

Abstract
Mismatch repair is a highly conserved pathway responsible for correcting DNA polymerase errors incorporated during genome replication. MutL is a mismatch repair protein known to coordinate several steps in repair that ultimately results in strand removal following mismatch identification by MutS. MutL homologs from bacteria to humans contain well-conserved N-terminal and C-terminal domains. To understand the contribution of the MutL N-terminal domain to mismatch repair, we analyzed 14 different missense mutations in Bacillus subtilis MutL that were conserved with missense mutations identified in the human MutL homolog MLH1 from patients with hereditary nonpolyposis colorectal cancer (HNPCC). We characterized missense mutations in or near motifs important for ATP binding, ATPase activity, and DNA binding. We found that 13 of the 14 missense mutations conferred a substantial defect to mismatch repair in vivo, while three mutant alleles showed a dominant negative increase in mutation frequency to wild-type mutL. We performed immunoblot analysis to determine the relative stability of each mutant protein in vivo and found that, although most accumulated, several mutant proteins failed to maintain wild-type levels, suggesting defects in protein stability. The remaining missense mutations located in areas of the protein important for DNA binding, ATP binding, and ATPase activities of MutL compromised repair in vivo. Our results define functional residues in the N-terminal domain of B. subtilis MutL that are critical for mismatch repair in vivo.
INTRODUCTION
Accurate DNA replication is critical for the maintenance of chromosomal DNA in all living cells (18). To ensure the faithful duplication of genetic material, replicative polymerases have proofreading activity (3′ to 5′ exonuclease) capable of correcting base-pairing errors formed during DNA synthesis (for a review, see references 18 and 31). Despite the high fidelity of replicative DNA polymerases, base-pairing errors still occur, and these errors can lead to deleterious mutations (for a review, see reference 30). The mismatch repair (MMR) pathway enlists two highly conserved proteins, MutS and MutL, to identify and correct base-pairing errors and insertion and deletion loops formed during DNA synthesis (32, 44). In doing so, MMR is capable of increasing the fidelity of the DNA replication pathway several-hundred-fold in a variety of organisms (8, 16, 20). In addition to its role in correcting replicative errors, MMR proteins also recognize DNA lesions produced by the oxidative by-products of intracellular metabolism (6, 43), as well as lesions formed by exogenous sources of DNA damage (52). Moreover, in Gram-negative bacteria, MMR has been shown to function in antirecombination, which prevents recombination between DNA from divergent sources, providing a genetic barrier between bacterial species (41).
MMR is critically important for human health. MMR deficiencies allow for some bacterial pathogens to persist within their host (e.g., see reference 36), while other pathogens require MMR for colonization of the intestine or ascent to the bladder during urinary tract infections (7, 10). Furthermore, MMR defects in bacteria can accelerate the acquisition of resistance to antibiotic treatments (5, 11, 36). Defects in the MMR pathway in humans cause an increased risk for the development of sporadic cancers, including hereditary nonpolyposis colorectal cancer (HNPCC) and Turcot's syndrome (16, 24, 35, 37). These are a few examples that highlight the practical importance of MMR to health care.
In the Gram-positive bacterium Bacillus subtilis, MutS recognizes a mismatch and recruits MutL to the site of mismatch recognition (13, 46, 48). Like most bacteria and all eukaryotic organisms, B. subtilis lacks the canonical methylation-directed pathway characterized in Escherichia coli (9, 14, 15). In E. coli, Dam methylase and MutH represent the two proteins required for generating and sensing the methylation signal (33). In B. subtilis, MutH and Dam are not present and d(GATC) sequences are not methylated in vivo (12). The hallmark of the methylation-independent mismatch repair pathway is the presence of an endonuclease-containing MutL homolog (27, 28, 38). B. subtilis MutL contains two domains located at the N and C termini of the protein. Based on sequence homology with E. coli and other systems, the B. subtilis MutL N-terminal domain is a GHKL (DNA gyrase, Hsp90, bacterial histidine kinases, and MutL) family ATPase with a DNA binding cleft (2, 3, 21, 26). A structure for the N-terminal domain of B. subtilis mutL has not been solved. The C-terminal domain is comprised of the endonuclease active site, a binding site for the replication processivity clamp DnaN and a zinc binding loop (38). The N- and C-terminal domains are connected by a flexible linker (2, 3, 21, 26).
The C-terminal endonuclease active site is highly conserved from B. subtilis to humans and possesses the consensus active site motif DQHA(X)2E(X)4E (27, 28, 38). Strikingly, the endonuclease active site in B. subtilis MutL is identical to the site in Saccharomyces cerevisiae and the human MutL homologs PMS1 and PMS2 (27). Recently, the crystal structure was solved for the C-terminal domain of B. subtilis MutL (38). Mutation of residues found within the endonuclease active site, the zinc binding loop, or a site shown to bind DnaN within the C-terminal domain of MutL decreases or abolishes MMR activity in vivo (38, 39). It is important to note that E. coli MutL lacks the endonuclease active site, as well as endonuclease activity and the zinc binding loop (22, 27). Furthermore, mutation of the DnaN interaction site within the C-terminal domain of E. coli MutL causes only a mild effect on mutagenesis, while mutation of the same site in B. subtilis MutL increases mutagenesis substantially (38, 39). Therefore, the effect of altering critical residues in the C-terminal domain of B. subtilis MutL has been well characterized, while the N-terminal domain has remained virtually unexplored in Gram-positive bacteria.
In this work, we characterized the N-terminal domain of B. subtilis using a mutational approach. Since B. subtilis MutL shares amino acid sequence conservation with the human MutL homolog MLH1, we characterized 14 B. subtilis mutL alleles with missense mutations corresponding to potentially pathogenic human MLH1 polymorphisms found in patients diagnosed with HNPCC. We analyzed these variants for their protein stability, mutation rate, and ability to exert a negative effect on MMR in the presence of the wild-type mutL gene in vivo. We found that mutant MutL proteins with residue changes in or near motifs important for ATP binding, ATPase activity, or DNA binding showed a wide range of defects in MMR. Mutations located near the ATP binding site consistently conferred the most striking increases in mutagenesis. We found that three mutant proteins (MutL S40A, G63R, and G94S) failed to accumulate in vivo, which is consistent with the complete defect in MMR conferred by each corresponding allele. We also found that three missense mutations [mutL(E30A), mutL(N34H), and mutL(R176G)] conferred a dominant negative phenotype, supporting the notion that the corresponding mutation in human MLH1 would have a pathogenic role leading to genome instability in HNPCC patients. Together, our results identify residues in the N-terminal domain of B. subtilis MutL that are critical for MMR, and we test corresponding MLH1 missense mutations for MMR defects in a tractable bacterial system.
MATERIALS AND METHODS
Bacteriology.
Each B. subtilis mutL allele was constructed through site-directed mutagenesis (47) of the wild-type mutL gene and confirmed by DNA sequence analysis. All primers used in this study are available upon request. Integration at the ectopic amyE locus of the wild-type strain (PY79) was achieved by double crossover using standard procedures (25). To construct strains deficient for the native mutL allele, chromosomal DNA from LAS393 (mutL::spc) was used to transform each strain containing the amyE::PspacmutL allele. All strains used in this study are listed in Table 1. Unless otherwise stated, antibiotics and isopropylthio-β-galactoside (IPTG) were used at the following concentrations: 100 μg/ml spectinomycin (Spc), 5 μg/ml chloramphenicol (Cat), and 1 mM IPTG.
Table 1.
List of strains
| Strain | Characteristics | Source or reference |
|---|---|---|
| NJB1 | PY79 (SPβ0) | 46, 48 |
| NJB21 | amyE::Pspac-mutL+ (cat) | 46, 48 |
| NJB22 | amyE::Pspac-mutL(G18A) (cat) | This study |
| NJB23 | amyE::Pspac-mutL(P24L) (cat) | This study |
| NJB24 | amyE::Pspac-mutL(G63R) (cat) | This study |
| NJB25 | amyE::Pspac-mutL(S102R) (cat) | This study |
| NJB26 | amyE::Pspac-mutL(E30A) (cat) | This study |
| NJB27 | amyE::Pspac-mutL(N34H) (cat) | This study |
| NJB28 | amyE::Pspac-mutL(K80E) (cat) | This study |
| NJB29 | amyE::Pspac-mutL(G94S) (cat) | This study |
| NJB30 | amyE::Pspac-mutL(R176G) (cat) | This study |
| NJB31 | amyE::Pspac-mutL(G234V) (cat) | This study |
| NJB32 | amyE::Pspac-mutL(R256C) (cat) | This study |
| NJB33 | amyE::Pspac-mutL(S40A) (cat) | This study |
| NJB34 | amyE::Pspac-mutL(N60S) (cat) | This study |
| NJB35 | amyE::Pspac-mutL(A17V) (cat) | This study |
| NJB37 | amyE::Pspac-mutL+(cat) mutL::spc | This study |
| NJB38 | amyE::Pspac-mutL(G18A) (cat) mutL::spc | This study |
| NJB39 | amyE::Pspac-mutL(P24L0 (cat) mutL::spc | This study |
| NJB40 | amyE::Pspac-mutL(G63R) (cat) mutL::spc | This study |
| NJB41 | amyE::Pspac-mutL(S102R) (cat) mutL::spc | This study |
| NJB42 | amyE::Pspac-mutL(E30A) (cat) mutL::spc | This study |
| NJB43 | amyE::Pspac-mutL(N34H) (cat) mutL::spc | This study |
| NJB44 | amyE::Pspac-mutL(K80E) (cat) mutL::spc | This study |
| NJB45 | amyE::Pspac-mutL(G94S) (cat) mutL::spc | This study |
| NJB46 | amyE::Pspac-mutL(R176G) (cat) mutL::spc | This study |
| NJB47 | amyE::Pspac-mutL(G234V) (cat) mutL::spc | This study |
| NJB48 | amyE::Pspac-mutL(R256C) (cat) mutL::spc | This study |
| NJB49 | amyE::Pspac-mutL(S40A) (cat) mutL::spc | This study |
| NJB50 | amyE::Pspac-mutL(N60S) (cat) mutL::spc | This study |
| NJB51 | amyE::Pspac-mutL(A17V) (cat) mutL::spc | This study |
| LAS393 | mutL::spc | 46, 48 |
Mutation rates.
Each strain to be tested was grown overnight on LB agar plates with the appropriate antibiotics, and 1 mM IPTG was prepared just prior to use. IPTG was used to induce expression of each mutant protein regulated by a Pspac promoter from the ectopic amyE locus. Following overnight growth on solid medium, 3-ml liquid LB cultures containing 1 mM IPTG were inoculated with a single colony and grown for approximately 3 h to reach late exponential growth (optical density at 600 nm [OD600] between 1.0 and 1.2). A 1-ml sample of culture was removed and concentrated by centrifugation at 10,000 rpm. The supernatant was then aspirated carefully, as to not disturb the cell pellet. Subsequently, the cells were resuspended in 100 μl of a 0.85% saline solution. The cell suspension was then serially 106-fold diluted into 0.85% saline. To obtain a count of the total number of viable cells in each culture, 100 μl of the final dilution was plated on LB while 100 μl of the original cell resuspension was plated on LB agar with 100 μg/ml rifampin (Rif) to determine the number of Rifr CFU. Following overnight growth, both the LB and LB-Rif plates were scored for colony-forming units. Mutation rate analysis (17) was conducted using the Ma-Sandri-Sarkar maximum likelihood estimator (MSS-MLE) method as described previously (23).
Immunoblotting.
A single colony, obtained from overnight growth on LB agar plates with the appropriate antibiotics, was used to inoculate a 20-ml liquid LB culture. Cultures were incubated at 37°C to reach mid-exponential growth (OD600 of 0.6 to 0.9). Cells were concentrated by centrifugation for 10 min at 10,000 rpm, followed by aspiration of the supernatant immediately following centrifugation. Cell pellets were resuspended in 1 ml of lysis buffer [10 mM Tris-HCl (pH 7.0), 0.5 mM EDTA, 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), and 1× protease inhibitor cocktail from the Thermo Scientific protease inhibitor cocktail kit]. After resuspension, the samples were subjected to sonication for lysis. Sonication was conducted at 20 Hz for three 40-s treatments per sample. After sonication, cell extracts were centrifuged for 20 min at 6,000 rpm to remove insoluble debris. Concentrations of soluble protein extracts were determined using the Pierce BCA protein assay kit by monitoring the absorbance of 562-nm wavelengths in a Tecan plate reader.
Soluble extracts were resolved by SDS-PAGE (47) after loading 15 μg of total protein per extract. Following transfer to a nitrocellulose membrane, each membrane was blocked in phosphate-buffered saline–Tween (PBS-T) with 5% milk for 1 h, followed by incubation overnight with a 1:500 primary antibody dilution against MutL or DnaN in blocking buffer as described previously (29). Washing in blocking buffer removed excess antibody before incubation of the membrane for 2 h in secondary antibody (goat anti-rabbit immunoglobulin–horseradish peroxidase [HRP] conjugate). For secondary antibodies, we used a working dilution of 1:2,000 in PBS-T with 5% milk. Following incubation, the membrane was washed once in PBS-T with 5% milk, followed by two washes in PBS-T to remove excess antibody. HRP was detected using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific), followed by exposure to Blue Lite autorad film (GeneMate).
RESULTS AND DISCUSSION
Identification of N-terminal mutL missense mutations defective for MMR.
To characterize the N-terminal domain of B. subtilis MutL in MMR, we chose a mutational approach. In order to identify potential residues important for MutL function, we used the International Society for Gastrointestinal Hereditary Tumors (InSiGHT database) to identify missense mutations in the human MutL homolog MHL1 from patients with hereditary nonpolyposis colorectal cancer (HNPCC; www.insight-group.org/) that altered residues conserved in B. subtilis MutL. We chose to study the corresponding MLH1 missense mutations from HNPCC patients in B. subtilis for two reasons. First, very little is known about the N-terminal domain of B. subtilis MutL and B. subtilis MutL is well conserved with human MLH1 (Fig. 1A). Second, using B. subtilis as a model system, this approach allowed us to determine and quantify the severity of the defect to MMR caused by missense mutations corresponding to MLH1 polymorphisms identified in HNPCC patients. This is important because it allowed us to functionally assess the corresponding missense mutations in a genetically tractable bacterial system in vivo. Some studies have characterized human MLH1 and MSH6 polymorphisms in the corresponding S. cerevisiae genes, yielding important insights into the effects on MMR in a genetically tractable eukaryotic organism (e.g., see references 19, 50, and 51). To our knowledge, a bacterial system has not been used for this purpose. Therefore, we were interested in using B. subtilis as a rapid experimental system for analysis of human MLH1 missense mutations and to functionally characterize the N-terminal domain of B. subtilis MutL.
Fig 1.

Homology model of B. subtilis MutL N-terminal domain with the locations of amino acid substitutions indicated. (A) A sequence alignment of the N-terminal domains of B. subtilis MutL (BsMutL) and human MLH1 (hMLH1) is shown. We show the primary structure alignment relevant to the location of the missense mutations that were tested in this study. Colored residues correspond to the colors used in panel B. (B) A model of the MutL N-terminal domain dimer is shown. The amino acid residue changes are modeled on the left monomer. Residues predicted to be important for ATP binding, ATP hydrolysis, and DNA binding are colored in orange, red, and purple, respectively. Mutants with functions that are unclear based on homology are colored in green.
To this end, we cloned 14 B. subtilis mutL missense mutations that correspond to human MLH1 missense mutations (Table 2). We also show the location of each amino acid substitution on a model of the B. subtilis MutL N-terminal domain (Fig. 1B). Each B. subtilis mutant mutL allele was expressed ectopically from an IPTG-regulated promoter from the amyE locus in a strain background disrupted for the native mutL gene. We performed a titration of IPTG to determine the level required to express MutL to wild-type levels in vivo while also determining the mutation rate (Fig. 2). Expression of mutL with 25 μM IPTG yielded a low protein level and a high spontaneous mutation rate. Induced expression with 100 μM or 1 mM IPTG yielded protein levels similar to those of mutL expressed from its native promoter, and both concentrations yielded a mutation rate identical to that of the wild-type control strain PY79 (Fig. 2). Since induced expression with 1 mM IPTG resulted in a protein level more comparable to that of native MutL, we chose to express each mutant protein with 1 mM IPTG for the analysis that follows.
Table 2.
B. subtilis mutL missense mutation conserved in human MLH1a
| B. subtilis mutL missense mutation | Human MLH1 missense mutation | No. of reported occurrences in humansb | Potential functionc |
|---|---|---|---|
| A17V | A21V | 6 | ATP binding |
| G18A | G22A | 11 | ATP binding |
| P24L | P28L | 32 | ATP hydrolysis |
| E30A | E34A | 4 | ATP hydrolysis |
| N34H | N38H | 8 | ATP hydrolysis |
| S40A | S44A | 5 | ATP hydrolysis |
| N60S | N64S | 20 | Unclear |
| G63R | G67R | 74 | ATP binding |
| K80E | K84E | 20 | ATP hydrolysis |
| G94S | G98S | 8 | ATP binding |
| S102R | S106R | 7 | ATP binding |
| R176G | R182G | 14 | DNA binding |
| G234V | G244V | 6 | DNA binding |
| R256C | R265C | 53 | ATP binding |
A list of MLH1 mutations, the corresponding B. subtilis missense mutations, the reported incidence in HNPCC patients, and the possible MutL functions impaired by each mutation is shown.
The occurrence of each human MLH1 missense mutation was obtained from the International Society for Gastrointestinal Tumors (www.insight-group.org).
Fig 2.
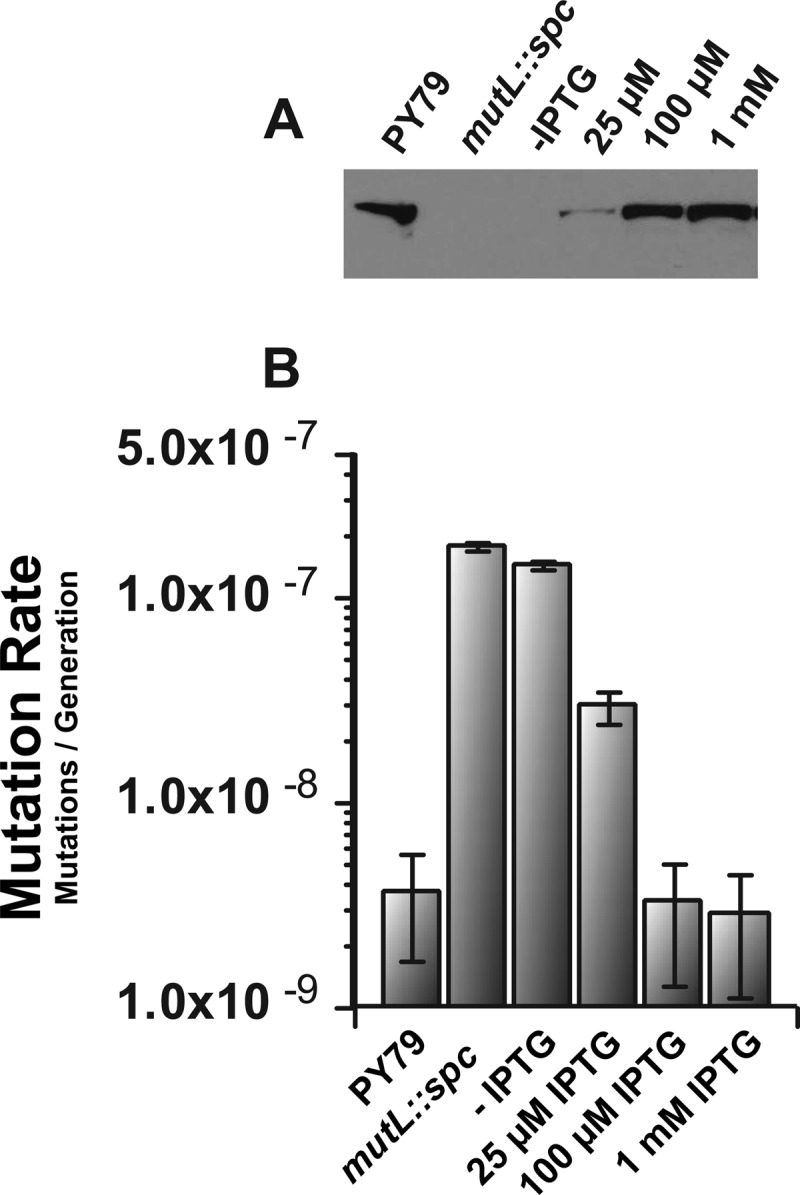
Ectopic MutL expression. (A) The levels of MutL protein (left to right) from the wild-type control strain PY79, a strain with the mutL::spc allele, and samples with mutL expressed from amyE with the native mutL gene disrupted (mutL::spc) with the indicated concentrations of IPTG are shown. All samples were derived from cell extracts prepared as described in Materials and Methods. The sample load was normalized to total protein in each extract. (B) The mutation rate corresponding to each concentration of IPTG used in panel A is shown. Error bars represent the 95% confidence interval.
In the mutation rate analysis described above and in the analysis described below, we determined the mutation rate by performing a fluctuation analysis using rifampin resistance as an indicator for spontaneous mutagenesis. Rifampin resistance in B. subtilis is conferred by one of several nucleotide substitutions in the rpoB gene (13, 34), encoding the beta subunit of RNA polymerase (4). To assess the proficiency of each mutL allele in MMR, we used the Ma-Sandri-Sarkar maximum likelihood estimator (MSS-MLE) method for calculation mutation rates (see Materials and Methods) (23). Prior to calculation of mutation rates for each mutant mutL allele, we compared the mutation rates calculated for the wild-type strain (PY79) and the wild-type control (mutL+), expressing the only cellular copy of mutL from the ectopic amyE locus with the native mutL gene disrupted (mutL::spc). We found that the wild-type and ectopically expressed mutL+ strains yielded mutation rates that were nearly identical, with rates of 3.9 × 10−9 and 3.8 × 10−9 Rifr mutations/generation, respectively (Fig. 2 and Table 3). Thus, the mutation rates conferred by mutL+ (PY79) and amyE::Pspac-mutL+ in a strain background disrupted for the native mutL locus (mutL::spc) were remarkably close, confirming that ectopic expression of functional mutL complements the mutL::spc allele (Fig. 2B and Table 3). We then tested a mutL-deficient strain (mutL::spc) and found a mutation rate of 192.1 × 10−9 mutations/generation (Table 3). Because mutL is required for MMR function, this mutation rate is representative of a complete loss of mismatch correction in B. subtilis and consistent with previously published data (13, 46).
Table 3.
Missense mutations in B. subtilis mutL cause defects in MMRa
| Genotype | Mutation rate × 10−9b | Relative mutation rate | Mutations per culture |
|---|---|---|---|
| Wild type (PY79) | 3.9 (2.0–5.8) | 1.0 | 2.5 |
| amyE::Pspac-mutL+ | 3.8 (2.0–5.6) | 1.0 | 2.3 |
| mutL::spc | 192.1 (184.1–200.1) | 50.1 | 117.2 |
| mutL(A17V) | 180.1 (173.1–187.2) | 47.0 | 126.6 |
| mutL(G18A) | 35.7 (29.9–41.4) | 9.3 | 19.8 |
| mutL(P24L) | 81.0 (74.8–87.2) | 21.1 | 54.1 |
| mutL(E30A) | 139.6 (132.0–147.2) | 36.4 | 83.4 |
| mutL(N34H) | 177.0 (169.3–184.8) | 46.2 | 110.3 |
| mutL(S40A) | 149.0 (142.5–155.4) | 38.9 | 112.3 |
| mutL(N60S) | 2.5 (0.9–3.9) | 0.7 | 1.4 |
| mutL(G63R) | 145.9 (137.3–154.5) | 38.1 | 76.1 |
| mutL(K80E) | 150.7 (142.8–158.5) | 39.3 | 89.2 |
| mutL(G94S) | 160.7 (153.5–168.0) | 41.9 | 107.0 |
| mutL(S102R) | 24.9 (20.1–29.7) | 6.5 | 15.3 |
| mutL(R176G) | 35.0 (30.3–39.6) | 9.1 | 26.2 |
| mutL(G234V) | 98.1 (91.5–104.6) | 25.6 | 65.0 |
| mutL(R256C) | 141.5 (134.0–148.9) | 36.9 | 87.4 |
The mutation rate, the relative mutation rate, and the mutations conferred per culture for each mutL allele examined are shown. Mutation rates and mutations per culture (17) were calculated by FALCOR (Fluctuation AnaLysis CalculatOR) using the MSS-MLE method (23). Calculations are based on data collected through rifampin resistance by fluctuation analysis for 14 to 50 independent cultures per strain. The relative mutation rate reflects normalization of mutation rates to that obtained for Pspac mutL+.
The error shown in the parentheses reflects the 95% confidence interval.
With this assay established, we examined 14 different mutL alleles expressed from the amyE locus with the native mutL gene disrupted (see genotype in Table 1). Each mutL allele was inserted at the amyE locus by double-crossover integration (see Materials and Methods), and each novel B. subtilis mutL allele was sequenced prior to use. We performed immunoblot analysis to determine the relative accumulation of each mutant protein in vivo. This experiment was performed because an HNPCC-associated missense mutation in human MLH1 could cause an MMR defect by simply destabilizing the protein in vivo, resulting in lower steady-state protein levels, compromising MMR in vivo. The absence of the immunoreactive band from each blot prepared from the mutL::spc strain (Fig. 2A and 3A and B) indicates that the antiserum we used was specific for MutL. The majority of the mutant MutL proteins accumulated to wild-type levels in vivo, including MutL with the A17V, G18A, P24L, E30A, N60S, S102R, or R176G change (Fig. 3). MutL proteins corresponding to N34H, K80E, G234V, or R256C were reduced in vivo with levels clearly lower than the wild-type control (Fig. 3). We also found that several mutant proteins failed to be detected by immunoblot analysis. These mutant proteins include MutL with the S40A, G94S, or G63R substitutions. Based on this result, we suggest that the MutL S40A, G96S, and G63R proteins are unstable and degraded in vivo (Fig. 3).
Fig 3.

Expression level of mutant MutL proteins in vivo. An immunoblot with the relative level of each mutant MutL protein in vivo is shown. (A and B) Whole-cell extracts probed for MutL or as a loading control DnaN on a duplicate blot prepared from the same extract are shown. The mutL+ represents the wild-type mutL allele expressed from amyE with 1 mM IPTG, the same expression condition used for each mutant protein tested.
We then determined mutation rates for each of the 14 mutL alleles that were cloned (Table 3). The three mutants that failed to accumulate in vivo conferred a mutation rate comparable to the control strain defective for mutL (mutL::spc). Of the 11 mutL alleles that accumulated, we found that 10 conferred mutation rates significantly higher than the control expressing mutL+ under identical growth conditions (Table 3). We tested missense mutations in or near motifs important for ATP binding, ATPase activity, and DNA binding in the MutL N-terminal domain (Fig. 1B and Table 3).
mutL alleles with mutations hypothesized to impair ATP hydrolysis [mutL(E30A), mutL(N34H), mutL(S40A), mutL(K80E), and mutL(G94S)] showed, on average, the highest increase in the mutation rate relative to missense mutations in or adjacent to motifs important for ATP binding or DNA binding (Table 3). Mutations that impaired ATP hydrolysis were likely to affect protein stability as judged by protein accumulation in vivo (Fig. 3). Of the ATPase mutants, only the mutL(E30A) mutant accumulated to levels equivalent to mutL+. In both E. coli and B. subtilis, MutL is a weak ATPase, with the B. subtilis protein showing the lower rate of hydrolysis (kcat = 0.3 min−1) (38). In B. subtilis, the mechanistic role of ATPase activity in MMR is unclear; however, ATP binding by MutL is required for activation of B. subtilis endonuclease activity (38). MutL endonuclease activity requires that MutL is bound to ATP, as MutL ADP fails to nick DNA (38). In E. coli, ATP hydrolysis by MutL is critical for MMR in vivo, although the precise role is unclear, as an ATPase-deficient MutL mutant shows wild-type levels of MutH incision and UvrD loading in vitro (26, 42, 49). Even though the mechanistic role of MutL ATPase activity during MMR is not fully understood, it is clear that the integrity of this site is important for functional MMR in E. coli (26, 42, 49), S. cerevisiae (e.g., see reference 51), B. subtilis (Table 3), and humans (40). One possibility is that, following ATP hydrolysis, MutL changes conformation, altering protein contacts important for a step in repair following endonuclease cleavage of the DNA.
Increased mutation rates were also conferred by five mutant alleles with missense mutations near the site of ATP binding. These alleles include mutL(A17V), mutL(G18A), mutL(P24L), mutL(G63R), and mutL(S102R). However, the relative mutation rates caused by these missense mutations varied more than the mutant proteins with substitutions near the motif important for ATP hydrolysis. The relative mutation rates ranged from 6.5 to 47 (Table 3). The G63R mutant showed a complete defect in MMR, consistent with the failure of the mutant protein to accumulate in vivo (Fig. 3). Missense mutations in the DNA binding cleft of the N-terminal domain conferred an increase in the mutation rate compared to the mutL+ strain. As with alleles hypothesized to be altered for ATP binding, the two missense mutations constructed in conserved DNA binding cleft residues varied in the mutation rate. The mutL(G234V) mutation conferred a relative mutation rate of 25.6-fold, and the mutL(R176G) missense mutation caused a relative mutation rate of only 9.1-fold compared with ectopically expressed mutL+. Based on these results we speculate that DNA binding in the N-terminal cleft may not be as important for MMR in vivo as DNA binding in the C-terminal domain of B. subtilis MutL.
mutL(E30A), mutL(N34H), and mutL(R176G) are dominant negative to native mutL.
A dominant negative effect caused by a mutL mutant would provide further insight into its functional ability. This would distinguish between a mutant protein that is unfolded and/or inactive relative to a protein that can cause a defect in the pathway when the wild-type protein is present. Additionally, it could provide insight into a pathogenic mutation in the corresponding MLH1 gene, which would cause an increase in mutation frequency in the presence of a wild-type allele in a diploid organism (24). Thus, we quantified the dominant negative effect of each mutant allele by determining the mutation rates of B. subtilis strains expressing both the mutant and wild-type mutL alleles (Table 4). In each strain tested, the native mutL locus remained intact and the missense mutants were expressed at the ectopic amyE locus from the Pspac promoter using the same level of IPTG as above. This will ensure that the mutant MutL proteins are expressed at the same level as the natively expressed wild-type MutL.
Table 4.
mutL(E30A), mutL(N34H), and mutL(R176G) show a dominant negative increase in mutation frequency to native mutLa
| Genotype | Mutation rate × 10−9b | Relative mutation rate | Mutations per culture |
|---|---|---|---|
| wild type (PY79) | 3.7 (1.8–5.4) | 1.0 | 2.2 |
| amyE::PspacmutL+ | 3.5 (1.5–5.3) | 1.0 | 2.0 |
| mutL::spc | 159.0 (151.1–167.0) | 45.4 | 93.5 |
| mutL(A17V) | 4.1 (1.8–6.3) | 1.2 | 2.9 |
| mutL(G18A) | 2.3 (0.8–3.6) | 0.7 | 1.3 |
| mutL(P24L) | 3.7 (1.5–5.7) | 1.1 | 2.3 |
| mutL(E30A) | 10.8 (7.5–14.1) | 3.1 | 7.9 |
| mutL(N34H) | 102.3 (95.7–108.8) | 29.2 | 68.2 |
| mutL(S40A) | 2.6 (0.8–4.1) | 0.7 | 1.5 |
| mutL(N60S) | 2.7 (0.4–4.4) | 0.8 | 1.2 |
| mutL(G63R) | 2.3 (0.7–3.6) | 0.7 | 1.4 |
| mutL(K80E) | 6.2 (3.5–8.9) | 1.8 | 4.0 |
| mutL(G94S) | 1.6 (0.4–2.4) | 0.5 | 0.9 |
| mutL(S102R) | 5.0 (2.5–7.5) | 1.4 | 3.1 |
| mutL(R176G) | 48.5 (41.1–55.8) | 13.8 | 22.3 |
| mutL(G234V) | 5.2 (2.3–7.8) | 1.5 | 2.6 |
| mutL(R256C) | 2.9 (0.8–4.6) | 0.8 | 1.7 |
The mutation rate, relative mutation rate, and mutation per culture determined for each mutant allele when expressed in the presence of the wild-type mutL gene are shown. Mutation rates and mutations per culture were calculated by FALCOR (Fluctuation AnaLysis CalculatOR) using the MSS-MLE method (17, 23). Calculations were based on data collected through rifampin resistance by fluctuation analysis for 14 to 50 independent cultures per strain. Relative mutation rate reflects normalization of mutation rates to that obtained for Pspac mutL+.
The error shown in parentheses reflects the 95% confidence interval.
We found that three missense mutants conferred a dominant-negative increase in mutation rate to native mutL. Two of these mutants, the E30A and N34H mutants, are hypothesized to affect ATP hydrolysis. The E30A mutant protein conferred a slight increase in mutation rate, with a relative rate of 3.1-fold compared to that for the wild-type control. However, the N34H mutant yielded a relative rate of 29.2-fold. These data suggest that both the mutL(E30A) and mutL(N34H) mutants are dominant to the wild-type mutL allele, causing a partial defect in the MMR pathway. We find these data interesting since a dominant negative phenotype has been shown in HNPCC alleles and E. coli mutL bearing missense mutations affecting ATP hydrolysis (1, 45). The third mutant, the mutL(R176G) mutant, is a conserved DNA binding residue. It has a relative dominant negative mutation rate of 13.8-fold (Table 4). The missense mutation corresponding to R176G in S. cerevisiae mlh1 (R182G) has been tested functionally and has also been found to be dominant negative, placing our data in good agreement with prior work (50). We speculate that the dominant negative results correspond to mutant MutL proteins forming mixed dimers with MutL+, decreasing mismatch repair activity in vivo, or that the mutant MutL is able to compete with wild-type MutL for sites of repair, decreasing MMR efficiency. We suggest the latter, since each dominant negative allele caused a partial decrease in MMR and not a complete loss in the pathway.
Conservation of MutL function between B. subtilis and eukaryotic systems.
Several of the missense mutations that we tested in B. subtilis MutL have been examined in S. cerevisiae mlh1 or in human MLH1 (50, 51). A previous study found that the S. cerevisiae mlh1 missense mutations corresponding to B. subtilis mutL(P24L) and mutL(N60S) result in an intermediate increase in the mutation rate, while the mutL(R256C) mutant causes an increase in the mutation rate lower than those for the mutL(P24L) and mutL(N60S) mutant alleles (51). The mutL(K80E) allele results in a mutation rate close to that of a strain lacking mlh1 (51). In B. subtilis, we found that the mutL(P24L) allele causes an intermediate increase in the mutation rate, the mutL(N60S) allele is wild type for mutagenesis, and the mutL(K80E) and mutL(R256C) alleles confer an increase in the mutation rate comparable to that for a strain lacking mutL. In addition, our analysis found that mutL(K80E) and mutL(R256C) result in low protein levels in vivo, explaining our finding that the mutation rate conferred by these alleles approaches that of a mutL-deficient strain. We also found that the MutL S40A, G63R, and G94S proteins failed to accumulate in vivo. Studies of the corresponding human mutant protein revealed effects on expression for MLH1 G67R and R265C, corresponding to B. subtilis mutL(G63R) and mutL(R256C), respectively (50). Thus, two of the mutants that we found to have low or no accumulation in B. subtilis were shown to decrease in accumulation following expression in human cell culture.
The mutL(N60S) missense mutation has a different effect on MMR in B. subtilis than the corresponding missense mutation in S. cerevisiae mlh1 (N64S), which caused a substantial defect in the MMR pathway (51). In addition, we found that the S40A mutant did not accumulate in vivo and that the corresponding change in human MLH1 showed greater than 75% expression, suggesting a difference in stability between the B. subtilis and human mutant proteins (50). Although we did observe some differences in the behavior of B. subtilis MutL relative to S. cerevisiae or human MLH1, most of the mutant proteins that we examined showed similar results on mutagenesis and protein accumulation. With these results, we suggest that B. subtilis MutL serves as an effective model for functional analysis of conserved mutL missense mutations identified in less experimentally tractable eukaryotic organisms.
ACKNOWLEDGMENTS
We are indebted to Jeanne Stuckey (University of Michigan, Life Sciences Institute) for help with modeling the N-terminal domain of B. subtilis MutL. We thank Jiaxing Li and Brian Walsh for initiating construction of the mutL mutants. We thank members of the Simmons lab for critical reading of the manuscript. We also thank Matthew Chapman and Blaise Boles for their comments on this study and the comments from two anonymous reviewers.
This work was supported by a grant from the Wendy Will Case Cancer Fund and by grant MCB1050948 from the National Science Foundation to L.A.S. A 2011 undergraduate summer fellowship from the Department of Molecular, Cellular, and Developmental Biology at the University of Michigan partially supported N.J.B. during this study.
Footnotes
Published ahead of print 27 July 2012
REFERENCES
- 1. Aronshtam A, Marinus MG. 1996. Dominant negative mutator mutations in the mutL gene of Escherichia coli. Nucleic Acids Res. 24:2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ban C, Junop M, Yang W. 1999. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell 97:85–97 [DOI] [PubMed] [Google Scholar]
- 3. Ban C, Yang W. 1998. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell 95:541–552 [DOI] [PubMed] [Google Scholar]
- 4. Boor KJ, Duncan ML, Price CW. 1995. Genetic and transcriptional organization of the region encoding the beta subunit of Bacillus subtilis RNA polymerase. J. Biol. Chem. 270:20329–20336 [DOI] [PubMed] [Google Scholar]
- 5. Chopra I, O'Neill AJ, Miller K. 2003. The role of mutators in the emergence of antibiotic-resistant bacteria. Drug Resist. Updat. 6:137–145 [DOI] [PubMed] [Google Scholar]
- 6. Colussi C, et al. 2002. The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr. Biol. 12:912–918 [DOI] [PubMed] [Google Scholar]
- 7. Cooper LA, Simmons LA, Mobley HL. 2012. Involvement of mismatch repair in the reciprocal control of motility and adherence of uropathogenic Escherichia coli. Infect. Immun. 80:1969–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cox EC, Degnen GE, Scheppe ML. 1972. Mutator gene studies in Escherichia coli: the mutS gene. Genetics 72:551–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Culligan KM, Meyer-Gauen G, Lyons-Weiler J, Hays JB. 2000. Evolutionary origin, diversification and specialization of eukaryotic MutS homolog mismatch repair proteins. Nucleic Acids Res. 28:463–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davies BW, et al. 2011. DNA damage and reactive nitrogen species are barriers to Vibrio cholerae colonization of the infant mouse intestine. PLoS Pathog. 7:e1001295 doi:10.1371/journal.ppat.1001295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denamur E, et al. 2002. High frequency of mutator strains among human uropathogenic Escherichia coli isolates. J. Bacteriol. 184:605–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dreiseikelmann B, Wackernagel W. 1981. Absence in Bacillus subtilis and Staphylococcus aureus of the sequence-specific deoxyribonucleic acid methylation that is conferred in Escherichia coli K-12 by the dam and dcm enzymes. J. Bacteriol. 147:259–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dupes NM, et al. 2010. Mutations in the Bacillus subtilis beta clamp that separate its roles in DNA replication from mismatch repair. J. Bacteriol. 192:3452–3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eisen JA. 1998. A phylogenomic study of the MutS family of proteins. Nucleic Acids Res. 26:4291–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eisen JA, Hanawalt PC. 1999. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat. Res. 435:171–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fishel R, et al. 1993. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis cancer. Cell 75:1027–1038 [DOI] [PubMed] [Google Scholar]
- 17. Foster PL. 2006. Methods for determining spontaneous mutation rates. Methods Enzymol. 409:195–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Friedberg EC, et al. 2006. DNA repair and mutagenesis, 2nd ed American Society for Microbiology, Washington, DC [Google Scholar]
- 19. Gammie AE, et al. 2007. Functional characterization of pathogenic human MSH2 missense mutations in Saccharomyces cerevisiae. Genetics 177:707–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ginetti F, Perego M, Albertini AM, Galizzi A. 1996. Bacillus subtilis mutS mutL operon: identification, nucleotide sequence and mutagenesis. Microbiology 142(Pt 8):2021–2029 [DOI] [PubMed] [Google Scholar]
- 21. Guarné A, Junop MS, Yang W. 2001. Structure and function of the N-terminal 40 kDa fragment of human PMS2: a monomeric GHL ATPase. EMBO J. 20:5521–5531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guarné A, et al. 2004. Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. EMBO J. 23:4134–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hall BM, Ma CX, Liang P, Singh KK. 2009. Fluctuation AnaLysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25:1564–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamilton SR, et al. 1995. The molecular basis of Turcot's syndrome. N. Engl. J. Med. 332:839–847 [DOI] [PubMed] [Google Scholar]
- 25. Hardwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. John Wiley & Sons, Chichester, United Kingdom [Google Scholar]
- 26. Junop MS, Yang W, Funchain P, Clendenin W, Miller JH. 2003. In vitro and in vivo studies of MutS, MutL and MutH mutants: correlation of mismatch repair and DNA recombination. DNA Repair (Amst) 2:387–405 [DOI] [PubMed] [Google Scholar]
- 27. Kadyrov FA, Dzantiev L, Constantin N, Modrich P. 2006. Endonucleolytic function of MutLalpha in human mismatch repair. Cell 126:297–308 [DOI] [PubMed] [Google Scholar]
- 28. Kadyrov FA, et al. 2007. Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J. Biol. Chem. 282:37181–37190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klocko AD, et al. 2011. Mismatch repair causes the dynamic release of an essential DNA polymerase from the replication fork. Mol. Microbiol. 82:648–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kool ET. 2001. Hydrogen bonding, base stacking, and steric effects in dna replication. Annu. Rev. Biophys. Biomol. Struct. 30:1–22 [DOI] [PubMed] [Google Scholar]
- 31. Kunkel TA. 1992. DNA replication fidelity. J. Biol. Chem. 267:18251–18254 [PubMed] [Google Scholar]
- 32. Kunkel TA, Erie DA. 2005. DNA mismatch repair. Annu. Rev. Biochem. 74:681–710 [DOI] [PubMed] [Google Scholar]
- 33. Lahue RS, Au KG, Modrich P. 1989. DNA mismatch correction in a defined system. Science 245:160–164 [DOI] [PubMed] [Google Scholar]
- 34. Nicholson WL, Schuerger AC, Setlow P. 2005. The solar UV environment and bacterial spore UV resistance: considerations for Earth-to-Mars transport by natural processes and human spaceflight. Mutat. Res. 571:249–264 [DOI] [PubMed] [Google Scholar]
- 35. Nyström-Lahti M, et al. 2002. Functional analysis of MLH1 mutations linked to hereditary nonpolyposis colon cancer. Genes Chromosomes Cancer 33:160–167 [PubMed] [Google Scholar]
- 36. Oliver A, Canton R, Campo P, Baquero F, Blazquez J. 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288:1251–1254 [DOI] [PubMed] [Google Scholar]
- 37. Peltomäki P. 2005. Lynch syndrome genes. Fam. Cancer 4:227–232 [DOI] [PubMed] [Google Scholar]
- 38. Pillon MC, et al. 2010. Structure of the endonuclease domain of MutL: unlicensed to cut. Mol. Cell 39:145–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pillon MC, Miller JH, Guarne A. 2011. The endonuclease domain of MutL interacts with the beta sliding clamp. DNA Repair (Amst) 10:87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raschle M, Dufner P, Marra G, Jiricny J. 2002. Mutations within the hMLH1 and hPMS2 subunits of the human MutLalpha mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSalpha. J. Biol. Chem. 277:21810–21820 [DOI] [PubMed] [Google Scholar]
- 41. Rayssiguier C, Thaler DS, Radman M. 1989. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 342:396–401 [DOI] [PubMed] [Google Scholar]
- 42. Robertson AB, Pattishall SR, Gibbons EA, Matson SW. 2006. MutL-catalyzed ATP hydrolysis is required at a post-UvrD loading step in methyl-directed mismatch repair. J. Biol. Chem. 281:19949–19959 [DOI] [PubMed] [Google Scholar]
- 43. Russo MT, et al. 2004. The oxidized deoxynucleoside triphosphate pool is a significant contributor to genetic instability in mismatch repair-deficient cells. Mol. Cell. Biol. 24:465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schofield MJ, Hsieh P. 2003. DNA mismatch repair: molecular mechanisms and biological function. Annu. Rev. Microbiol. 57:579–608 [DOI] [PubMed] [Google Scholar]
- 45. Shcherbakova PV, Kunkel TA. 1999. Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol. Cell. Biol. 19:3177–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Simmons LA, Davies BW, Grossman AD, Walker GC. 2008. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol. Cell 29:291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Simmons LA, Kaguni JM. 2003. The DnaAcos allele of Escherichia coli: hyperactive initiation is caused by substitution of A184V and Y271H, resulting in defective ATP binding and aberrant DNA replication control. Mol. Microbiol. 47:755–765 [DOI] [PubMed] [Google Scholar]
- 48. Smith BT, Grossman AD, Walker GC. 2001. Visualization of mismatch repair in bacterial cells. Mol. Cell 8:1197–1206 [DOI] [PubMed] [Google Scholar]
- 49. Spampinato C, Modrich P. 2000. The MutL ATPase is required for mismatch repair. J. Biol. Chem. 275:9863–9869 [DOI] [PubMed] [Google Scholar]
- 50. Takahashi M, et al. 2007. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res. 67:4595–4604 [DOI] [PubMed] [Google Scholar]
- 51. Wanat JJ, Singh N, Alani E. 2007. The effect of genetic background on the function of Saccharomyces cerevisiae mlh1 alleles that correspond to HNPCC missense mutations. Hum. Mol. Genet. 16:445–452 [DOI] [PubMed] [Google Scholar]
- 52. Young LC, Hays JB, Tron VA, Andrew SE. 2003. DNA mismatch repair proteins: potential guardians against genomic instability and tumorigenesis induced by ultraviolet photoproducts. J. Investig. Dermatol. 121:435–440 [DOI] [PubMed] [Google Scholar]