consisted of 10 mM Tris, 50 mM KCl, 1 mM dithiothreitol (DTT), 2.5% glycerol, 5 mM MgCl2, 50 ng of poly(dI·dC)/μl, 0.05% NP-40 (pH 7.5)] (25), with the following specifications: purified CifR protein (16 nM, unless otherwise specified) was coincubated with the probe (15 fmol) for 30 min at room temperature in a volume of 10 μl. EBH (1 mM) or unlabeled competitor DNA of the same sequence (300 pmol) was added where indicated. The probes used here are listed in Table S1 in the supplemental material. For Western blot detection of CifR-His bound to DNA, the EMSA protein was transferred to a nitrocellulose membrane and examined as described below.
RNA isolation.
Strains were diluted 1:100 from overnight cultures into fresh LB medium, supplemented with 1 mM EBH, as indicated, and grown at 37°C with shaking. RNA was harvested from 1 ml of triplicate samples grown to an optical density at 600 nm (OD600) of ∼1.0. Samples were pelleted and resuspended in 100 μl of 2 mg of lysozyme/ml in TE buffer (10 mM Tris-HCl, 1 mM EDTA [pH 8.0]), followed by incubation at room temperature for 5 min to lyse the cells. The RNA was extracted using the RNeasy kit (Qiagen) according to the manufacturer's instructions and tested for DNA contamination by PCR.
cDNA synthesis and qRT-PCR.
cDNA was synthesized from 1 μg of total RNA using the QuantiTect reverse transcription kit (Qiagen) according to the prescribed protocols. Quantitative reverse transcriptase PCR (qRT-PCR) was performed using cDNA, primers designed to the cif, cifR, and rplU genes, a transcriptional control (21), and Maxima SYBR green qPCR Master Mix (Fermentas). A Bio-Rad iCycler was used to perform the reactions and data analysis was performed using CFX manager software (Bio-Rad). The data are expressed as picograms of input cDNA of the gene of interest relative to the rplU gene transcript control.
Sputum sample isolation and transcriptional analysis.
Respiratory sputum samples were collected from four individuals with CF and immediately frozen in a dry-ice–ethanol bath, followed by storage at −80°C for no more than 2 weeks. The static coculture control was performed as described previously (26) at a multiplicity of infection of 100:1 and harvested after 6 h. To isolate total sputum RNA, samples were thawed on ice, and the pellet was resuspended in TE buffer and homogenized by passage through 16-, 20-, and 24-gauge syringe needles. Lysozyme (3 mg/ml) was then added, and samples were incubated at room temperature for 5 min with intermittent vortexing. Subsequent RNA isolation steps were performed using a Qiagen RNeasy kit according to the manufacturer's instructions. A Promega RQ1 RNase-Free DNase kit (catalog no. M6101) was used to remove contaminating DNA. For cDNA synthesis, 450 ng of total RNA was used as a template in a cDNA synthesis reaction using random primers [(NS)5], RNaseOUT (Invitrogen, catalog no. 10777-019), and Superscript III. cDNA synthesis reactions were performed using the SSIII protocol. Absence of DNA was confirmed by cycle threshold differences between RT and no-RT controls. qRT-PCRs were performed using Bio-Rad SsoFast EvaGreen Supermix according to the manufacturer's protocol. The annealing temperatures for rplU, cif, and cifR were 60, 63.7, and 63.7°C, respectively.
5′RACE.
RNA for 5′ rapid amplification of cDNA ends (5′RACE) was isolated from P. aeruginosa cultured in LB with EBH. The 5′RACE was performed using a FirstChoice RLM-RACE kit (Invitrogen) according to the manufacturer's instructions. RACE amplification products were ligated into pGEM-T vector (Promega), transformed into DH5α E. coli and selected on medium containing 150 μg of ampicillin/ml and 40 μg of X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside)/ml. A minimum of three positive clones were sequenced for each transcript.
Western detection of Cif from P. aeruginosa PA14 and clinical isolates.
P. aeruginosa was subcultured 1:100 from overnight cultures into 5 ml of LB, grown at 37°C with shaking to an OD600 of 1.0, and then immediately pelleted and frozen at −80°C. For the epoxide induction assay, 1 mM EBH, cyclohexene oxide (CO), styrene oxide (SO), (S)-2-(4-nitrophenyl) oxirane (SNO), (R)-2-(4-nitrophenyl) oxirane (RNO), epoxyhexane (EH), trans-2,3-epoxysuccinate (TES), cis-2,3-epoxysuccinate (CES), or glycidol (G) dissolved in dimethyl sulfoxide (DMSO) were added to culture medium prior to inoculation. Cell pellets were resuspended in 1× sodium dodecyl sulfate (SDS) loading buffer (21) and boiled for 10 min before resolving them by SDS-PAGE. Western analysis was performed as described previously (10, 21). Membranes were probed with Cif antiserum (26). Cif protein expression was determined by quantification of pixel density of the Cif band relative to a nonspecific band using ImageJ.
Cloning of CifR-3C-Decahis expression construct.
Generation of a CifR expression construct containing a 3C-cleavable C-terminal decahistidine tag was performed via PCR amplification of the cifR gene from P. aeruginosa PA14 genomic DNA with the primers BspHI CifR F and BspHI CifR R and Phusion High-Fidelity polymerase (Finnzymes). The PCR product was digested with BspHI and ligated with T4 ligase (NEB) into an NcoI-digested and phosphatase-treated pET16b vector. Ligated plasmid was transformed into E. coli Top10, and transformants were selected on LB supplemented with 150 μg of ampicillin/ml. Positive clones were verified by sequencing. This construct generated a CifR protein that possesses a carboxy-terminal decahistidine tag, preceded by a cleavage site (LEVLFQGP) for human rhinovirus 3C (HRV-3C) protease.
CifR protein purification.
CifR protein was purified by immobilized metal affinity chromatography (IMAC), with subsequent removal of the polyhistidine affinity tag. Rosetta 2 (DE3) (Novagen) E. coli transformed with the CifR expression plasmid was grown in 4 liters of 2×YT broth supplemented with 100 μg of ampicillin/ml and 34 μg of chloramphenicol/ml at 37°C. Expression of CifR protein was then induced at an OD600 of 0.6 by addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to 100 μM, and cultures were incubated overnight at 16°C. Cells were harvested from the medium by centrifugation at 5,000 × g for 15 min at 4°C. After removal of the supernatant, the cell pellets were resuspended in 25 ml of lysis buffer per liter of culture volume. The lysis buffer consisted of 50 mM Tris (pH 8.5), 150 mM NaCl, 2 mM MgCl2, 1 mM ATP, 25 U of benzonase (Novagen)/ml, and one EDTA-free complete protease inhibitor cocktail tablet (Roche) per 50 ml. The cells were lysed using a French press, and the lysate was clarified by centrifugation for 1 h at 4°C in a Ti 45 rotor (Beckman) at 40,000 rpm, which generates an average relative centrifugal force of 125,171 × g and a k-factor of 168.1. The supernatant was then passed over a 5-ml column of Ni-Sepharose resin (GE Healthcare) that had been preequilibrated with IMAC buffer consisting of 50 mM Tris (pH 8.5), 500 mM NaCl, and 1 mM DTT supplemented with 20 mM imidazole (pH 8.5). After a wash with 10 column volumes of IMAC buffer supplemented with 77 mM imidazole to remove the unbound material, CifR protein was eluted from the resin over a 15-column-volume gradient running from 248 to 324 mM imidazole in IMAC buffer. The fractions were pooled, concentrated, and dialyzed into size-exclusion buffer containing 25 mM Tris (pH 8.5), 150 mM NaCl, 0.1 mM DTT, and 0.1 mM ATP. The protein concentration was determined by Bradford assay (Bio-Rad), and HRV-3C protease was added to a mass ratio of 1:10 (protease to CifR). Cleavage of the decahistidine tag proceeded overnight at 4°C. The HRV-3C protease possesses a noncleavable histidine tag, which is subsequently used to remove it from the sample, along with any uncleaved CifR protein, by passage over a 5-ml column of Ni-Sepharose resin (GE). The flowthrough was collected, and cleaved CifR protein was further purified using size-exclusion chromatography (SEC) with a HiLoad Superdex 200 Prep-Grade 26/60 column (GE Healthcare) using size-exclusion buffer. The mature CifR protein was concentrated using a stirred-cell concentrator with a 3,000-molecular-weight cutoff (Pierce) and dialyzed into 10 mM Tris (pH 8.5), 50 mM KCl, 1 mM DTT, and 5 mM MgCl2.
RESULTS
Evidence for two CifR binding sites.
We have previously reported that purified CifR was capable of binding to the intergenic region between the cifR gene and morB, the first gene in the cif operon (Fig. 1A) (25). CifR binding to the intergenic region was previously demonstrated by EMSA using a purified hexahistidine-tagged CifR protein (25). For the present study, we generated a new CifR expression system using a pET16b expression vector containing a C-terminal cleavable decahistidine tag. This new construct allowed us to more easily isolate pure, concentrated, CifR protein that does not contain extraneous peptides. Analytical SEC suggests this protein (predicted size, 21.75 kDa) is a monomer in solution (Fig. 1B), unlike the related TetR which forms a dimer (31).
Fig 1.

CifR, a monomer in solution, binds to the intergenic region between the cifR gene and morB operon. (A) Depiction of the operon containing the cif gene. The cifR gene is divergently transcribed from this operon, which includes a gene coding for a predicted morphinone reductase (morB), a putative MFS transporter (PA14_26110), as well as the epoxide hydrolase-encoding cif gene. The double-stranded DNA (dsDNA) fragment used in Fig. 1C is indicated and labeled as the “intergenic DNA fragment.” (B) SEC of CifR shows a peak at 228.0 ml, corresponding to a predicted molecular mass of 26.9 kDa. Since the predicted molecular mass of recombinantly expressed CifR protein is 21.9 kDa, these data indicate that CifR is monomeric in solution. The size markers used are indicated in the figure. (C) EMSA of CifR with a biotinylated intergenic region dsDNA fragment reveals increased binding with increasing concentrations of CifR (2-fold from 5 to 512 nM). A second, slower-migrating band appears with higher concentrations of CifR. The CifR-DNA interaction can be effectively disrupted by coincubation with 1 mM EBH (rightmost lane).
Incubation of this protein with the intergenic region resulted in a detectable shift of the biotinylated DNA fragment by EMSA (Fig. 1C). This interaction could be disrupted upon addition of the Cif substrate EBH (Fig. 1C, rightmost lane). This finding is consistent with previous work showing that CifR responds to this epoxide through release of DNA, as observed by EMSA, and via transcriptional derepression, as shown by in vivo studies (25). Interestingly, upon addition of increasing concentrations of CifR, we observed two migrating species, the slower of which became the dominant band at the highest concentrations of CifR (Fig. 1C). The detection of two bands by EMSA suggested that there may be more than one CifR binding site in this region.
Identification of two separable CifR binding sites within the cifR-morB intergenic region.
To test the hypothesis that CifR was binding to two sites in the intergenic space between the cifR gene and the morB operon, we generated a set of three overlapping biotinylated DNA fragments spanning this region (Fig. 2A). EMSA analysis showed that CifR was capable of EBH-responsive binding to the two DNA fragments most proximal to the cifR gene (regions 2 and 3) but could not bind to the scrambled region 3 sequence DNA fragment or the region 1 DNA fragment (Fig. 2B). Only one shifted species was observed for either of the region 2 or region 3 DNA fragments, even with excess CifR (see Fig. S1 in the supplemental material), suggesting the presence of a single CifR binding site on each fragment.
Fig 2.

CifR binds to two distinct sites in the region between the cifR and morB genes. (A) Sequence of cifR-morB intergenic region. Predicted translational start sites for cifR (left) and morB (right) are indicated in boldface. Black bars indicate DNA fragments used in EMSA analyses and competition studies. (B) EMSA with DNA fragments spanning the intergenic region revealed CifR bound to two DNA fragments (regions 2 and 3). No binding is observed in the presence of 1 mM EBH or with a biotinylated scrambled sequence. (C) EMSA with two nonoverlapping DNA fragments exhibits CifR binding. No binding is seen with excess unlabeled competitor of identical sequence or 1 mM EBH. A “+” indicates the addition of the specified component.
To verify that CifR recognized two separable binding sites, smaller biotinylated DNA fragments were designed that contained no overlapping regions. These oligonucleotides were designated “cifR proximal” and “morB proximal” (Fig. 2A). Both of these probes could be bound by CifR, and the binding was abrogated by the addition of 1 mM EBH or a 200-fold molar excess unlabeled DNA fragment of the same sequence, indicating the interaction was sensitive to the inducer of cif gene expression (Fig. 2C). These data support our hypothesis that CifR binds to two sites in the cifR-morB intergenic region.
Determination of the relative affinities of CifR for each binding site.
Given that we had initially observed differences in binding ability using the region 2 and region 3 probes (see Fig. S1 in the supplemental material), we tested whether there were differences in the relative affinities of CifR for both the cifR proximal and the morB proximal 36-bp fragments, using a previously reported method (24). Using a gradient of unbound biotinylated DNA fragments as the reference (Fig. 3A and C), we analyzed the percentage of DNA bound by increasing concentrations of CifR and quantifying band intensity. Using this method, we determined that CifR has apparent affinities of 37 ± 9 nM and 437 ± 65 nM, respectively, for the cifR proximal and morB proximal sites. These data show that the CifR-DNA interactions at both sites are in line with characterized TetR family DNA-binding proteins (see Discussion).
Fig 3.

Apparent Kd determination by EMSA. (A) EMSA with the cifR proximal binding site. (Left side) Examples of DNA fragment gradient; (right side) DNA fragments with increasing concentrations of CifR. (B) Affinity of CifR for cifR proximal site. The data are plotted as the log complex/free biotinylated DNA versus CifR concentration. The apparent equilibrium is at 37 nM. (C) Representative EMSA with the morB proximal binding site. (D) Affinity of CifR for morB proximal binding site. The apparent equilibrium for CifR to this binding site is 437 nM. These differences are statistically significant (P < 0.01).
CifR binds a ∼27-bp fragment of DNA to repress transcription.
We surmised that CifR recognized and bound a region smaller than 36 bp, like many other regulators in this family (33). Thus, we decided to further define the binding region for CifR by designing several smaller DNA fragments from the cifR and morB proximal oligonucleotide sequences. These smaller DNA fragments did not show any shift by EMSA (data not shown); however, combinations of these probes, fused by PCR, allowed us to narrow down the binding site to an approximate region of <30 bp for each site.
Using this sequence information, we designed unlabeled DNA fragments (to obviate any potential interference from biotin) of 27 bp to test for interactions with CifR using two approaches. First, using an uncleaved decahistidine-tagged CifR, we performed an EMSA and detected CifR-bound to DNA by Western blot analysis using an anti-His antibody (Fig. 4A). With this technique we were able to visualize CifR bound to the 36-bp intergenic sequences, as well as the new 27-bp fragments. No binding was observed with a scrambled sequence control (Fig. 4A), indicating CifR recognized a specific sequence within that region. Next, we verified this interaction by testing whether a 200-fold excess of unlabeled 27-bp fragment was capable of competing CifR from a biotin-labeled 36-bp fragment. As can be seen in Fig. 4B, both of the unlabeled 36- and 27-bp fragments are capable of competing for CifR binding. However, scrambled versions of the 27-bp sequence do not compete CifR binding from the biotinylated 36-bp DNA fragments. In addition, Western analyses with smaller DNA fragments (16 to 25 bp) resulted in a noticeable reduction in binding by CifR (data not shown). These data suggest that CifR is capable of binding these 27-bp fragments (Fig. 4C).
Fig 4.

CifR binds two 27-bp regions overlapping the −10 and +1 sites of the morB and cifR promoters. (A) Western detection of decahistidine-labeled CifR on an EMSA with unbiotinylated DNA fragments. (B) Competition-based EMSA. The CifR/36-bp DNA interaction was competed with a 200-fold molar excess of unlabeled 36-bp, 27-bp, or scrambled 27-bp DNA fragment. (C) Map of intergenic region. The 5′untranslated region (5′UTR) and translational start sites are indicated with arrows. The putative CifR binding site (boxed), conserved sequences (boldface), and the predicted −10 and −35 promoter motifs (gray text, underlined) are indicated for each promoter.
Characterization of the cifR gene and morB operon promoters.
Given the location of the CifR repression sites identified in the intergenic region between the cifR gene and the morB operon, it became necessary to provide a context for these data in relationship to the respective transcriptional start sites. Therefore, we utilized 5′RACE to map the start sites for the cifR gene and morB operon transcripts, as indicated in Fig. 4C.
We next used the promoter prediction software, BPROM (SoftBerry), to identify the −10 and −35 sites for each sequence. As can be seen in Fig. 4C, there is significant overlap of these two promoter sequences, with the −35 position of each transcript separated by only one base from the −10 position of the opposite transcript. With this information, we were able to determine that the 27-bp regions likely bound by CifR overlap the −10 and +1 sites of each transcript (Fig. 4C).
Characterization of the CifR recognition sequence.
While analyzing the promoters of each of these sequences, we identified a 18-bp sequence located between the −10 and +1 sites of each transcript that showed a high degree of conservation on the template strand (Fig. 4C and Fig. 5A). We hypothesized that this sequence, located within the 27-bp binding region, was recognized by CifR. To test this hypothesis in vitro, we generated biotinylated versions of the 36 nucleotide fragments that contained a 6-bp scramble in the center of the 18-bp conserved sequence (designated “6-mix”, Fig. 5A). We then performed an EMSA comparing the native sequence to the 6-mix sequence (Fig. 5B). Although CifR was able to bind the native sequence of each of these binding sites, we did not observe any binding to the probes with the 6-bp alteration, suggesting that CifR recognizes a specific sequence for binding.
Fig 5.

Mutation of conserved sequence alters CifR binding in vitro and in vivo. (A) Comparison of template strand, cifR proximal, and morB proximal binding site-conserved sequences. Nucleotide differences between the two binding sites are boxed. Mutated sequences are listed below with changes highlighted (gray, underlined). (B) EMSA with mutated DNA fragments. A “+” indicates the addition of the specified component. (C) Western detection of Cif in strains grown with or without EBH. 6-mix indicates P. aeruginosa with a 6-bp sequence change in the morB promoter, indicated above (Fig. 5A). (D) Quantitative RT-PCR of cif gene transcription in strains with or without EBH. The data are expressed as picograms input RNA relative to a transcriptional control (rplU). Significance: a, statistically significant difference between the wild type (WT) and 6-mix in the absence of EBH (P = 0.0008); b, statistically significant difference between the WT and the cifR deletion strain (P = 0.0002); and c, statistically significant difference in cif transcription in the cifR deletion strain with or without EBH (P = 0.0003). (E) qRT-PCR of cifR transcription in WT and the 6-mix mutant strain. Significance: a, statistically significant difference between WT and 6-mix (P = 0.0317); ns, not significant.
The EMSA analysis provided important information regarding CifR sequence recognition. However, these data could not lend insight into how each of these sequences modulate transcription of the cifR gene and the morB operon in vivo. Our next step was to verify the physiological role of these binding sites by altering them in vivo and analyzing changes in cifR and cif gene expression. We approached this analysis by introducing the same 6-bp changes from the EMSA oligonucleotides onto the chromosome by homologous recombination. Unfortunately, despite repeated attempts to introduce a cifR proximal 6-mix mutation, we were unable to isolate this mutant. We were successful at introducing the morB proximal 6-mix mutation.
We first analyzed Cif expression in the morB proximal 6-mix mutant by Western analysis. This analysis was done from cell pellets as a measure of Cif protein production, although secreted Cif shows a similar trend (see Fig. S2 in the supplemental material). Figure 5C shows that, compared to the wild type, the 6-mix mutant has an increase in Cif protein levels, suggesting that mutating this 6-bp region does have an effect on Cif protein expression in vivo. This mutant does not appear to express the Cif protein as highly as a cifR deletion mutant (Fig. 5C).
Next we quantified gene expression changes in the presence or absence of 1 mM EBH by qRT-PCR (Fig. 5D). The 6-mix mutant showed a 5-fold increase in cif gene expression relative to the wild type. There is a further 4-fold increase in expression upon addition of EBH to wild-type levels, supporting our hypothesis that CifR responds to the presence of this epoxide. Interestingly, the cifR deletion mutant also exhibited a small, but significant 2-fold increase in cif gene expression in the presence of EBH, indicating additional, non-CifR-dependent, epoxide-mediated regulation of cif gene transcription.
Since the CifR repression site between the −10 and +1 of morB is very close to the predicted −35 promoter element of the divergently transcribed cifR gene, we decided to test the expression of the cifR gene in this 6-mix mutant background (Fig. 5E). Compared to the wild type, there is a 7.5-fold increase in cifR transcript in the 6-mix mutant. Again, upon the addition of 1 mM EBH, this difference is eliminated. Together, these data show that CifR represses transcription of both transcripts via the morB proximal site in vivo and suggest that CifR binding at both sites is required for complete repression by CifR. In addition, any transcriptional differences in expression of the cif gene-containing operon and the cifR gene due to mutation of the binding site are eliminated by addition of epoxide. This shows that the primary mechanism of repression at this locus is likely the same for both transcripts and is mediated by CifR and its epoxide effector.
Characterization of cif and cifR gene expression in CF sputum.
We have shown CifR expression appears to be important for Cif modulation under in vitro conditions, but CifR expression has not been demonstrated in RNA extracted from patient samples. Previously, we had detected cif transcript from the sputum of CF patients (25). We decided to test whether we could detect cifR gene expression in sputum and whether this could be correlated to cif gene expression. For this analysis, we compared gene expression between bacterial RNA isolated from sputum to that observed in P. aeruginosa grown on CFBE41o− human-derived airway epithelial cells. The cif and cifR transcripts were detected in all four sputum samples tested (Fig. 6). The relative abundance of both transcripts varied from sample to sample, suggesting that expression is variable between patients. In general, greater cif gene transcription correlated with greater cifR gene transcription, similar to the relationship we have described in vitro. Surprisingly, cifR gene expression was consistently higher in the sputum samples than in the coculture control, suggesting derepression of this gene in the sputum samples.
Fig 6.
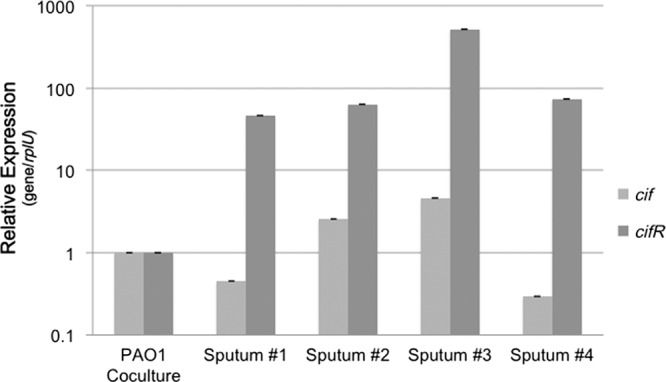
Comparison of cif and cifR gene expression in CF sputum. Quantification of the cif gene (light gray) and cifR gene (dark gray) transcripts in sputum from four CF patients was performed. Expression is normalized to the levels observed in P. aeruginosa PAO1 cocultured on CFBE41o− airway epithelial cells.
P. aeruginosa isolates from CF patients maintain Cif and CifR expression and/or regulation over time.
Often over the course of CF lung infection, P. aeruginosa adapts to its environment through phenotypic and genetic changes. These adaptations can include a transition to mucoidy, the loss of quorum sensing, and downregulation of several virulence factors (16, 17).
In our transcriptional analysis of sputum samples, we observed variability in cif gene expression and relatively high levels in cifR gene expression. We have previously reported similar variability in cif gene expression among clinical isolates (26). We suspected that this variability was due to a temporal loss of Cif expression. We decided to test whether we could link changes in Cif expression to changes in the newly characterized promoter region of the cifR gene. For the present study, we analyzed Cif expression from 36 clinical strains of P. aeruginosa that were isolated from eight CF patients during their lifetimes (5).
As shown in Fig. 7A, Cif protein expression (in the absence of the epoxide inducer) was variable, even among clinical isolates collected from the same patient at the same time point (see Table S2 in the supplemental material). In general, the clinical isolates displayed lower production of Cif relative to the laboratory strain P. aeruginosa PA14 (PA14, Fig. 7A; see also Table S2 in the supplemental material). It is important to note that Cif protein was expressed in all strains tested. Cif expression did not decrease over time, suggesting there was no strong negative selection against expression of this virulence factor as strains adapted to the CF lung. Colony morphology also appeared to have no bearing on Cif expression since classic, mucoid, and dwarf isolates had representatives of high and low expression (see Table S2 in the supplemental material).
Fig 7.
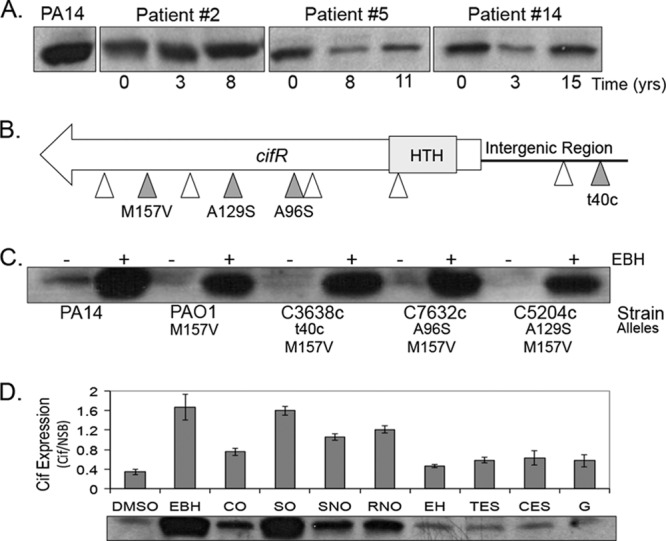
Cif expression in clinical isolates varies over time. (A) Example of cell-associated Cif protein expression from clinical isolates compared to the lab strain P. aeruginosa PA14 (PA14, left) as determined by Western blotting. Numbers indicate the time in years from when the first P. aeruginosa isolate was harvested. These experiments were performed in the absence of the epoxide inducer. (B) Map of mutations, identified in 36 isolates screened. The predicted DNA-binding region is indicated in gray (HTH, helix turn helix). White arrows indicate synonymous mutations in the coding sequence or a mutation in the intergenic region not assigned a function. Gray arrows indicate nonsynonymous mutations or those that might impact gene expression. (C) Cif protein expression with (+) or without (−) EBH in P. aeruginosa PA14, P. aeruginosa PAO1, and isolates harboring the mutations described in panel B. t40c is a noncoding mutation that changes the final nucleotide in the morB proximal binding site. (D) Cif protein expression in wild-type P. aeruginosa PA14 in the presence of epoxides shown as Cif signal intensity relative to a nonchanging, nonspecific band (NSB, not shown). DMSO, negative control; CO, cyclohexene oxide; SO, styrene oxide; SNO, (S)-nitrophenyl oxirane; RNO, (R)-nitrophenyl oxirane; EH, epoxyhexane; TES, trans-epoxysuccinate; CES, cis-epoxysuccinate; G, glycidol.
Next, we attempted to link the differences in expression to mutations in the intergenic region between cifR and the morB operon or in the cifR gene sequence. These data are summarized in Fig. 7B. Sequence analysis of the 36 isolates identified a number of point substitutions in the specified sequences. Many of these were synonymous substitutions or did not correspond to nucleotide changes likely to affect the promoters. All isolates contained an M157V amino acid sequence variant present in P. aeruginosa PAO1 that does not inhibit CifR function (Fig. 7C and see Table S2 in the supplemental material). One mutation was identified in the final base (5′-3′) of the morB proximal CifR binding site, but this did not correspond to an increase in Cif protein levels (see Table S2 in the supplemental material). We did identify two nonsynonymous alanine-to-serine mutations in the CifR sequence within the predicted ligand-binding domain. These mutations did not appear to ablate CifR repression (see Table S2 in the supplemental material and Fig. 7C), but we decided to test whether these mutations in CifR could negatively impact the ability of CifR to respond to an epoxide stimulus. Western analysis of clinical isolates grown in the presence of EBH all showed a sharp increase in Cif protein production, similar to P. aeruginosa PA14, suggesting no impairment of ligand response, even at lower concentrations of the inducer (Fig. 7C and see Fig. S3A in the supplemental material). These data support the hypothesis that both Cif and CifR expression and the function of the CifR protein are maintained in lung isolates of P. aeruginosa over time.
Effect of epoxides on Cif induction.
Since we could not link cif and cifR gene expression changes to mutation, we suspected that CifR was responding to an epoxide inducer in the lung. Our group has reported on the ability of the Cif protein to hydrolyze epoxides, including EBH and cis-stilbene oxide (3, 25, 26). Although these observations have allowed us to verify the epoxide hydrolase activity of the Cif protein, these substrates are unlikely to be found in the context of CF lung infection. In addition, no other epoxide except for EBH has been reported to induce Cif protein expression in vivo (25). We suspect that a better understanding of which kinds of epoxides are able to induce Cif protein expression through CifR may provide insight into the identity of the inducer(s) of Cif protein expression in the lung. Thus, we characterized the ability of the several different nonendogenous epoxides to induce Cif protein expression. These data are summarized in Fig. 7D.
As previously shown, Cif protein expression is strongly induced in the presence of EBH relative to a DMSO control (Fig. 7D). The additional epoxides were able to induce expression to various degrees (Fig. 7D). SO was capable of increasing Cif protein levels to a degree similar to EBH. Others, such as CO, RNO, and SNO, were able to moderately induce Cif expression, while the remaining epoxides exhibited little induction. We tested a selection of these epoxides with the clinical isolates carrying the alanine-to-serine substitutions and observed no alteration in epoxide-mediated induction (see Fig. S3B in the supplemental material). Thus, these data indicate that CifR is capable of responding to multiple, distinct epoxide ligands.
DISCUSSION
Over the past decade, the study of P. aeruginosa pathogenesis has shifted from virulence factor identification to the characterization of virulence factor modulation and expression during infection. Much work has been done in recent years describing the regulators and regulatory pathways involved in lung infection such as LasR, RhlR, MucA, and cyclic-di-GMP signaling, since these systems are often the first perturbed or mutated during chronic infection (11, 13, 17, 39). We characterized here the modulation of the virulence factor, Cif, by its regulator, CifR.
We have previously reported the discovery of CifR, a TetR-family regulator that is encoded by a gene that is adjacent to, and divergently transcribed from, the cif gene-containing operon and that binds directly to the intergenic space between the cifR gene and the morB operon (25). In the present study we show CifR binds to two distinct sites within this intergenic region, overlapping the −10 and +1 promoter elements for both the cifR gene and the morB operon. Binding at a similar promoter-proximal site has been shown recently for another TetR-family regulator, RamR, of Salmonella enterica (4), in contrast to other family members that have been shown to bind at a location more distal to the transcriptional start (33, 35). However, these repressors have all been shown to bind at a single site to mediate repression of the operon as either a dimer or a dimer of dimers (4, 33, 35), whereas CifR binds at two distinct operators, separated by 28 bp, to control the expression of divergently transcribed loci. The auto-regulation exhibited by CifR, common in the TetR family, is believed to be a feedback control mechanism that ensures optimal repressor concentration (33). Mutagenesis of the operator proximal to morB resulted in an increase in expression of both the cif and cifR transcripts, suggesting that both sites are required for complete repression of each of these genes. Although the morB proximal binding does not directly overlap with the cifR promoter, the −35 position is separated by 1 base from the −10 position of the predicted morB promoter (Fig. 4C). CifR bound to the morB promoter region may provide enough steric interference to prevent RNA polymerase binding the cifR promoter. Alternatively, CifR may work at these two binding sites cooperatively to repress both transcripts, and mutating one of these binding results in the loss of such cooperativity, although we have no in vitro data to support this possibility. Indeed, our data suggest that CifR is a monomer in solution.
Analysis of the CifR binding sequence revealed that CifR binds a 27-bp region of DNA with high affinity. Other TetR-family regulators, such as AcrR of E. coli and QacR of Staphylococcus aureus, have operators of similar lengths (24 and 28 bp, respectively) (37, 40). These regulators bind their operators as dimers of dimers, whereas TetR binds a smaller, 15-bp sequence as one dimer (33). The nature of CifR-DNA binding is the subject of ongoing study.
We also determined the apparent Kd of CifR for the cifR and morB proximal binding sites to be 35 and 512 nM, respectively. The 35 nM affinity of CifR for the cifR proximal binding site is consistent with other TetR-family proteins, such as AcrR (20.2 nM) and CmeR (88 nM) in their affinity for their binding sites (23, 35). The apparent dissociation constant for the morB proximal site is higher than that of the cifR proximal site, which may be attributable to differences in the sequences in these sites (Fig. 4C and 5A). Changes in the sequence of the RamR binding site of S. enterica also resulted in dramatic differences in DNA binding (4), highlighting the importance of sequence conservation. Finally, genome-wide sequence analysis revealed no other predicted CifR binding sites, suggesting that CifR may only regulate the morB operon and the cifR gene via these sites.
Analysis of cif gene expression under laboratory conditions showed that CifR-mediated repression was important for maintaining low-level expression under noninducing conditions. We decided to test whether this was true in the context of the CF lung by attempting to detect cifR transcript in CF sputum and correlating that to cif gene transcription. These data show that the cifR gene is expressed, suggesting this regulator may be derepressed in the lung. Increases in cif gene expression corresponded to increases in the cifR gene transcriptional level showing that the regulatory relationship observed in vitro is likely paralleled in vivo and furthermore suggesting that there may be an epoxide inducer in the lung.
The sputum sample transcriptional data suggested that cif gene transcription appears variable. Previous expression analysis of a few clinical isolates had also shown that cif gene expression is variable (26), indicating heterogeneity in the lung environment similar to what is seen with other virulence factors (12, 16). We attempted to address these questions by exploiting a collection of clinical isolates of P. aeruginosa from a longitudinal study of CF patients. We observed changes in Cif protein expression from these isolates; however, the expression did not decrease but instead remained variable over time. This variability could not be linked to mutations in the promoter or cifR gene sequences, since many of the mutations were present in all isolates from the same patient. In addition, colony morphology could not be linked to changes in expression. These data may suggest that there are other, yet-to-be-discovered regulatory factors involved in cif gene and Cif protein expression. Supporting this hypothesis, our cifR deletion strain exhibits a small but significant increase (2-fold) in cif gene transcription in the presence of EBH (Fig. 5D), suggesting the presence of another epoxide-sensitive regulator. Ongoing research is aimed at identifying additional factors involved in cif gene regulation.
We have previously shown that Cif is an epoxide hydrolase and CifR responds to the epoxide EBH by a reduction in DNA binding, thus permitting transcription (25, 26). In the present study we attempted to test the diverse types of epoxide signals potentially bound by CifR through the identification of epoxides capable of inducing Cif expression (i.e., inducing the loss of CifR-DNA binding). There were clear differences in the ability of each of these epoxides to induce Cif protein expression, with some much better at inducing expression than others. It is possible that this result is due to differences in the CifR ligand site or membrane permeability of these compounds. Either way, P. aeruginosa expresses Cif and CifR in the lung, perhaps in response to an epoxide trigger, which must be able to traverse the bacterial membrane and target CifR. These epoxide findings are important, since they will facilitate future research into the Cif enzyme substrate in the context of the human lung.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Speert Lab for their generous donation of strains. We also thank Pinar Gurel and Aaron White for assistance with protein purification and bioinformatics, respectively. We thank the Translational Research Core (TRC) for providing the sputum samples.
This study was supported by an Immunology Training Grant (T32 AI007363), a Renal Function and Disease training grant (T32 DK007301) to A.E.B. and C.D.B., a Molecular Microbiology and Pathogenesis training grant (T32-AI007519) to C.D.B., and a National Institutes of Health (NIH) grant (R01 AI091699) to D.R.M. and G.A.O. The TRC is supported by the National Institute of General Medical Sciences of the NIH under award P20GM103413.
The content here is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Published ahead of print 27 July 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Bahl CD, MacEachran DP, O'Toole GA, Madden DR. 2010. Purification, crystallization, and preliminary X-ray diffraction analysis of Cif, a virulence factor secreted by Pseudomonas aeruginosa. Acta Crystallogr. Sect. F Struct. Biol. Crystallogr. Commun. 66:26–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bahl CD, Madden DR. 2012. Pseudomonas aeruginosa Cif defines a distinct class of alpha/beta epoxide hydrolases utilizing a His/Tyr ring-opening pair. Protein Pept. Lett. 19:186–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bahl CD, et al. 2010. Crystal structure of the cystic fibrosis transmembrane conductance regulator inhibitory factor Cif reveals novel active-site features of an epoxide hydrolase virulence factor. J. Bacteriol. 192:1785–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baucheron S, et al. 2012. Binding of the RamR repressor to wild-type and mutated promoters of the RamA gene involved in efflux-mediated multidrug resistance in Salmonella enterica serovar Typhimurium. Antimicrob. Agents Chemother. 56:942–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Behrends V, et al. Metabolic adaptations of Pseudomonas aeruginosa during cystic fibrosis chronic lung infections. Environ. Microbiol., in press [DOI] [PubMed] [Google Scholar]
- 6. Bendiak GN, Ratjen F. 2009. The approach to Pseudomonas aeruginosa in cystic fibrosis. Semin. Respir. Crit. Care Med. 30:587–595 [DOI] [PubMed] [Google Scholar]
- 7. Bomberger JM, et al. 2009. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog. 5:e1000382 doi:10.1371/journal.ppat.1000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bomberger JM, et al. 2010. A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PLoS Pathog. 7:e1001325 doi:10.1371/journal.ppat.1001325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boucher RC. 2007. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu. Rev. Med. 58:157–170 [DOI] [PubMed] [Google Scholar]
- 10. Caiazza NC, O'Toole GA. 2004. SadB is required for the transition from reversible to irreversible attachment during biofilm formation by Pseudomonas aeruginosa PA14. J. Bacteriol. 186:4476–4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ciofu O, Mandsberg LF, Bjarnsholt T, Wassermann T, Hoiby N. 2010. Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 156:1108–1119 [DOI] [PubMed] [Google Scholar]
- 12. Ciofu O, Riis B, Pressler T, Poulsen HE, Hoiby N. 2005. Occurrence of hypermutable Pseudomonas aeruginosa in cystic fibrosis patients is associated with the oxidative stress caused by chronic lung inflammation. Antimicrob. Agents Chemother. 49:2276–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. D'Argenio DA, et al. 2007. Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol. Microbiol. 64:512–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harrison F. 2007. Microbial ecology of the cystic fibrosis lung. Microbiology 153:917–923 [DOI] [PubMed] [Google Scholar]
- 15. Hazlett LD. 2004. Corneal response to Pseudomonas aeruginosa infection. Prog. Retin. Eye Res. 23:1–30 [DOI] [PubMed] [Google Scholar]
- 16. Hogardt M, Heesemann J. 2012. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int. J. Med. Microbiol. 300:557–562 [DOI] [PubMed] [Google Scholar]
- 17. Hogardt M, Heesemann J. 2012. Microevolution of Pseudomonas aeruginosa to a chronic pathogen of the cystic fibrosis lung. Curr. Top. Microbiol. Immunol. [Epub ahead of print.] doi:10.1007/82_2011_199 [DOI] [PubMed] [Google Scholar]
- 18. Kadurugamuwa JL, Beveridge TJ. 1997. Natural release of virulence factors in membrane vesicles by Pseudomonas aeruginosa and the effect of aminoglycoside antibiotics on their release. J. Antimicrob. Chemother. 40:615–621 [DOI] [PubMed] [Google Scholar]
- 19. Kerr KG, Snelling AM. 2009. Pseudomonas aeruginosa: a formidable and ever-present adversary. J. Hosp. Infect. 73:338–344 [DOI] [PubMed] [Google Scholar]
- 20. Ko YH, Pedersen PL. 2001. Cystic fibrosis: a brief look at some highlights of a decade of research focused on elucidating and correcting the molecular basis of the disease. J. Bioenerg. Biomembr. 33:513–521 [DOI] [PubMed] [Google Scholar]
- 21. Kuchma SL, et al. 2010. c-di-GMP-mediated repression of swarming motility by Pseudomonas aeruginosa: the pilY1 gene and its impact on surface-associated behaviors. J. Bacteriol. 92:2950–2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lau GW, Hassett DJ, Ran H, Kong F. 2004. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol. Med. 10:599–606 [DOI] [PubMed] [Google Scholar]
- 23. Li M, et al. 2007. Crystal structure of the transcriptional regulator AcrR from Escherichia coli. J. Mol. Biol. 374:591–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Y, Jiang Z, Chen H, Ma WJ. 2004. A modified quantitative EMSA and its application in the study of RNA-protein interactions. J. Biochem. Biophys. Methods 60:85–96 [DOI] [PubMed] [Google Scholar]
- 25. MacEachran DP, Stanton BA, O'Toole GA. 2008. Cif is negatively regulated by the TetR family repressor CifR. Infect. Immun. 76:3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacEachran DP, et al. 2007. The Pseudomonas aeruginosa secreted protein PA2934 decreases apical membrane expression of the cystic fibrosis transmembrane conductance regulator. Infect. Immun. 75:3902–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maniatis T, Sambrook J, Fritsch EF. 1989. Molecular cloning: a laboratory manual, 2nd ed CSHL Press, Cold Spring Harbor, NY [Google Scholar]
- 28. Mathee K, et al. 2008. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U. S. A. 105:3100–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mena KD, Gerba CP. 2009. Risk assessment of Pseudomonas aeruginosa in water. Rev. Environ. Contam. Toxicol. 201:71–115 [DOI] [PubMed] [Google Scholar]
- 30. Mittal R, Aggarwal S, Sharma S, Chhibber S, Harjai K. 2009. Urinary tract infections caused by Pseudomonas aeruginosa: a minireview. J. Infect. Public Health 2:101–111 [DOI] [PubMed] [Google Scholar]
- 31. Orth P, Schnappinger D, Hillen W, Saenger W, Hinrichs W. 2000. Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system. Nat. Struct. Biol. 7:215–219 [DOI] [PubMed] [Google Scholar]
- 32. Rahme LG, et al. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899–1902 [DOI] [PubMed] [Google Scholar]
- 33. Ramos JL, et al. 2005. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69:326–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodriguez-Rojas A, Oliver A, Blazquez J. Intrinsic and environmental mutagenesis drive diversification and persistence of Pseudomonas aeruginosa in chronic lung infections. J. Infect. Dis. 205:121–127 [DOI] [PubMed] [Google Scholar]
- 35. Routh MD, Su CC, Zhang Q, Yu EW. 2009. Structures of AcrR and CmeR: insight into the mechanisms of transcriptional repression and multi-drug recognition in the TetR family of regulators. Biochim. Biophys. Acta 1794:844–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sato H, Frank DW. 2004. ExoU is a potent intracellular phospholipase. Mol. Microbiol. 53:1279–1290 [DOI] [PubMed] [Google Scholar]
- 37. Schumacher MA, et al. 2002. Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. EMBO J. 21:1210–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl. Environ. Microbiol. 72:5027–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith EE, et al. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. U. S. A. 103:8487–8492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Su CC, Rutherford DJ, Yu EW. 2007. Characterization of the multidrug efflux regulator AcrR from Escherichia coli. Biochem. Biophys. Res. Commun. 361:85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Williams BJ, Dehnbostel J, Blackwell TS. Pseudomonas aeruginosa: host defence in lung diseases. Respirology 15:1037–1056 [DOI] [PubMed] [Google Scholar]
- 42. Ye S, MacEachran DP, Hamilton JW, O'Toole GA, Stanton BA. 2008. Chemotoxicity of doxorubicin and surface expression of P-glycoprotein (MDR1) is regulated by the Pseudomonas aeruginosa toxin Cif. Am. J. Physiol. Cell Physiol. 295:C807–C818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yorgey P, Rahme LG, Tan MW, Ausubel FM. 2001. The roles of mucD and alginate in the virulence of Pseudomonas aeruginosa in plants, nematodes, and mice. Mol. Microbiol. 41:1063–1076 [DOI] [PubMed] [Google Scholar]
- 44. Zhao WH, Hu ZQ. Beta-lactamases identified in clinical isolates of Pseudomonas aeruginosa. Crit. Rev. Microbiol. 36:245–258 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.