

Abstract
The prototypical type II fatty acid synthesis (FAS) pathway in bacteria utilizes two distinct classes of β-ketoacyl synthase (KAS) domains to assemble long-chain fatty acids, the KASIII domain for initiation and the KASI/II domain for elongation. The central role of FAS in bacterial viability and virulence has stimulated significant effort toward developing KAS inhibitors, particularly against the KASIII domain of the β-acetoacetyl-acyl carrier protein (ACP) synthase FabH. Herein, we show that the opportunistic pathogen Pseudomonas aeruginosa does not utilize a FabH ortholog but rather a new class of divergent KAS I/II enzymes to initiate the FAS pathway. When a P. aeruginosa cosmid library was used to rescue growth in a fabH downregulated strain of Escherichia coli, a single unannotated open reading frame, PA5174, complemented fabH depletion. While deletion of all four KASIII domain-encoding genes in the same P. aeruginosa strain resulted in a wild-type growth phenotype, deletion of PA5174 alone specifically attenuated growth due to a defect in de novo FAS. Siderophore secretion and quorum-sensing signaling, particularly in the rhl and Pseudomonas quinolone signal (PQS) systems, was significantly muted in the absence of PA5174. The defect could be repaired by intergeneric complementation with E. coli fabH. Characterization of recombinant PA5174 confirmed a preference for short-chain acyl coenzyme A (acyl-CoA) substrates, supporting the identification of PA5174 as the predominant enzyme catalyzing the condensation of acetyl coenzyme A with malonyl-ACP in P. aeruginosa. The identification of the functional role for PA5174 in FAS defines the new FabY class of β-ketoacyl synthase KASI/II domain condensation enzymes.
INTRODUCTION
Fatty acid synthesis (FAS) is a primary metabolic pathway central to both eukaryotes and bacteria. A wide range of cellular processes are dependent on fatty acids, from the biosyntheses of essential cellular structural components (membrane phospholipids, lipoproteins, and lipoglycans), cofactors (lipoate and biotin), and energy storage reserves to diffusible secondary metabolites (quorum-sensing [QS] signal molecules, siderophores, and biosurfactants). Although common to bacteria, plants, and animals, fatty acids are assembled using two distinct types of biosynthetic machinery. In the mammalian FAS I type, fatty acids are built using a single multisubunit polypeptide that serves as the acyl carrier protein (ACP) tether and source of enzymatic activities for acyl chain elongation (77, 78). Plants and bacteria, with the exception of some actinomycetes (27, 71), utilize the disassociated FAS II pathway for de novo fatty acid production (17, 49, 86). In this system, multiple discrete enzymes assemble the nascent fatty acid chain on a freely diffusible phosphopantetheinylated ACP. The disassociated and modular organization of bacterial FAS II enables the synthesis of diverse fatty acid products utilized by multiple pathways, a versatility that is unmatched in FAS I systems, where the scope is more limited to long-chain saturated fatty acids (86).
While the architecture of the FAS enzymatic machinery differs considerably, the overall catalytic strategy is similar among both FAS I and FAS II model organisms (78, 86). Fatty acids are assembled two carbons at a time through iterative rounds of Claisen-type condensations between malonyl-ACP donor and nascent acyl acceptor. In the bacterial FAS II prototype pathway of Escherichia coli, the initial condensation between malonyl-ACP and acetyl coenzyme A (acetyl-CoA) to form β-acetoacetyl-ACP is catalyzed by the initiating enzyme β-ketoacyl ACP synthase III (FabH) (84). The β-keto carbonyl is next reduced to a hydroxyl group by the NADPH-dependent β-ketoacyl-ACP reductase (FabG) (2), dehydrated by β-hydroxyacyl-ACP dehydratase (FabZ/FabA) to form trans-2-enoyl-ACP (42, 54), and then reduced to a saturated acyl-ACP by NADH-dependent enoyl-ACP reductase (FabI) (4). The acyl-ACP intermediate is condensed with malonyl-ACP by the elongation β-ketoacyl ACP synthases I/II (FabB/FabF) (31), the product of which reenters the reduction cycle for another round of elongation. In E. coli, unsaturated fatty acids are produced from trans-2-decenoyl-ACP by the cis-/trans-enoyl-ACP isomerase activity of FabA (42). The cis-3-decenoyl-ACP is then specifically extended by the FabB elongase to make unsaturated fatty acids (69). Intermediates from the FAS pathway also provide the β-hydroxyl myristoyl-ACP and lauroyl-/myristoyl-ACP needed for lipopolysaccharide biosynthesis (64).
The divergence between mammalian and bacterial FAS pathways, coupled with the multifaceted role in both the viability and virulence of many pathogenic bacteria, makes fatty acid inhibition an attractive strategy for the development of new antimicrobial agents (8, 32, 37, 92). Much effort has been devoted toward understanding bacterial FAS enzymes and cognate inhibitors, particularly for the initiating condensing enzyme FabH (9, 44, 56, 91). Unlike bacterial FAS II which has discrete initiating and elongating β-ketoacyl synthases (KAS), both the priming and the subsequent elongation condensations are catalyzed by the same KAS domain in FAS I systems using first acetyl-ACP and then subsequently β-ketoacyl-ACP acceptor. Thus, there is no equivalent KAS enzymatic activity in mammalian cells that directly utilizes acetyl-CoA, and the KASIII domain of FabH is almost exclusively a bacterium-specific domain (12). A dedicated protein for fatty acid initiation allows FabH to serve as a posttranscriptional regulatory checkpoint for de novo fatty acid flux. In E. coli, FabH is subject to stringent feedback regulation by long-chain acyl-ACP so as to coordinate the rate of initiation with that of elongation (36). FabH is highly conserved, possessing an invariant triad of active site residues (28), as well as being essential across a diverse spectrum of both Gram-positive and Gram-negative bacteria that includes E. coli (47), Streptomyces coelicolor (67), and Lactococcus lactis (47). The uniqueness of the KASIII domain, conservation among FabH orthologs, and regulatory role in controlling FAS flux all suggest potential for broad-spectrum and bacterial selective inhibition.
It has recently been reported that de novo FAS can be bypassed by exogenous fatty acid uptake from serum in certain Gram-positive pathogens, including Streptococcus (6). However, other Gram-positive bacteria such as Staphylococcus are not subject to bypass of FAS by exogenous fatty acids due to differences in FAS regulatory mechanisms (58). The essentiality of the FAS pathway has not been questioned in Gram-negative bacteria, in large part due to the dependence of lipopolysaccharide biogenesis on FAS precursors (59). To this end, we initiated a program to study FAS initiation in the opportunistic nosocomial human pathogen Pseudomonas aeruginosa. P. aeruginosa is a versatile Gram-negative pathogen, associated with infections in burn patients, in the respiratory tracts of cystic fibrosis patients, and more generally among the immunocompromised (43). We hypothesized that inhibition of FabH-mediated FAS initiation in P. aeruginosa would not only prevent growth through depleting fatty acid pools for membrane phospholipid and lipopolysaccharide biogenesis but also suppress quorum sensing, since all three major QS signal molecules contain fatty acid moieties (87). We herein provide evidence that P. aeruginosa does not utilize a FabH KASIII ortholog as the primary means for FAS initiation like other bacteria studied thus far but instead uses the unannotated open reading frame (ORF) PA5174. Depletion of the PA5174 gene product elicited a pleiotropic growth and QS-defective phenotype. The levels of siderophores and numerous virulence factors decreased, which is consistent with a general role of PA5174 in FAS initiation. The identification of the functional role for PA5174, the inaugural member of a highly divergent class of KASI/II domain enzymes, defines the new FabY class of β-ketoacyl synthase condensation enzymes present in P. aeruginosa.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
E. coli strains were both K-12 descendants (EPI100-TiR and the reference strain BW25113), while all P. aeruginosa strains were derived from the reference strain PAO1 (80). E. coli and P. aeruginosa were grown in LB-Miller medium at 37°C unless otherwise noted. Antibiotic markers were selected with gentamicin (Gent) (100 μg/ml in P. aeruginosa or Chromobacterium violaceum and 10 μg/ml in E. coli), carbenicillin (Carb) (150 μg/ml in P. aeruginosa and 100 μg/ml in E. coli), chloramphenicol (Cam) (20 μg/ml in E. coli), and kanamycin (Kan) (25 μg/ml in E. coli). The bacterial strains and plasmids used in this study are listed in Table 1.
Table 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Relevant genotype or phenotypea | Source or reference |
|---|---|---|
| E. coli strains | ||
| EPI100-T1R | Phage T1 resistant [F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara leu)7697 galU galK λ− rpsL nupG tonA] | Epicentre |
| TMY19 | EPI100-T1R fabH::camR gutA::kanR araC PBAD-fabHSe; Kanr Camr; d-glucitol negative; l-arabinose conditional growth | This study |
| BW25113 | E. coli Κ-12 wild type [Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ− rph-1 Δ(rhaD-rhaB)568 hsdR514] | CGSC7636b |
| TMY32 | BW25113 fabH::camR [pET-PA5174]; Kanr Camr | This study |
| P. aeruginosa strains | ||
| NB52019 | P. aeruginosa PAO1prototroph K767 | K. Poole |
| TMT01 | NB52019 ΔPA3333 (fabH2) using pJLM34 | This study |
| TMT02 | NB52019 ΔPA0999 (pqsD) using pTMT107 | This study |
| TMT12 | NB52019 ΔPA3286 using pTMT111 | This study |
| TMT15 | NB52019 ΔPA0998 (pqsC) using pTMT114 | This study |
| TMT16 | NB52019 ΔPA0998 ΔPA0999 ΔPA3333 ΔPA3286 | This study |
| TMT38 | NB52019 PA5174::gentR fabHEc using pTMT123; Gentr | This study |
| TMT39 | NB52019 PA5174::gentR fabH′ using pTMT124; Gentr | This study |
| TMT41 | TMT39 (pZEN-PA5174); Gentr Carbr | This study |
| NB56503 | Chromobacterium violaceum ATCC 31532 | ATCC |
| TMT45 | NB56503 cviI::gentR using pTMT127; Gentr; HSL negative | This study |
| Plasmids | ||
| pWEB | ColE1 cos ori; Carbr Neor host vector for cosmid library | Epicentre |
| pTMYcos1 to pTMYcos17 | pWEB-NB52019 genome library selected in E. coli TMY19c | This study |
| pWEB-PA5174 | pWEB with the PA5174 gene; Carbr | This study |
| pKD4 | oriRλ kanR; Kanr | 18 |
| pKD4-PBAD | pKD4 with the l-arabinose-inducible araC PBAD cassette | This study |
| pKD3 | oriRλ camR; Camr | 18 |
| pUCGM | pUC19 vector with the Gentr cassette for the PCR template | This study |
| pET24b(+) | IPTG-inducible T7 promoter for protein expression; Kanr | Novagen |
| pET-PA5174 | pET24b(+) with PA5174 and C-terminal His tag; Kanr | This study |
| pEX18ApGW | Gene replacement conjugation vector; oriT+sacB+; Carbr | 15 |
| pJLM34 | pEX18ApGW-ΔPA3333 (in-frame deletion) | This study |
| pTMT107 | pEX18ApGW-ΔPA0999 (in-frame deletion) | This study |
| pTMT111 | pEX18ApGW-ΔPA3286 (in-frame deletion) | This study |
| pTMT114 | pEX18ApGW-ΔPA0998 (in-frame deletion) | This study |
| pTMT123 | pEX18ApGW-PA5174::gentR fabHEc; Gentr | This study |
| pTMT124 | pEX18ApGW-PA5174::gentR fabH′ fragment; Gentr | This study |
| pZEN | E. coli (pMB1 ori)-P. aeruginosa (pAK1900 ori) shuttle vector with lacIq and IPTG-inducible lacO PTRC; Carbr | This study |
| pZEN-PA5174 | pZEN with the PA5174 gene | This study |
| pTMT127 | pEX18ApGW- cviI::gentR | This study |
Carbr, carbenicillin resistant; Camr, chloramphenicol resistant; Kanr, kanamycin resistant; fabHEc, fabH from E. coli; fabHSe, fabH from S. enterica.
Strain CGSC7636 at the Coli Genetic Stock Center (CGSC).
Insert coordinates listed in Table S2 in the supplemental material.
Construction of P. aeruginosa deletion strains.
The conjugation vector pEX18ApGW encoding carbenicillin resistance (Carbr), oriT transfer origin, and sucrose counterselection (sacB) was utilized for all P. aeruginosa gene deletion constructs (15). Unmarked, in-frame internal gene deletion cassettes were made using ∼1-kb PCR amplicons (P1/P2 and P3/P4) of homologous flanking DNA to each KASIII domain-containing open reading frame (PA0998, PA0999, PA3286, and PA3333). Approximately 90 bp was retained at both the N and C termini to mitigate polar effects. Deletion cassettes (∼2 kb) were assembled by overlap PCR and inserted into pEX18ApGW by either standard DNA restriction/ligation (T4 DNA ligase) or by using the Gateway system cloning protocol (Invitrogen). The PA5174-E. coli fabH exchange vector pTMT123 was constructed by assembly of 5 DNA fragments (linearized pEX18ApGW [HindIII/KpnI] with PCR products using primer pairs for upstream flanking DNA [PA5174 P1/P2], Gentr cassette [Gent for PA5174/rev], FabH of E. coli [fabH Ec for/rev], and downstream flanking DNA [PA5174 P3/P4]) using the In-fusion system (Clontech). An internal DNA fragment of E. coli fabH in pTMT123 was removed by digestion (MfeI/SphI), end repaired, and self ligated to generate the PA5174 knockout vector pTMT124. The pEX18ApGW cviI::gentR vector was likewise assembled from 4 DNA fragments (linearized pEX18ApGW [HindIII/KpnI] with PCR products of primer pairs cviI P1/P2, Gent for cviI/rev, and cviI P3/P4) using the In-fusion system. Primers are listed in Table S1 in the supplemental material.
The vectors were mobilized in trans using either E. coli SM10 (76) or E. coli Stellar cells (Clontech) transformed with the helper plasmid pRK2013 (24) as the conjugation-proficient donor strains. The E. coli donor and P. aeruginosa recipient (106 CFU in 10 μl for each strain) were cospotted on LB agar and incubated at 37°C for 8 h to overnight. The mating mixtures were then scraped off the plate, resuspended, and plated on Pseudomonas isolation agar (PIA) to counterselect E. coli and with antibiotic to select for integrated vector. Merodiploid colonies were confirmed by PCR, outgrown for 4 h in LB at 37°C, and then spread on LB agar containing 7% sucrose (LB–7% sucrose agar) (without NaCl) to counterselect colonies harboring unresolved plasmid. Mutant alleles were checked in P. aeruginosa by colony PCR using primers annealing outside the targeted region as well as confirming anticipated antibiotic sensitivity/resistance. The quadruple knockout strain TMT16 was constructed stepwise by 4 successive rounds of gene deletion. The PA5174::gentR deletion strain TMT39 was back complemented by electroporation (14) of the shuttle vector pZEN-PA5174, which was made by ligation of the PA5174 gene (amplified with primers pZEN for NdeI/PA5174 pZEN rev XbaI) into a similarly restricted pZEN vector. The C. violaceum cviI::gentR (TMT45) reporter strain was directly isolated by streaking the pTMT127 mating mixture to single colonies at 28°C on sucrose LB agar containing Gent and Carb. The cviI deletion was confirmed in unpigmented candidate colonies (due to loss of homoserine lactone signaling [51]) by PCR analysis.
Construction and cosmid complementation of the fabH inducible E. coli strain TMY19.
The l-arabinose (l-Ara)-inducible FabH strain TMY19 was constructed in E. coli EPI100 in two steps (Fig. 1). First, an integration cassette targeted to the d-glucitol transporter operon (gutAEB) was constructed by cloning the Salmonella enterica subsp. enterica (ATCC 19430) fabHSe (the Se superscript indicates that the fabH gene was from S. enterica) ortholog (amplified with primers fabH Se for EcoRI/fabH Se rev KpnI) in front of the PBAD promoter in pKD4-PBAD. The resulting plasmid (containing araC-PBADfabHSe) was used as the template for PCR amplification of the integration cassette (primers gutAEB pKD4 P1/gutAEB pKD4 P2). The 40 bp of homology to the d-glucitol operon on the termini of the integration cassette directed site-specific recombination using the Red recombinase system (18). Insertion of fabHSe at the gut locus was confirmed by PCR analysis as well as by loss of d-glucitol catabolism using MacConkey-sorbitol indicator plates (52). The resulting strain, merodiploid in fabH orthologs, was then subjected to a second round of Red recombinase-mediated gene disruption using a fabHEc::camR (the Ec superscript indicates that the fabH gene was from E. coli) cassette (amplified using primers fabH Ec KO P1/fabH Ec KO P2 with pKD3 as the template). The l-Ara conditional strain TMY19 was maintained on LB agar plus l-arabinose (0.2%) to enable initiation of fatty acid biosynthesis by heterologous expression of the integrated PBAD-fabHSe cassette.
Fig 1.

Cosmid complementation strategy for identification of FabH-type activity in P. aeruginosa PAO1. The E. coli strain TMY19 has a chloramphenicol cassette (camR) inserted within fabH (b1091) and needs l-Ara supplementation to induce expression of the PBAD-regulated S. enterica fabH ortholog (inserted within the d-glucitol phosphotransferase operon) in order to sustain wild-type growth rates. The operon is transcriptionally silent under standard culture conditions in the absence of d-glucitol (52). A sheared library of wild-type P. aeruginosa PAO1 genomic DNA was ligated into the pWEB cosmid vector, packaged, and transduced by phage into E. coli TMY19. Transductants were directly selected on unsupplemented LB agar to identify cosmids (pTMYcos1 to pTMYcos17) that complement depletion of FabH.
The pWEB cosmid cloning kit (Epicentre) was used to introduce a library of pWEB cosmids harboring P. aeruginosa PAO1 genomic DNA into E. coli TMY19. Cosmids were directly introduced into strain TMY19 that had been pregrown in medium containing 0.2% l-Ara by lambda phage transduction as instructed by the manufacturer. Transductants were isolated on unsupplemented LB agar (100 μg/ml Carb) for pWEB selection. Aliquots were plated in parallel on LB agar with l-Ara as the positive control, along with aliquots of bacteria that were not infected to monitor the spontaneous induction background. The plates were incubated at 37°C for 32 h before comparison. A total of 17 larger colonies that stood out from the background growth due to leakiness of the PBAD promoter were selected for further analysis. Cosmids (pTMYcos1 to pTMYcos17) were isolated, and junction sites were sequenced using pWebup seq/pWebdown seq (seq stands for sequencing) primers (cosmid summary listed in Table S2 in the supplemental material). Purified cosmids were then retransformed by electroporation into pTMY19 to confirm phenotypic rescue. The unidentified open reading frame PA5174, common to all E. coli TMY19 growth-rescuing cosmids and immediately proximal to the vector junction site, was amplified and cloned into pWEB using the In-fusion system (Clontech). The plasmid pWEB-PA5174 was then electroporated into strain TMY19 to evaluate complementation.
FabH complementation in E. coli BW25113.
The PA5174 open reading frame was cloned into the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible expression vector pET24b(+) (Novagen) using the PCR product of primers pET24-PA5174 for/rev. The expression plasmid pET-PA5174 was transformed into E. coli BW25113. The Red recombinase system was used to delete the fabH gene using the fabHEc::camR cassette (see above) in order to generate strain TMY32.
Growth analysis.
For E. coli and P. aeruginosa strains, starter cultures were made by scraping cells off LB agar plates that had been supplemented with appropriate selection antibiotics and/or inducers. Cultures were typically resuspended to ∼2 × 105 CFU/ml in LB and distributed into clear 96-well flat-bottom untreated microplates (Costar 3370). The plates were incubated at 37°C, and the optical density at 600 nm (OD600) was recorded every 10 min on a Spectramax plate reader with intermittent shaking.
Fatty acid composition analysis.
Bacteria were streaked on LB agar plates and incubated overnight at 37°C. Biomass was scraped from the surface and suspended in phosphate-buffered saline (PBS). The cell pellet was thoroughly washed three times with PBS. Lipids were saponified, methylated, extracted, and washed according to the Sherlock microbial identification system (Microbial ID, Inc., Newark, DE). Fatty acid methyl ester (FAME) composition was determined by gas chromatography with flame ionization detection. Structure assignments were made by comparison of retention times to those of authentic FA standards.
Macromolecular labeling of fatty acids.
Colonies were scraped from LB agar plates that had been grown overnight and resuspended to an OD600 of 0.1 (∼107 CFU/ml) in M9 minimal salts medium supplemented with glycerol (0.4% [vol/vol]), uridine (80 μg/ml), and tryptic soy broth (7% [vol/vol]). Cultures were incubated with shaking at 37°C for 3 h to return the cells back to the exponential growth phase before being back diluted to an OD600 of 0.1 with prewarmed supplemented minimal medium. Aliquots of 50 μl were then added to 50 μl of prewarmed medium in 96-well round-bottom microplates (catalog no. 351177; BD Falcon) that contained either [3H]acetate (3.47 Ci/mmol) (catalog no. NET003H; Perkin Elmer) for fatty acid or [3H]uridine (25.5 Ci/mmol) (catalog no. NET174; Perkin Elmer) for RNA precursor radiolabeling (final concentration of 10 μCi/ml for each). After being incubated for 30 min at 37°C, wells were quenched with 100 μl of ice-cold 20% trichloroacetic acid. The plates were held at 4°C overnight to precipitate macromolecules, and the precipitates were collected by filtration (Perkin Elmer UniFilter-96 GF/C). The filters were washed extensively with water, dried, and sealed, and then 40 μl of scintillant (Perkin Elmer Betaplate Scint) was added prior to counting (Perkin Elmer MicroBeta Trilux).
Extraction and assay of quorum-sensing signal molecules.
Cultures were grown in LB medium for 24 h at 37°C with shaking (250 rpm) in baffled flasks. Under these conditions, all cultures reached stationary phase at least 8 h before harvesting and had similar final cell densities. Bacteria were pelleted by centrifugation (3,000 × g, 5 min, room temperature [RT]), and the culture supernatants were decanted and passed through a sterile filter (0.45 μm). Quorum-sensing signal molecules were twice extracted from 10 ml of supernatant for each sample using either an equal volume of ethyl acetate for homoserine lactones (HSL) (74) or an equal volume of acidified ethyl acetate for 2-heptyl-3-hydroxy-4(1H)-quinolone (PQS) (25). The organic extracts were concentrated to dryness using a nitrogen bubbler, and the residue was resuspended in 100 μl of ethyl acetate (HSL) or methanol (PQS). The extracts were analyzed by thin-layer chromatography (TLC). For the rhl-dependent acylated HSL, 15 μl of each sample was spotted on Partisil LKC18 (Whatman) reverse-phase TLC plates, while 3 μl of each sample was spotted on reverse-phase octadecyl Si-C18 (Baker) TLC plates for the las-dependent 3-oxo HSL. The plates were developed with 60:40 (vol/vol) methanol (MeOH)-H2O as described previously (74). The 3-oxo acyl HSL-inducible β-galactosidase bioreporter strain Agrobacterium tumefaciens NL4(pZLR4) ATCC BAA-240 was used in an agar overlay to visualize 3-oxo HSL spots (10), while the acyl HSL spots were detected with the reporter C. violaceum strain TMT45 as has been described for similar C. violaceum reporter strains (51). PQS content was assessed by normal-phase TLC on activated silica 60 F254 plates (Merck) that had been pretreated with a 5% KH2PO4 soak before development with a 95:5 (vol/vol) dichloromethane-methanol mobile phase. The plates were visualized by transillumination with UV light (25). Authentic acyl- and 3-oxo-acyl-HSL (Sigma, Cayman Chemical, or Santa Cruz Biotechnology) and PQS (Sigma) standards were used for compound identification by comparing migration distances (Rf values).
Rhamnolipid production on SW agar.
Rhamnolipid secretion was assessed using SW agar indicator plates as described previously (61). Briefly, cultures were pregrown in SW minimal medium containing glycerol {20 g/liter glycerol, 0.7 g/liter KH2PO4, 0.9 g/liter Na2HPO4, 2 g/liter NaNO3, 0.4 g/liter MgSO4 · H2O, 0.1 g/liter CaCl2 · 2H2O, and 2 ml/liter of a trace element solution [2 g/liter FeSO4 · 7H2O, 1.5/liter g MnSO4 · H2O, and 0.6 g/liter (NH4)6Mo7O24 · 4H2O]}. Wells were cut into the surface of SW agar plates (0.2 g cetyltrimethylammonium bromide [Sigma], 0.005 g methylene blue [MP Biomedical], and 12 g agar to 1 liter of the above medium) and filled with 10 μl of stationary-phase culture. The plates were incubated at 37°C for 48 h and then placed at 4°C overnight to intensify the development of the blue halo complexation zone before photographing.
Pseudomonas exoproduct assays.
Rhamnolipids were extracted from supernatants produced by 24-h cultures grown in LB at 37°C with shaking (250 rpm). Aliquots (1 ml) of supernatant were acidified by adding 100 μl of 20% citric acid, and the aqueous phase was then twice extracted with an equal volume of chloroform-methanol (2:1 [vol/vol]). Pooled organic fractions were concentrated using a nitrogen bubbler, and the residue was resuspended in 1 ml of methanol. Rhamnose content was measured using the anthrone colorimetric assay (93) and quantitated using l-rhamnose (Sigma) as the standard. The elastase proteolytic activity in culture supernatants, grown as described for the rhamnose assay, was determined using the elastin Congo red assay (20). After a 3-h digestion period at 37°C (100 mM Tris, 1 mM CaCl2 [pH 7.5] with 20 mg of elastin Congo red substrate [Sigma] plus 100 μl of spent supernatant), suspensions were clarified by centrifugation (16,000 × g, 5 min, RT), and the absorbance at 495 nm was measured to determine the amount of liberated dye. To measure pyocyanin production, Pseudomonas cultures were grown in glycerol alanine minimal medium [1% (vol/vol) glycerol, 6 g/liter l-alanine, 2 g/liter MgSO4, 0.1 g K2HPO4, 18 mg/liter FeSO4, 1 mg/liter biotin] at 37°C with shaking for 42 h (70). Pyocyanin-containing aliquots (5 ml) of clarified supernatants were extracted with chloroform (3 ml), separated, and then back extracted into the aqueous phase by the addition of 1 ml of 0.2 N HCl (23). The absorbance of the pink aqueous phase was measured at 520 nm and quantified using a pyocyanin (Cayman Chemical) standard curve.
Pseudomonas swarming assay.
Swarming assays were performed on modified M8 minimal medium agar supplemented with MgSO4 (1 mM), glucose (0.2%), Casamino Acids (0.5%), and solidified with 0.4% agar (7). The plates were dried for 2 h at room temperature, inoculated, and incubated overnight at 37°C before imaging.
Siderophore secretion assay.
The CAS-LB plates were prepared by a modified version of the published protocol (72). A 10× blue dye solution consisting of 1 mM chrome azurol S (CAS) (Acros), 2 mM cetyltrimethylammonium bromide, and 500 μM FeCl3 · 6H2O was sterilized by autoclaving. A 10-ml aliquot was added to 100 ml of molten LB-Miller agar (1.5%) immediately before being poured. Plates were dried for 1 h at RT prior to inoculation.
Recombinant PA5174 protein expression and purification.
Recombinant PA5174 protein was obtained by transforming E. coli BL21(DE3)(pLysS) (Novagen) with the His tag expression vector pET-PA5174. Colonies were inoculated into 1 liter of LB medium and grown at 37°C with shaking until mid-exponential growth (OD600 of 0.6). Protein production was induced with 1 mM IPTG, and expression was allowed to continue for 3 h. The biomass was then harvested by centrifugation (5,000 × g, 10 min, RT) and stored at −80°C. The pellet was subjected to 2 rounds of freeze-thaw cycles and then incubated in lysis solution (1× Bugbuster [Novagen], 5 kU/ml recombinant lysozyme, 25 U/ml benzonuclease) with gentle shaking at room temperature for 20 min. The solution was clarified by centrifugation (15,000 × g, 30 min, 4°C), and the supernatant was gently shaken with nickel-nitrilotriacetic acid (Ni-NTA) His bind resin (1 h on ice). The slurry was loaded into an empty column, washed with 50 ml of binding buffer (50 mM Tris, 500 mM NaCl, 10 mM imidazole [pH 7.5]), 20 ml of wash buffer (binding buffer with 50 mM imidazole total), and eluted (binding buffer plus 200 mM imidazole total) in 1-ml fractions. Fractions containing protein of the expected size (∼70 kDa) by SDS-polyacrylamide gel electrophoresis (PAGE) were pooled and concentrated (Amicon Ultra, 30-kDa molecular size cutoff; Millipore). The sample was further purified by size exclusion chromatography using a Superdex200 16/60 column with an isocratic elution buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM d/l-dithiothreitol, and 10% glycerol). Protein fractions with PA5174 were aliquoted and flash frozen at −80°C.
Recombinant PA5174 and FabH enzyme assays.
A continuous, coupled spectrophotometric assay was used to measure the β-acetoacetyl synthase activity of P. aeruginosa PA5174 and E. coli FabH. Acetyl-CoA and malonyl-ACP were added as substrates, and activity was monitored by NADPH consumption using the coupled E. coli β-ketoacyl-ACP reductase (FabG) as has been described previously (56). A final 100-μl reaction mixture consisting of 0.1 M K2HPO4/KH2PO4 buffer (pH 7.0), 50 μM holo-ACP, and 5 mM dithiothreitol (DTT) was incubated in a 96-well clear Costar plate at 30°C for 5 min to obtain fully reduced holo-ACP. The E. coli FabD malonyl-CoA:ACP transacylase (80 ng) was added along with 100 μM malonyl-CoA and incubated for another 15 min to generate malonyl-ACP in situ. Next, 100 μM NADPH, 2 μg of E. coli FabG, and 100 μM acetyl-CoA was mixed in before adding PA5174 or E. coli FabH to initiate the reaction. The UV absorption at 340 nm was monitored on a PHERAstar microplate reader.
Conformation-sensitive urea-PAGE was used to separate and analyze acyl-ACP products (68). Radiolabeled malonyl-ACP was produced in situ using the same procedure as described above, except a total of 40 μM malonyl-CoA (10 μM [2-14C]malonyl-CoA) was added to the 20-μl reaction mixture. For each saturated straight-chain acyl-CoA acceptor tested, 200 μM acyl-CoA (C2 to C16 in length; Sigma), along with 0.1 μg of PA5174 (final concentration of 70 nM) was added, and the reaction mixtures were incubated at room temperature for 1 h. The products were separated by 0.5 M urea–16% PAGE and visualized by film autoradiography. Where indicated, β-ketoacyl reaction products were further reduced to acyl-ACP using 1 μg each of E. coli FabG/FabI/FabA plus 200 μM each of the NADH and NADPH cofactors.
Bioinformatics analysis.
The Basic Local Alignment Search Tool (BLAST) (http://blast.ncbi.nlm.nih.gov/) search algorithm was used to identify E. coli FabH (b1091) KASIII domain orthologs in the sequenced P. aeruginosa PAO1 genome (80). Likewise, the open reading frames with stand-alone KASI/II domains in other whole-genome sequenced Pseudomonas species were identified by BLAST analysis using the KASI/II domain proteins of the reference strain P. aeruginosa PAO1 (PA5174/PA1373/PA1609/PA2965). All KASI/II domain-containing protein sequences (48 total sequences from 4 P. aeruginosa strains, 7 other Pseudomonas species, Azotobacter vinelandii DJ, and E. coli K-12 MG1655) were aligned using Clustal Omega v1.0.3 with mBed-like clustering guide tree and mBed-like iteration with default settings for iteration number (75). The phylogenetic tree was inferred using the neighbor-joining option with partial deletion for gap treatment with the MEGA5 program (82). The bootstrap consensus tree was inferred from 1,000 replicates with distances computed using the Poisson correction method.
RESULTS
P. aeruginosa does not use a KASIII domain for fatty acid initiation.
Multiple P. aeruginosa FAS genes (fabAB [38], fabDG-acpfabF [46], and fabI and fabZ [40]) have been identified based on sequence similarity to orthologs in E. coli, suggesting a conserved FAS pathway. Unlike the situation in E. coli, however, the P. aeruginosa fabH gene is noticeably absent from any FAS-associated cluster and must reside in a unique chromosomal location. Analysis of the genome of P. aeruginosa PAO1 for ORFs encoding proteins similar to the experimentally verified E. coli FabH (b1091) by BLAST search yielded four candidate KASIII-containing orthologs (Fig. 2). All four proteins (PA3333 [FabH2], PA3286, PA0998 [PqsC], and PA0999 [PqsD]) possess 25 to 30% amino acid identity to E. coli FabH and contain at least two of the three signature Cys-His-Asn active site residues that form the catalytic triad (19). While PA0998 and PA0999 belong to the Pseudomonas quinolone signal (PQS) operon (22), a dual function or side activity capable of masking the ΔFabH phenotype could not be ruled out, so all candidates were subjected to deletion analysis. In-frame deletions were constructed singly or sequentially in the same genetic background to generate the KASIII-less P. aeruginosa strain TMT16. The fatty acid compositions of lipid extracts from all of the strains were similar regardless of whether the KASIII proteins were deleted singly or in combination (see Table S3 in the supplemental material) and were similar to published values (11). The growth curves of the entire KASIII domain deletion strain set were nearly superimposable (Fig. 2C). Collectively, the data indicate that P. aeruginosa does not primarily utilize a KASIII protein for fatty acid initiation but rather has a unique FAS initiation protein.
Fig 2.

Fatty acid β-ketoacyl acyl carrier protein (ACP) synthase III (KASIII) domain-containing proteins in P. aeruginosa PAO1. (A) The E. coli K-12 MG1655 FabH sequence (b1091) was used to query the P. aeruginosa PAO1 genome for similar protein sequences using the BLAST search tool. Four candidate P. aeruginosa FabH-encoding open reading frames at 3 distinct genomic loci all containing the KASIII conserved domain were identified: PA3333 (white) (FabH2, E-value = 8e−66), PA3286 (white) (E-value = 3e−25), PA0998 (white) (PqsC, E-value = 3e−17), and PA0999 (gray) (PqsD, E-value = 2e−68). (B) Alignment of putative active site regions with the Cys112-His244-Asn274 catalytic triad of E. coli FabH (FabH Ec) (indicated by asterisks). Identical amino acid residues are shaded with a black background, and partially conserved substitutions are shaded in gray. (C) Growth curves of P. aeruginosa strains with all KASIII candidate genes (wild type [▲]) or with KASIII candidate genes deleted singly (PA0999 [■], PA3286 [□], PA3333 [○], PA0998 [△]) or in combination (TMT16 [●]) in LB broth at 37°C.
PA5714 complements FabH depletion in E. coli.
In order to identify P. aeruginosa candidate proteins with FabH-type enzymatic activity, we constructed a regulated FabH strain in E. coli (Fig. 1). We first inserted an araC-PBAD-fabHSe cassette within the d-glucitol (gutAEB) catabolism operon, utilizing the S. enterica FabH ortholog to minimize integration at the native fabH locus (47) and loss of d-glucitol metabolism as a convenient phenotypic marker to screen recombinant clones for site-specific integration. Using l-arabinose (l-Ara)-supplemented media, the E. coli fabH gene was next disrupted to create the TMY19 strain, which is dependent on l-Ara to realize growth rates comparable to that of the wild type. A random cosmid library of P. aeruginosa PAO1 genomic DNA was introduced into strain TMY19 and then plated on unsupplemented media in order to find cosmids capable of complementing the fabH depleted growth defect. Cosmids from 17 large colonies with enhanced growth were sequenced and back transformed into TMY19 to confirm the rescue phenotype (results summarized in Table S2 in the supplemental material). Of the 17 cosmids, 14 at least partially rescued growth in the absence of L-Ara supplementation. All of the complementing cosmids harbored P. aeruginosa genomic inserts that started immediately upstream of the unannotated ORF PA5174 and extended 20 to 40 kb downstream (Fig. 3A). PA5174 is predicted to encode a 634-amino-acid protein belonging to the KASI/II domain family. The strong bias for selection of inserts with PA5174 juxtaposed to the cosmid backbone immediately suggested that expression of PA5174 by plasmid-based promoters was necessary for complementation. To confirm that PA5174 alone was responsible for complementation, we cloned the PA5174 gene without its native promoter region into the pWEB cosmid so as to be under the control of the T7 phage promoter. The growth characteristics of the resulting strain TMY19(pWEB-PA5174) were similar to those seen after L-Ara supplementation of the parent strain (Fig. 3B); PA5174 is thus by itself sufficient to complement FabH and restore initiation of FAS.
Fig 3.

Complementation of FabH depletion in E. coli K-12 by a P. aeruginosa cosmid library. (A) The plasmid-genomic DNA insert junction sites of cosmids that rescued growth of the FabH-depleted E. coli strain TMY19. All 14 sites were located in the immediate 5′ intergenic region of the unknown open reading frame PA5174 and contained 24 to 42 kb of downstream DNA. All inserts except for pTMYcos13 (asterisk) placed PA5174 proximal to the T7 promoter on the pWEB vector backbone. The number of base pairs intervening between the respective cosmid-junction site and the start codon of PA5174 is indicated in parentheses. Cosmids selected for further growth analysis are underlined. (B) Growth curves of E. coli strain TMY19 grown in various conditions at 37°C. E. coli TMY19 was grown in LB only (▲) or in LB plus 0.2% l-arabinose inducer (△). E. coli TMY19 was grown in LB plus 0.2% l-arabinose inducer with cosmid pTMY2 (●), cosmid pTMY10 (■), cosmid pTMY17 (○) or plasmid pWEB-PA5174 (□). (C) Growth curves of the IPTG-inducible PA5174 E. coli strain TMY32 with fabH deleted in LB (in the absence of IPTG [■] or in the presence of 1 mM IPTG [△]) were measured at 37°C in LB and compared to that of the parent wild-type strain BW25113 (●).
Since FabH expression is not fully repressed in uninduced E. coli TMY19, it was necessary to verify that PA5174 could completely replace fabH in E. coli and alone support the cellular demand for de novo FAS. The PA5174 gene was cloned into an IPTG-inducible plasmid (pET-PA5174) and transformed into E. coli BW25113. Deletion of the fabH gene was attempted using the Red recombinase system (18) with a fabH::camR integration cassette. No fabH disrupted mutants were obtained in wild-type BW25113, consistent with the essential function of FabH in E. coli (47), but viable colonies were obtained in the presence of plasmid-encoded PA5174. Growth analysis confirmed complete fabH-PA5174 cross complementation (Fig. 3C), nominating PA5174 as the P. aeruginosa FabH functional ortholog.
PA5174 deletion in P. aeruginosa attenuates growth by decreasing de novo fatty acid synthesis.
The genomic context indicates that PA5174 is likely a monocistronic gene (Fig. 3A), with minimal anticipated polarity concerns upon deletion. Since no loss-of-function transposon disruption mutants have been reported (no insertions in P. aeruginosa PAO1 [88] and only an extreme C-terminal transposon mutant in P. aeruginosa PA14 [48]), PA5174 likely has an essential cellular role. To test this, allelic exchange vectors that either replaced PA5174 with a gentamicin resistance-E. coli fabH (pTMT123) cassette or with a gentamicin resistance-E. coli fabH nonfunctional fragment (pTMT124) were constructed. Cointegrants were passively resolved, and the sacB+ integration vector was counterselected with sucrose. Allelic exchange with fabH gave rise to hyperpigmented blue-green gentamicin-resistant mutants (TMT38 [PA5174::fabH]), while the nonfunctional fabH fragment (TMT39 [ΔPA5174]) resulted in small, translucent colonies that were gentamicin resistant and took 2 days to appear. Analysis of the growth characteristics in liquid LB confirmed a markedly attenuated growth phenotype for the ΔPA5174 strain (doubling time increased from 19 to 62 min in the absence of PA5174). Complementation with either E. coli fabH (TMT38) or PA5174 plasmid (TMT41) retained growth rates comparable to that of the wild type (Fig. 4A).
Fig 4.

Growth characteristics and complementation of the P. aeruginosa ΔPA5174 strain. (A) Growth of the P. aeruginosa PAO1 parent strain (●), ΔPA5174 strain (■), PA5174 back-complemented with plasmid (TMT41 [△]), and the E. coli FabH-complemented strain (TMT38 [□]) in LB at 37°C. Basal expression of PA5174 on the plasmid is sufficient for full complementation in the absence of IPTG induction. (B) The rate of radiolabeled precursor incorporation by FAS ([3H]acetate [black bars]) and RNA ([3H]uridine [white bars]) biosynthetic pathways was measured after a 30-min incubation period in supplemented M9 minimal medium. The percent incorporated label was normalized to the value of the wild-type (Wt) P. aeruginosa PAO1 control. Error bars represent the standard deviations from the means calculated from three independent experiments.
Since a defect in FAS upon PA5174 deletion was suspected, the FAS de novo flux was next measured by macromolecular labeling using radiolabeled precursors to monitor FAS ([3H]acetate substrate) and RNA ([3H]uridine substrate) pathway activity. Both RNA and FAS activity were lower in the ΔPA5174 mutant (Fig. 4B), in part reflecting slower overall metabolic activity. However, the larger decrease in the rate of label incorporation for FAS in comparison to RNA indicates particular attenuation specific to FAS. Both FAS and RNA pathway activity returned to near wild-type levels in the strain complemented with PA5174 (TMT41). Collectively, the data support a central role for the PA5174 gene product in FAS initiation in P. aeruginosa, analogous to the function of FabH in E. coli.
The rhl QS system is muted by deletion of PA5714.
The rhl QS signal molecules N-butanoyl-l-homoserine lactone (C4-HSL) and N-hexanoyl-l-homoserine lactone (C6-HSL) are synthesized from S-adenosylmethionine and β-butanoyl-ACP/β-hexanoyl-ACP, respectively (65). Since both molecules are formed from β-acetoacetyl-ACP, the putative product of PA5174, muted signaling in the cognate C4-HSL/C6-HSL-responsive rhl QS regulatory circuit would be expected in the ΔPA5174 strain if PA5174 encodes the major FAS initiation enzyme. Acyl-HSL was extracted from culture supernatants and separated by TLC. The acyl-HSL spots were detected using an agar overlay of a C. violaceum reporter strain which produces a purple chromophore in response to acyl-HSL (51). The amount of acyl-HSL in the ΔPA5174 strain was nearly undetectable in comparison to that in the wild type (Fig. 5A). Production of acyl-HSL actually increased beyond wild-type levels in the PA5174 complemented strain (TMT41), confirming a tight association between PA5174 expression and acyl-HSL synthesis.
Fig 5.

Characterization of rhl quorum sensing and dependent exoproducts produced in P. aeruginosa ΔPA5174 strain. (A) The acylated HSL fraction was extracted from the supernatants of stationary-phase cultures grown in LB broth at 37°C for 24 h and separated by reverse-phase TLC. The levels of acylated HSL were estimated using a soft agar overlay of the C. violaceum reporter strain TMT45 and compared to standards (C4-HSL [20 nmol] and C6-HSL [0.2 nmol]). The cvi QS system of C. violaceum detects C6-HSL with the highest sensitivity (51), limiting quantitative comparisons to within the same HSL acyl chain length. (B) Cultures of P. aeruginosa were spotted into wells cut into the agar of SW plates. Blue halos indicate zones of ion-pairing complexation between the methylene blue dye, cetyltrimethylammonium bromide, and secreted rhamnolipids. (C) Rhamnose levels in stationary-phase culture supernatants were measured using the colorimetric anthrone assay. (D) Swarming motility was evaluated on M8 agar plates supplemented with Casamino Acids and glucose after overnight incubation at 37°C.
We next looked at levels of rhamnolipids among the PA5174 strain panel. Rhamnolipids are secreted surfactant glycolipids formed by acylation of l-rhamnose using the FAS pathway intermediate β-hydroxydecanoyl-ACP (93). Since rhamnolipids depend on the rhl QS system for expression (66) and are themselves assembled from FAS pathway intermediates, the levels of rhamnolipids should reflect defects in FAS initiation. To detect rhamnolipids, cultures were spotted into wells cut into SW agar indicator plates (61). Blue halos in the agar indicate zones of ion-pairing complexation between the methylene blue dye, cetyltrimethylammonium bromide, and secreted rhamnolipids. While the wild-type strain and the PA5174-complemented strain produced prominent halos, no halo was observed with the ΔPA5174 mutant strain (Fig. 5B). Rhamnolipid quantification by colorimetric detection of rhamnose showed a 5-fold decrease in the ΔPA5174 mutant while there was a 3-fold increase in the complemented strain relative to the wild type (Fig. 5C). Swarming motility on semisolid agar was also evaluated in order to estimate rhamnolipid secretion, as the surface wetting properties of the amphiphilic glycolipid are thought to be critical for breaking the surface tension that otherwise hinders flagellar propulsion (45). Strains were spotted in the middle of swarming plates, and the degree of spreading was compared after overnight incubation (Fig. 5D). Consistent with the rhamnolipid assays, the ΔPA5174 strain was nonmotile, while both the wild-type and complemented strains formed short, flowering tendrils characteristic of some P. aeruginosa PAO1 isolates (83).
Both las and PQS QS-dependent exoproducts are downregulated by deletion of PA5714.
Together with the rhl system, the las and Pseudomonas quinolone signal molecules form the QS network of P. aeruginosa (33, 41, 87). The las and PQS signal molecules both utilize β-ketoacyl medium-chain fatty acid metabolites to acylate HSL to make N-(3-oxododecanoyl)-l-HSL (3-oxo-C12-HSL) (40) and to synthesize 2-heptyl-3-hydroxy-4-quinolone (PQS) (62), respectively. The 3-oxo-acyl-HSL fraction was extracted from culture supernatants, separated by TLC, and visualized using the inducible β-galactosidase reporter strain Agrobacterium tumefaciens NL4(pZLR4) (10). While spot intensities were nearly equivalent between the wild type and the ΔPA5174 strain, 3-oxo-acyl-HSL levels were increased in the plasmid-complemented strain (Fig. 6A). The ΔPA5174 mutant apparently can synthesize 3-oxo-acyl-HSL to wild-type levels in LB liquid culture after 24 h of incubation despite the FAS defect. The secreted virulence factor elastase (lasB) is in large part induced by the las QS system (30). Unlike the levels of the 3-oxo-acyl-HSL inducer, elastase activity was almost undetectable in the ΔPA5174 strain, while plasmid complementation with PA5174 restored elastase to 2-fold above the wild type (Fig. 6B).
Fig 6.

Characterization of las and PQS-dependent quorum sensing in the P. aeruginosa ΔPA5174 strain. (A) The 3-oxo-HSL produced by the las quorum-sensing system was extracted from the supernatants of stationary-phase cultures grown in LB broth at 37°C for 24 h and separated by reverse-phase TLC. The levels of 3-oxo-HSL were estimated using a soft agar overlay of the A. tumefaciens NTL4(pZLR4) β-galactosidase reporter strain and compared to standards ([3-oxo-C6-HSL [1 pmol], 3-oxo-C8-HSL [0.2 pmol], 3-oxo-C10-HSL [20 pmol], and 3-oxo-C12-HSL [100 pmol]). The tra QS system of A. tumefaciens detects 3-oxo-C8-HSL with the highest sensitivity, limiting quantitative comparisons to within the same HSL acyl chain length (74). (B) The elastase activity (LasB) in culture supernatants was measured using a Congo red-elastin dye release assay. (C) The amount of PQS was compared by direct UV illumination of culture supernatant extracts separated by normal-phase TLC on silica 60 F254 plates. Synthetic PQS standard (20 nmol) and P. aeruginosa TMT15 (ΔpqsC) were used as the positive and negative controls, respectively. (D) Pyocyanin was extracted from supernatants obtained from cultures grown in glycerol alanine minimal media for 42 h at 37°C, acidified, and quantified by absorbance at 520 nm. Values (in μg/ml of supernatant) were determined using a standard curve generated with purified pyocyanin.
The uncoupling of 3-oxo-HSL inducer concentration and elastase activity in the ΔPA5174 strain suggested that another participant involved in lasB induction was absent. PQS-mediated signaling, which itself is subject to hierarchal control by the las QS system (87), also contributes to lasB induction (60). We thus analyzed the levels of PQS in supernatant extracts (Fig. 6C). The PQS level was reduced to a trace in the supernatant extract when PA5174 was deleted, similar in quantity to the level in the PQS negative-control ΔpqsC strain. In contrast, PQS production was markedly enhanced in the complemented strain. PQS signaling is absolutely necessary for production of the phenazine secondary metabolite pyocyanin (29). Assay of pyocyanin levels in culture extracts mirrored the respective PQS levels in each of the strains, ranging from a trace in ΔPA5174 to a 5-fold increase in the plasmid-complemented strain (Fig. 6D). The pyocyanin levels measured from cultures grown in liquid broth are consistent with the characteristic blue-green hyper- and hypopigmentation initially observed on agar.
PA5174 deletion in P. aeruginosa reduces production of siderophores.
The dominant siderophore produced by fluorescent pseudomonads, including P. aeruginosa, is the iron chelator pyoverdine (85). Since pyoverdine is coregulated by QS systems (79) and is assembled on a myristate fatty acid residue (21, 34), we investigated whether siderophore secretion was attenuated in the ΔPA5174 strain by streaking on LB-chrome azurol S (CAS) indicator plates (72). Zones that change from blue to yellow-orange indicate where the siderophores have sequestered Fe3+ away from the blue CAS-Fe3+ complex. While the wild-type and plasmid-complemented P. aeruginosa strains generated prominent zones of clearing, the ΔPA5174 strain did not produce a zone, indicating a lack of relative siderophore production (Fig. 7). In addition to pyoverdine, a second siderophore (pyochelin) and PQS can also chelate Fe3+ (5). The Fe3+ sequestration zone produced by the PQS− control strain TMT02 (ΔpqsD) was similar to that of the wild type (Fig. 7). Thus, the absence of dedicated siderophores in the ΔPA5174 strain is responsible for the difference observed in the Fe3+ sequestration zone.
Fig 7.
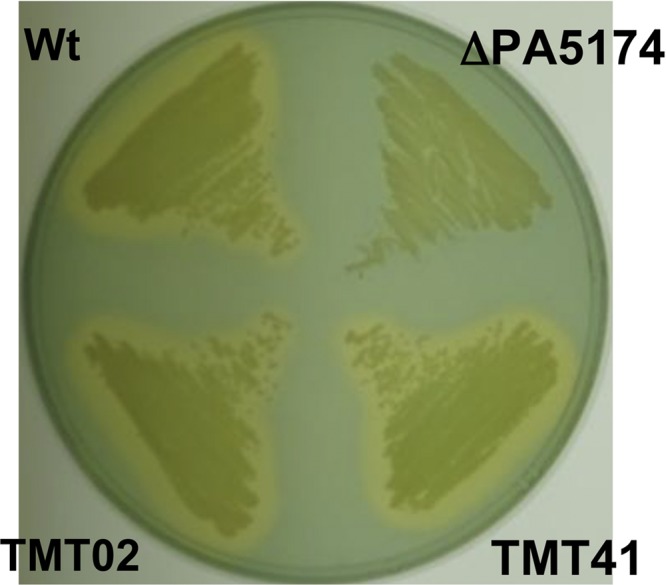
Siderophore secretion in P. aeruginosa ΔPA5174. The wild-type P. aeruginosa PAO1 (Wt), TMT39 (ΔPA5174), ΔPA5174(pZEN-PA5174) (TMT41), and ΔpqsD PQS− (TMT02) strains were streaked onto LB agar plates containing CAS and incubated at 37°C for 24 h. Zones of yellow-orange clearing indicate sequestration of Fe3+ away from CAS- Fe3+ complexes by secreted siderophores.
Characterization of recombinant PA5174 β-ketoacyl ACP synthase activity.
Recombinant PA5174 was expressed in E. coli and purified by His tag affinity chromatography in order to characterize in vitro enzymatic activity. Using a FabG coupled continuous spectrophotometric assay to monitor NADPH consumption via reduction of β-ketoacyl-ACP product (56), the specific activities of PA5174 and E. coli FabH with malonyl-ACP and acetyl-CoA as substrates were 13.4 and 7.4 pmol/min/ng under the same assay conditions (Fig. 8A). The FAS initiation activity of PA5174 is on par with E. coli FabH and with previously reported data (13, 63).
Fig 8.

Enzymatic activity and acyl-CoA acceptor substrate specificity of recombinant PA5174. (A) The continuous FabG coupled assay was initiated with 15 nM FabH (●) or PA5174 (○), and the reaction mixture was incubated at 30°C. Reaction progress was monitored by UV absorbance at 340 nm to assay NADPH consumption. Background absorbance (no enzyme addition) was subtracted from each curve before plotting. (B) Discontinuous conformation-sensitive urea-PAGE was employed to separate acyl-ACP products with incorporated [2-3H]malonyl-ACP substrate. Gels were sequentially imaged using Coomassie blue staining and autoradiography. All lanes contained [2-3H]malonyl-ACP and acetyl-CoA substrates. Lanes: 1, substrates only; 2, E. coli FabH; 3, PA5174; 4, E. coli FabH plus FabG/FabA/FabI; 5, PA5174 plus FabG/FabA/FabI. The positions of malonyl-ACP (malonyl), β-acetoacetyl-ACP or acetyl-ACP (β-acac/ac), and β-butyryl-ACP (β-butyryl) are labeled. (C) The acyl-CoA substrate specificity of PA5174 was assayed using [2-3H]malonyl-ACP substrate alone (control [Con]) or with various acyl-CoA acceptors (C2 to C16). Reaction products were separated by urea-PAGE, stained, and sequentially imaged. Under these assay conditions, the [2-3H]β-acetoacetyl-ACP reaction product using C2-CoA as the acceptor is not adequately separated from the [2-3H]acetyl-ACP side product produced by decarboxylation of [2-3H]malonyl-ACP unless further reduced by FabG/FabA/FabI as in lanes 4 and 5 of panel B.
We next examined the reaction product of PA5174 using conformation-sensitive urea-PAGE with [2-14C]malonyl-ACP (68). The putative β-acetoacetyl-ACP product of PA5714 migrates faster than the starting substrate [2-14C]malonyl-ACP and to the same extent as the E. coli FabH standard reaction (Fig. 8B). To further identify the product, the FAS reduction cycle enzymes from E. coli (FabG/FabA/FabI) were included in the reaction mixture. The radioactive acyl-ACP band migrated faster on the gel, indicating that the product of PA5174 is a substrate for FabG/FabA/FabI. The electrophoretic mobility of the reduced product is identical to that of the E. coli FabH butyryl-ACP standard. The lack of additional bands indicates that PA5174 cannot efficiently carry out the next round of condensation for chain elongation using acyl-ACP as a substrate, as do canonical KASI/II domain FAS condensing enzymes.
The substrate specificity of PA5174 enzyme was probed using various straight-chain saturated acyl-CoA (C2 to C16) acceptors. Reaction products were analyzed by conformation-sensitive urea-PAGE as described above using [2-14C]malonyl-ACP. PA5174 utilized short-chain acyl-CoA as substrates, including acetyl-CoA, butyryl-CoA, and hexanoyl-CoA, but did not use any of the longer-chain acyl-CoA substrates (Fig. 8C). A second radioactive band migrating slightly ahead of the malonyl-ACP substrate was apparent for the long-chain substrates. However, this band is likely [2-14C]acetyl-ACP, which is produced through decarboxylation of [2-14C]malonyl-ACP. Decarboxylation upon extended reaction times has previously been noted for all three types of KAS enzymes (1, 13) and becomes apparent in the absence of suitable acyl-CoA acceptors. All of the in vitro characteristics of PA5174 are entirely consistent with its assignment as the FAS initiation enzyme in P. aeruginosa, catalyzing the formation of β-acetoacetyl-ACP from acetyl-CoA and malonyl-ACP.
DISCUSSION
The enzymatic pathway of FAS type II biosynthesis in bacteria has been well defined in large part using E. coli and Salmonella as model organisms (17, 86). While exceptions in the overarching catalytic strategy to assemble fatty acids remain rare, novel FAS enzymes with little sequence similarity to their functional counterparts in the canonical FAS type II pathway are becoming increasingly common, as organisms beyond members of the family Enterobacteriaceae are subjected to detailed study (59). For instance, P. aeruginosa encodes a second enoyl-ACP reductase (FabV) which bears no sequence similarity to FabI (94). In this report, we provide evidence that FAS initiation in P. aeruginosa is also unique in using the KASI/II domain-containing protein PA5174. The viability of a P. aeruginosa mutant that lacks all KASIII domain-containing proteins (Fig. 2C), the attenuation of growth due to the FAS defect in the ΔPΑ5174 mutant (Fig. 4A), intergeneric cross complementation between P. aeruginosa PA5174 and E. coli fabH (Fig. 3 and 4), the pleiotropic phenotype stemming from depletion of fatty acid-dependent metabolites (Fig. 5 to 7), and the in vitro enzymatic characterization (Fig. 8) all support the conclusion that PA5174 encodes the predominant β-acetoacetyl-ACP synthase of P. aeruginosa. We propose distinguishing β-ketoacyl-ACP synthases with KASI/II domains and a preference for short-chain acyl-CoA acceptor substrates as the new FabY class of condensing enzymes.
The viability of the ΔfabY mutant indicates that an alternate secondary mechanism for FAS initiation must exist. In a fabH mutant of Lactococcus lactis, growth can be restored by overexpression of FabF (55). A suppression mechanism whereby acetyl-ACP is generated either through FabF-mediated decarboxylation of malonyl-ACP or acetyl-CoA:ACP transacylation was proposed. Acetyl-ACP, in turn, could then be used as the acceptor substrate (albeit a poor one) in a condensation reaction with malonyl-ACP to generate essential β-acetoacetyl-ACP pools. There are three KASI/II candidates in P. aeruginosa PAO1 (FabB, FabF, and FabF2 [Fig. 9]), as well as four KASIII enzymes (Fig. 2A), to supply either of the putative decarboxylation/transacylation activities required to bypass FabY. Alternatively, any of the KASIII proteins could have an intrinsic secondary FabH-type activity that becomes biologically relevant only in the absence of FabY, as deletion of all KASIII proteins in a wild-type background was aphenotypic with respect to growth (Fig. 2C).
Fig 9.

Phylogenetic tree and distribution of KASI/II domain-containing proteins among pseudomonads. All open reading frames within each genome having significant similarity to P. aeruginosa PAO1 KASI/II proteins (E-value < 1e−10) and that are not a subdomain of a polyketide synthase multidomain protein were included in the analysis. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The tree is drawn to scale, with branch lengths in the units of number of amino acid substitutions per site. Analyses were conducted by using MEGA5 (82). The three main KASI/II groups (FabB, FabF, and FabY) are indicated, and all members within each cluster share at least partial genomic synteny. Locus tag abbreviations: PA, P. aeruginosa PAO1 (orange); PSPA7, P. aeruginosa PA7 (orange); PA14, P. aeruginosa UCBPP-PA14 (orange); PLES, P. aeruginosa LESB58 (orange); Pmen, P. mendocina ymp (blue); MDS, P. mendocina NK-01 (blue); Psefu, P. fulva 12-X (red), PST, P. stutzeri A1501 (black), PputGB1, P. putida GB-1 (black); Psyr, P. syringae pv. syringae B728a (black); PFLU, P. fluorescens SBW25 (black); Avin, Azotobacter vinelandii DJ (purple); b, E. coli (green).
Other KASI/II proteins could theoretically utilize an acetyl-CoA acceptor in place of acetyl-ACP to make β-acetoacetyl-ACP, particularly since FabY now demonstrates that substrate specificity of a KASI/II domain is not necessarily confined to acyl-ACP acceptor substrates. Phylogenetic analysis of KASI/II domain enzymes in pseudomonads, however, would seem to make this the least likely of all the possible FabY bypass mechanisms (Fig. 9). There are three KASI/II domain protein clusters conserved among multiple Pseudomonas species (FabB, FabF, and FabY). The other KASI/II proteins are more narrowly distributed, suggesting strain-specific auxiliary cellular roles outside primary FAS metabolism. The FabB and FabF clusters (as defined by their respective E. coli orthologs; green labels) tightly cluster and have representatives from all 12 Pseudomonas strains analyzed, consistent with their role in FAS elongation. The FabY cluster is highly divergent from its FabF/FabB KASI/II counterparts, having the least similar sequence and being ∼200 amino acids longer. The sequence divergence likely reflects acquisition of short-chain acyl-CoA substrate selectivity, a catalytic activity not shared with distantly related KASI/II proteins. Interestingly, FabY is also more narrowly distributed at the species level, being confined to all sequenced P. aeruginosa strains (orange labels), Pseudomonas fulva (red labels), Pseudomonas mendocina (blue labels), and Azotobacter vinelandii (purple labels). The A. vinelandii genome is included with Pseudomonas, as its taxonomic distinction within a separate genus is debatable based on 16S rRNA analysis (57, 73). Indeed, the conservation of FabY supports the notion that P. aeruginosa is more closely related to A. vinelandii than to other Pseudomonas species. The A. vinelandii genome is also noteworthy in that there are no KASIII orthologs in the sequenced genome (unpublished observation), and as with the quadruple knockout P. aeruginosa strain TMT16, FAS initiation must be entirely KASIII independent. Conversely, there are no FabY orthologs in the genomes of Pseudomonas stutzeri, Pseudomonas putida, Pseudomonas fluorescens, and Pseudomonas syringae (all black labels), indicating that FabY was either relatively recently acquired by, or only retained in, the P. aeruginosa-specific branch after speciation of the pseudomonad lineage. How the other Pseudomonas species initiate FAS is unclear, but it is likely that a KASIII domain protein may be involved. In Pseudomonas syringae pv. tabaci 6605, a FabH ortholog (orf3) located within the flagellin glycosylation island has been identified (81). Deletion of the monocistronic orf3 had no effect on protein glycosylation, but a role in FAS initiation was suggested based on a defect in HSL QS that is reminiscent of the ΔFabY phenotype observed here in P. aeruginosa. Once more, a KASIII ortholog embedded in a flagellin glycosylation island is absent in P. aeruginosa PAO1 (b-type flagellin) (3) but is present in P. putida, P. fluorescens, and P. syringae (unpublished observation). The presence of a putative FabH ortholog within an element subject to extensive rearrangement and horizontal gene exchange suggests a possible genetic means for establishing species-dependent FAS initiation pathways among pseudomonads.
Inhibition of FAS initiation in P. aeruginosa is a compelling strategy for developing new antimicrobial agents. A multitude of cellular products, including essential as well as virulence-associated factors, all depend on FAS initiation to directly provide biosynthetic precursors or are themselves subject to transcriptional regulation by fatty acid-containing signal molecules (Fig. 10). Deletion of FabY attenuated not only growth but also QS, siderophore secretion, and a host of virulence factors, many of which are by themselves attractive antibiotic development targets. Inhibition of iron acquisition by blocking siderophore production (53), and QS inhibition in particular (50), have garnered substantial interest as strategies for treating P. aeruginosa infections. The three dominant QS signal molecules from the las, rhl, and PQS systems, all of which contain fatty acid moieties, together form a complex, cell density-dependent regulatory circuit controlling the expression of a suite of virulence factors (87, 89). QS-regulated exoproducts include the toxin pyocyanin which induces oxidative stress in infected tissue, elastase which degrades elastin and other host matrix proteins, and rhamnolipids which have intrinsic cytolytic activity. QS signaling is also critical in orchestrating the transition from planktonic to multicellular biofilm growth associated within chronic infections (35). The levels of rhl acyl-HSL and PQS, as well as all associated exoproducts, were markedly depressed in the absence of FabY. The progression of QS in the ΔFabY mutant is likely delayed by the lack of fatty acid precursors, as has also been observed with decreased FAS enoyl-ACP reductase activity (39). The near wild-type amounts of the las 3-oxo-HSL, which sits atop the QS hierarchal signal cascade, suggests that pools become available as FAS demands for phospholipid/lipopolysaccharide biosynthesis wane during extended stationary-phase incubation. Conversely, all QS exoproducts were upregulated well beyond wild-type levels in the multicopy plasmid FabY-complemented strain. This was especially apparent for the PQS signal. Hypervirulence is associated with early and robust quorum signaling in transmissible epidemic clinical isolates (26). FabY and QS signal expression appears to be correlated in P. aeruginosa PAO1 and warrants future study into the regulatory mechanisms connecting fatty acid flux and QS-mediated virulence.
Fig 10.

Cellular products dependent on FabY (i.e., FabH-type) activity in P. aeruginosa PAO1. The β-acetoacetyl-ACP product of FabY is used as a primer for elongation by the FAS elongation machinery, whose products are directly incorporated into a multitude of essential cell components, cofactors, and quorum sensing (QS) signaling molecules. Some exoproducts, including rhamnolipids, are dependent on fatty acid precursors as well as being subject to transcriptional QS regulation. Molecular fragments originating from FabY enzymatic activity are shown in bold type (blue, red, or green). The depicted biotin pathway in P. aeruginosa PAO1 is based on the proposed bioC and/or bioH pathway of E. coli (16). E2, (α)-ketoacid dehydrogenase subunit E2-containing lipoylation domain.
The identification of FabY as the predominant FAS initiation enzyme in P. aeruginosa adds to the growing diversity in bacterial FAS pathways (59). Being narrowly confined among pathogens to P. aeruginosa, extending the spectrum of small-molecule inhibitors optimized against KASIII-type FabH targets to P. aeruginosa may prove to be challenging. However, simultaneous inhibition of the initiating and elongating KAS I/II enzyme family (FabB, FabF, and FabY) is potentially more feasible, given that a single domain carries out all KAS steps in the FAS pathway of P. aeruginosa. The KASI/II active site Cys-His-His triad is conserved in all 3 isozymes, including FabY, and the additional ∼200 amino acids of FabY are located outside the central KASI/II domain at the N and C termini (see Fig. S1 in the supplemental material). Insights into the structure and biochemistry of FabY, as well as the mechanism of FabY FAS bypass, will be necessary to fully evaluate the ultimate potential of FabY as a target for P. aeruginosa-specific FAS inhibitor development. In the accompanying manuscript (90), we report that the PA3286 KASIII domain-containing condensing enzyme initiates FAS in the absence of FabY through shunting of exogenous fatty acids from the β-oxidation degradation pathway into de novo FAS (90).
Supplementary Material
ACKNOWLEDGMENTS
We thank Herbert P. Schweizer (Colorado State University) for the pEX18ApGW plasmid and Carl Balibar (Novartis) for the pKD4-PBAD plasmid.
Footnotes
Published ahead of print 29 June 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Alberts AW, Bell RM, Vagelos PR. 1972. Acyl carrier protein. XV. Studies of β-ketoacyl-acyl carrier protein synthetase. J. Biol. Chem. 247:3190–3198 [PubMed] [Google Scholar]
- 2. Alberts AW, Majerus PW, Talamo B, Vagelos PR. 1964. Acyl-carrier protein. II. Intermediary reactions of fatty acid synthesis. Biochemistry 3:1563–1571 [DOI] [PubMed] [Google Scholar]
- 3. Arora SK, Bangera M, Lory S, Ramphal R. 2001. A genomic island in Pseudomonas aeruginosa carries the determinants of flagellin glycosylation. Proc. Natl. Acad. Sci. U. S. A. 98:9342–9347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bergler H, et al. 1994. Protein EnvM is the NADH-dependent enoyl-ACP reductase (FabI) of Escherichia coli. J. Biol. Chem. 269:5493–5496 [PubMed] [Google Scholar]
- 5. Bredenbruch F, Geffers R, Nimtz M, Buer J, Haussler S. 2006. The Pseudomonas aeruginosa quinolone signal (PQS) has an iron-chelating activity. Environ. Microbiol. 8:1318–1329 [DOI] [PubMed] [Google Scholar]
- 6. Brinster S, et al. 2009. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 458:83–86 [DOI] [PubMed] [Google Scholar]
- 7. Caiazza NC, Shanks RM, O'Toole GA. 2005. Rhamnolipids modulate swarming motility patterns of Pseudomonas aeruginosa. J. Bacteriol. 187:7351–7361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campbell JW, Cronan JE., Jr 2001. Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu. Rev. Microbiol. 55:305–332 [DOI] [PubMed] [Google Scholar]
- 9. Castillo YP, Perez MA. 2008. Bacterial beta-ketoacyl-acyl carrier protein synthase III (FabH): an attractive target for the design of new broad-spectrum antimicrobial agents. Mini. Rev. Med. Chem. 8:36–45 [DOI] [PubMed] [Google Scholar]
- 10. Cha C, Gao P, Chen YC, Shaw PD, Farrand SK. 1998. Production of acyl-homoserine lactone quorum-sensing signals by gram-negative plant-associated bacteria. Mol. Plant Microbe Interact. 11:1119–1129 [DOI] [PubMed] [Google Scholar]
- 11. Chao J, Wolfaardt GM, Arts MT. 2010. Characterization of Pseudomonas aeruginosa fatty acid profiles in biofilms and batch planktonic cultures. Can. J. Microbiol. 56:1028–1039 [DOI] [PubMed] [Google Scholar]
- 12. Chen Y, et al. 2011. Structural classification and properties of ketoacyl synthases. Protein Sci. 20:1659–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi KH, Heath RJ, Rock CO. 2000. Beta-ketoacyl-acyl carrier protein synthase III (FabH) is a determining factor in branched-chain fatty acid biosynthesis. J. Bacteriol. 182:365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Choi KH, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64:391–397 [DOI] [PubMed] [Google Scholar]
- 15. Choi KH, Schweizer HP. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol. 5:30 doi:10.1186/1471-2180-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cronan JE, Lin S. 2011. Synthesis of the alpha,omega-dicarboxylic acid precursor of biotin by the canonical fatty acid biosynthetic pathway. Curr. Opin. Chem. Biol. 15:407–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cronan JE, Rock CO. 1996. Biosynthesis of membrane lipids, p 612–636 In Neidhardt FC, et al. (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed, vol 1 American Society for Microbiology, Washington, DC [Google Scholar]
- 18. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davies C, Heath RJ, White SW, Rock CO. 2000. The 1.8 A crystal structure and active-site architecture of beta-ketoacyl-acyl carrier protein synthase III (FabH) from Escherichia coli. Structure 8:185–195 [DOI] [PubMed] [Google Scholar]
- 20. Diggle SP, et al. 2003. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol. Microbiol. 50:29–43 [DOI] [PubMed] [Google Scholar]
- 21. Drake EJ, Gulick AM. 2011. Structural characterization and high-throughput screening of inhibitors of PvdQ, an NTN hydrolase involved in pyoverdine synthesis. ACS Chem. Biol. 6:1277–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dubern JF, Diggle SP. 2008. Quorum sensing by 2-alkyl-4-quinolones in Pseudomonas aeruginosa and other bacterial species. Mol. Biosyst. 4:882–888 [DOI] [PubMed] [Google Scholar]
- 23. Essar DW, Eberly L, Hadero A, Crawford IP. 1990. Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications. J. Bacteriol. 172:884–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. U. S. A. 76:1648–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fletcher MP, Diggle SP, Camara M, Williams P. 2007. Biosensor-based assays for PQS, HHQ and related 2-alkyl-4-quinolone quorum sensing signal molecules. Nat. Protoc. 2:1254–1262 [DOI] [PubMed] [Google Scholar]
- 26. Fothergill JL, et al. 2007. Widespread pyocyanin over-production among isolates of a cystic fibrosis epidemic strain. BMC Microbiol. 7:45 doi:10.1186/1471-2180-7-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gago G, Diacovich L, Arabolaza A, Tsai SC, Gramajo H. 2011. Fatty acid biosynthesis in actinomycetes. FEMS Microbiol. Rev. 35:475–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gajiwala KS, et al. 2009. Crystal structures of bacterial FabH suggest a molecular basis for the substrate specificity of the enzyme. FEBS Lett. 583:2939–2946 [DOI] [PubMed] [Google Scholar]
- 29. Gallagher LA, McKnight SL, Kuznetsova MS, Pesci EC, Manoil C. 2002. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J. Bacteriol. 184:6472–6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gambello MJ, Iglewski BH. 1991. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J. Bacteriol. 173:3000–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garwin JL, Klages AL, Cronan JE., Jr 1980. Structural, enzymatic, and genetic studies of beta-ketoacyl-acyl carrier protein synthases I and II of Escherichia coli. J. Biol. Chem. 255:11949–11956 [PubMed] [Google Scholar]
- 32. Gerusz V. 2010. Recent advances in the inhibition of bacterial fatty acid biosynthesis. Annu. Rep. Med. Chem. 45:295–311 [Google Scholar]
- 33. Girard G, Bloemberg GV. 2008. Central role of quorum sensing in regulating the production of pathogenicity factors in Pseudomonas aeruginosa. Future Microbiol. 3:97–106 [DOI] [PubMed] [Google Scholar]
- 34. Hannauer M, et al. 2012. Biosynthesis of the pyoverdine siderophore of Pseudomonas aeruginosa involves precursors with a myristic or a myristoleic acid chain. FEBS Lett. 586:96–101 [DOI] [PubMed] [Google Scholar]
- 35. Haussler S. 2010. Multicellular signalling and growth of Pseudomonas aeruginosa. Int. J. Med. Microbiol. 300:544–548 [DOI] [PubMed] [Google Scholar]
- 36. Heath RJ, Rock CO. 1996. Regulation of fatty acid elongation and initiation by acyl-acyl carrier protein in Escherichia coli. J. Biol. Chem. 271:1833–1836 [DOI] [PubMed] [Google Scholar]
- 37. Heath RJ, White SW, Rock CO. 2002. Inhibitors of fatty acid synthesis as antimicrobial chemotherapeutics. Appl. Microbiol. Biotechnol. 58:695–703 [DOI] [PubMed] [Google Scholar]
- 38. Hoang TT, Schweizer HP. 1997. Fatty acid biosynthesis in Pseudomonas aeruginosa: cloning and characterization of the fabAB operon encoding beta-hydroxyacyl-acyl carrier protein dehydratase (FabA) and beta-ketoacyl-acyl carrier protein synthase I (FabB). J. Bacteriol. 179:5326–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hoang TT, Schweizer HP. 1999. Characterization of Pseudomonas aeruginosa enoyl-acyl carrier protein reductase (FabI): a target for the antimicrobial triclosan and its role in acylated homoserine lactone synthesis. J. Bacteriol. 181:5489–5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoang TT, Sullivan SA, Cusick JK, Schweizer HP. 2002. Beta-ketoacyl acyl carrier protein reductase (FabG) activity of the fatty acid biosynthetic pathway is a determining factor of 3-oxo-homoserine lactone acyl chain lengths. Microbiology 148:3849–3856 [DOI] [PubMed] [Google Scholar]
- 41. Juhas M, Eberl L, Tummler B. 2005. Quorum sensing: the power of cooperation in the world of Pseudomonas. Environ. Microbiol. 7:459–471 [DOI] [PubMed] [Google Scholar]
- 42. Kass LR, Brock DJ, Bloch K. 1967. Beta-hydroxydecanoyl thioester dehydrase. I. Purification and properties. J. Biol. Chem. 242:4418–4431 [PubMed] [Google Scholar]
- 43. Kerr KG, Snelling AM. 2009. Pseudomonas aeruginosa: a formidable and ever-present adversary. J. Hosp. Infect. 73:338–344 [DOI] [PubMed] [Google Scholar]
- 44. Khandekar SS, Daines RA, Lonsdale JT. 2003. Bacterial beta-ketoacyl-acyl carrier protein synthases as targets for antibacterial agents. Curr. Protein Pept. Sci. 4:21–29 [DOI] [PubMed] [Google Scholar]
- 45. Kohler T, Curty LK, Barja F, van Delden C, Pechere JC. 2000. Swarming of Pseudomonas aeruginosa is dependent on cell-to-cell signaling and requires flagella and pili. J. Bacteriol. 182:5990–5996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kutchma AJ, Hoang TT, Schweizer HP. 1999. Characterization of a Pseudomonas aeruginosa fatty acid biosynthetic gene cluster: purification of acyl carrier protein (ACP) and malonyl-coenzyme A:ACP transacylase (FabD). J. Bacteriol. 181:5498–5504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lai CY, Cronan JE. 2003. Beta-ketoacyl-acyl carrier protein synthase III (FabH) is essential for bacterial fatty acid synthesis. J. Biol. Chem. 278:51494–51503 [DOI] [PubMed] [Google Scholar]
- 48. Liberati NT, et al. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U. S. A. 103:2833–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lu YJ, Zhang YM, Rock CO. 2004. Product diversity and regulation of type II fatty acid synthases. Biochem. Cell Biol. 82:145–155 [DOI] [PubMed] [Google Scholar]
- 50. Martin CA, Hoven AD, Cook AM. 2008. Therapeutic frontiers: preventing and treating infectious diseases by inhibiting bacterial quorum sensing. Eur. J. Clin. Microbiol. Infect. Dis. 27:635–642 [DOI] [PubMed] [Google Scholar]
- 51. McClean KH, et al. 1997. Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology 143(Part 12):3703–3711 [DOI] [PubMed] [Google Scholar]
- 52. Meredith TC, Woodard RW. 2005. Identification of GutQ from Escherichia coli as a d-arabinose 5-phosphate isomerase. J. Bacteriol. 187:6936–6942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miethke M, Marahiel MA. 2007. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 71:413–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mohan S, Kelly TM, Eveland SS, Raetz CR, Anderson MS. 1994. An Escherichia coli gene (FabZ) encoding (3R)-hydroxymyristoyl acyl carrier protein dehydrase. Relation to fabA and suppression of mutations in lipid A biosynthesis. J. Biol. Chem. 269:32896–32903 [PubMed] [Google Scholar]
- 55. Morgan-Kiss RM, Cronan JE. 2008. The Lactococcus lactis FabF fatty acid synthetic enzyme can functionally replace both the FabB and FabF proteins of Escherichia coli and the FabH protein of Lactococcus lactis. Arch. Microbiol. 190:427–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nie Z, et al. 2005. Structure-based design, synthesis, and study of potent inhibitors of beta-ketoacyl-acyl carrier protein synthase III as potential antimicrobial agents. J. Med. Chem. 48:1596–1609 [DOI] [PubMed] [Google Scholar]
- 57. Ozen AI, Ussery DW. 2012. Defining the Pseudomonas genus: where do we draw the line with Azotobacter? Microb. Ecol. 63:239–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. 2011. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc. Natl. Acad. Sci. U. S. A. 108:15378–15383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Parsons JB, Rock CO. 2011. Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery? Curr. Opin. Microbiol. 14:544–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pesci EC, et al. 1999. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 96:11229–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pinzon NM, Ju LK. 2009. Improved detection of rhamnolipid production using agar plates containing methylene blue and cetyl trimethylammonium bromide. Biotechnol. Lett. 31:1583–1588 [DOI] [PubMed] [Google Scholar]
- 62. Pistorius D, et al. 2011. Biosynthesis of 2-alkyl-4(1H)-quinolones in Pseudomonas aeruginosa: potential for therapeutic interference with pathogenicity. ChemBioChem 12:850–853 [DOI] [PubMed] [Google Scholar]
- 63. Qiu X, et al. 2005. Crystal structure and substrate specificity of the beta-ketoacyl-acyl carrier protein synthase III (FabH) from Staphylococcus aureus. Protein Sci. 14:2087–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Raychaudhuri A, Jerga A, Tipton PA. 2005. Chemical mechanism and substrate specificity of RhlI, an acylhomoserine lactone synthase from Pseudomonas aeruginosa. Biochemistry 44:2974–2981 [DOI] [PubMed] [Google Scholar]
- 66. Reis RS, Pereira AG, Neves BC, Freire DM. 2011. Gene regulation of rhamnolipid production in Pseudomonas aeruginosa–a review. Bioresour. Technol. 102:6377–6384 [DOI] [PubMed] [Google Scholar]
- 67. Revill WP, Bibb MJ, Scheu AK, Kieser HJ, Hopwood DA. 2001. Beta-ketoacyl acyl carrier protein synthase III (FabH) is essential for fatty acid biosynthesis in Streptomyces coelicolor A3(2). J. Bacteriol. 183:3526–3530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rock CO, Cronan JE., Jr 1981. Acyl carrier protein from Escherichia coli. Methods Enzymol. 71(Part C):341–351 [DOI] [PubMed] [Google Scholar]
- 69. Rosenfeld IS, D'Agnolo G, Vagelos PR. 1973. Synthesis of unsaturated fatty acids and the lesion in fabB mutants. J. Biol. Chem. 248:2452–2460 [PubMed] [Google Scholar]
- 70. Schaber JA, et al. 2004. Analysis of quorum sensing-deficient clinical isolates of Pseudomonas aeruginosa. J. Med. Microbiol. 53:841–853 [DOI] [PubMed] [Google Scholar]
- 71. Schweizer E, Hofmann J. 2004. Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol. Mol. Biol. Rev. 68:501–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schwyn B, Neilands JB. 1987. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 160:47–56 [DOI] [PubMed] [Google Scholar]
- 73. Setubal JC, et al. 2009. Genome sequence of Azotobacter vinelandii, an obligate aerobe specialized to support diverse anaerobic metabolic processes. J. Bacteriol. 191:4534–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shaw PD, et al. 1997. Detecting and characterizing N-acyl-homoserine lactone signal molecules by thin-layer chromatography. Proc. Natl. Acad. Sci. U. S. A. 94:6036–6041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sievers F, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering - transposon mutagenesis in Gram-negative bacteria. Biotechnology 1:784–791 [Google Scholar]
- 77. Smith S. 1994. The animal fatty acid synthase: one gene, one polypeptide, seven enzymes. FASEB J. 8:1248–1259 [PubMed] [Google Scholar]
- 78. Smith S, Witkowski A, Joshi AK. 2003. Structural and functional organization of the animal fatty acid synthase. Prog. Lipid Res. 42:289–317 [DOI] [PubMed] [Google Scholar]
- 79. Stintzi A, Evans K, Meyer JM, Poole K. 1998. Quorum-sensing and siderophore biosynthesis in Pseudomonas aeruginosa: lasR/lasI mutants exhibit reduced pyoverdine biosynthesis. FEMS Microbiol. Lett. 166:341–345 [DOI] [PubMed] [Google Scholar]
- 80. Stover CK, et al. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964 [DOI] [PubMed] [Google Scholar]
- 81. Taguchi F, et al. 2006. A homologue of the 3-oxoacyl-(acyl carrier protein) synthase III gene located in the glycosylation island of Pseudomonas syringae pv. tabaci regulates virulence factors via N-acyl homoserine lactone and fatty acid synthesis. J. Bacteriol. 188:8376–8384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tremblay J, Deziel E. 2008. Improving the reproducibility of Pseudomonas aeruginosa swarming motility assays. J. Basic Microbiol. 48:509–515 [DOI] [PubMed] [Google Scholar]
- 84. Tsay JT, Oh W, Larson TJ, Jackowski S, Rock CO. 1992. Isolation and characterization of the beta-ketoacyl-acyl carrier protein synthase III gene (fabH) from Escherichia coli K-12. J. Biol. Chem. 267:6807–6814 [PubMed] [Google Scholar]
- 85. Visca P, Imperi F, Lamont IL. 2007. Pyoverdine siderophores: from biogenesis to biosignificance. Trends Microbiol. 15:22–30 [DOI] [PubMed] [Google Scholar]
- 86. White SW, Zheng J, Zhang YM, Rock CO. 2005. The structural biology of type II fatty acid biosynthesis. Annu. Rev. Biochem. 74:791–831 [DOI] [PubMed] [Google Scholar]
- 87. Williams P, Camara M. 2009. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 12:182–191 [DOI] [PubMed] [Google Scholar]
- 88. Winsor GL, et al. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39:D596–D600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Winstanley C, Fothergill JL. 2009. The role of quorum sensing in chronic cystic fibrosis Pseudomonas aeruginosa infections. FEMS Microbiol. Lett. 290:1–9 [DOI] [PubMed] [Google Scholar]
- 90. Yuan Y, Leeds JA, Meredith TC. 2012. Pseudomonas aeruginosa directly shunts β-oxidation degradation intermediates into de novo fatty acid biosynthesis. J. Bacteriol. 194:5185–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang HJ, Li ZL, Zhu HL. 2012. Advances in the research of beta-ketoacyl-ACP synthase III (FabH) inhibitors. Curr. Med. Chem. 19:1225–1237 [DOI] [PubMed] [Google Scholar]
- 92. Zhang YM, White SW, Rock CO. 2006. Inhibiting bacterial fatty acid synthesis. J. Biol. Chem. 281:17541–17544 [DOI] [PubMed] [Google Scholar]
- 93. Zhu K, Rock CO. 2008. RhlA converts beta-hydroxyacyl-acyl carrier protein intermediates in fatty acid synthesis to the beta-hydroxydecanoyl-beta-hydroxydecanoate component of rhamnolipids in Pseudomonas aeruginosa. J. Bacteriol. 190:3147–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhu L, Lin J, Ma J, Cronan JE, Wang H. 2010. Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob. Agents Chemother. 54:689–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.