

Abstract
In Escherichia coli, RecA–single-stranded DNA (RecA-ssDNA) filaments catalyze DNA repair, recombination, and induction of the SOS response. It has been shown that, while many (15 to 25%) log-phase cells have RecA filaments, few (about 1%) are induced for SOS. It is hypothesized that RecA's ability to induce SOS expression in log-phase cells is repressed because of the potentially detrimental effects of SOS mutagenesis. To test this, mutations were sought to produce a population where the number of cells with SOS expression more closely equaled the number of RecA filaments. Here, it is shown that deleting radA (important for resolution of recombination structures) and increasing recA transcription 2- to 3-fold with a recAo1403 operator mutation act independently to minimally satisfy this condition. This allows 24% of mutant cells to have elevated levels of SOS expression, a percentage similar to that of cells with RecA-green fluorescent protein (RecA-GFP) foci. In an xthA (exonuclease III gene) mutant where there are 3-fold more RecA loading events, recX (a destabilizer of RecA filaments) must be additionally deleted to achieve a population of cells where the percentage having elevated SOS expression (91%) nearly equals the percentage with at least one RecA-GFP focus (83%). It is proposed that, in the xthA mutant, there are three independent mechanisms that repress SOS expression in log-phase cells. These are the rapid processing of RecA filaments by RadA, maintaining the concentration of RecA below a critical level, and the destabilizing of RecA filaments by RecX. Only the first two mechanisms operate independently in a wild-type cell.
INTRODUCTION
Regulation of DNA transactions is critical to the maintenance and duplication of chromosomes in all organisms. Any operation to a chromosome involving DNA replication or recombination must be precise and accurate or genetic information will be altered. Homologous recombination plays important roles in helping to repair broken replication forks and other types of DNA damage in an error-free manner (11). RadA, RecA, and RAD51 (homologs in Archaea, Bacteria, and Eucarya, respectively) participate in these functions through their ability to form structurally similar protein-DNA helical filaments (5, 49, 51, 64). The abilities of RecA to catalyze repair and recombination and induce the SOS response all stem from the ability of RecA to form a nucleoprotein filament (23, 32). The regulation of this protein-DNA filament is extremely important as it has been shown that too much or too little recombination can be detrimental to an organism (24, 41).
As mentioned above, in Escherichia coli and other bacteria (17) the RecA-DNA filament also plays a key role as a regulator of the SOS response (26; reviewed in references 27 and 59). RecA initiates the SOS response by polymerizing on single-stranded DNA (ssDNA) produced by the processing of DNA damage. This RecA-DNA filament is an allosteric effector of LexA autoproteolysis (26, 42). Depletion of the LexA repressor increases transcription of at least 40 genes that help repair and mutagenize the DNA and inhibit cell division (8).
RecA-dependent homologous recombination occurs minimally in about 15% of log-phase cells. This is based on the analysis of hybrid dif sites (53). Since there is a bias in the resolution of Holliday structures, formed as a result of broken replication forks, to the noncrossover configuration, this percentage could be higher (12, 57). An independent method to assess the number of recA loading events using RecA-green fluorescent protein (RecA-GFP) foci indicated that approximately 13% of exponential-phase cells grown in minimal medium have RecA structures (45). It is thought that the recombination/loading event identified via hybrid sites and the RecA-GFP foci represent places where RecA is binding to and helping to repair broken replication forks. However, in the absence of externally applied DNA damage, about 1% of a log-phase population of cells is induced for SOS expression (34, 39). Similar patterns of RecA-GFP focus production and the lack of corresponding SOS expression have also been seen in Bacillus subtilis (2). Given the discrepancy between the percentage of cells with evidence of RecA-mediated recombination and the percentage of cells expressing SOS, it is hypothesized that the ability of the RecA filaments at broken forks or other sites of internal damage to induce SOS is repressed. The rationale for a specific mechanism for this repression is that the cell would prefer not to invoke some of the more dire consequences of SOS induction, such as increased expression of mutagenic polymerases, if it is repairing normal, housekeeping types of DNA damage. In the current work, we address this hypothesis by determining what factors may contribute to the repression of SOS induction at these housekeeping RecA filaments.
SOS is induced when the LexA repressor interacts with the RecA-DNA filament and its rate of autoproteolysis is increased (26). Electron-micrographic studies suggest that LexA binds in the groove of the RecA-DNA filament (63). Biochemical studies showing that LexA competes with duplex DNA to bind a RecA-ssDNA filament suggest there is a competition between the abilities to do recombination and induce the SOS response (21, 44, 63). If the interaction of the LexA protein with a filament is rate limiting, then increasing the length, amount, and/or stability of the filament is likely to increase the likelihood of interactions with LexA and production of an SOS response.
RecA's activity is controlled at several levels. As first proposed by Mount (36), recA is transcriptionally regulated by lexA as part of the SOS response (reviewed in reference 18). Exponentially grown cells are estimated to have about 15,000 molecules of RecA (54), and this level can increase 10-fold during an SOS response (8). RecA requires either RecFOR or RecBCD to load onto gapped DNA and at the ends of double-stranded DNA (dsDNA), respectively (reviewed in reference 40). DinI, RecX, UvrD, RdgC, PsiB, and RecOR have been shown to affect the stability of the RecA-ssDNA filament either in vivo, in vitro, or both (reviewed in reference 10). It is known that RecA filaments are dynamic. Subunits can add to either end with a net addition to the 3′ end or a net dissociation from the 5′ end (4, 19, 25; reviewed in reference 10). RecX is known to destabilize RecA filaments by capping the 3′ end of the growing filament, preventing further additions (15, 16, 43, 58). Other proteins, such as exonuclease III (XthA), have been shown to indirectly affect RecA-GFP focus formation as they degrade substrates that RecA could potentially bind (6).
Recent studies have analyzed recA mutants constitutive for SOS in the absence of external DNA damage (i.e., recA4142 mutants) and characterized the requirements for this SOS expression (28, 29). It was found that SOS expression in recA4142 (F217Y) mutants was dependent on its initial level of transcription, recBCD, ruvAB, recJ, and xonA. The data are consistent with the model that the loading of RecA4142 occurs at reversed replication forks (28). Furthermore, it was found that recX and xthA repressed the level of SOS constitutive expression in the recA4142 mutant (29).
Since constitutive SOS expression in recA4142 mutants requires ruvAB, it is possible that it additionally requires other proteins involved in processing RecA-mediated recombinational intermediates. radA (sms) was originally isolated for its role in radiation resistance in E. coli (14). Further studies of radA mutants have revealed a genetic redundancy with recG and ruvABC, suggesting a role for RadA in stabilizing or processing branched DNA or blocked forks (1, 31, 55).
In the current work, three independent factors that repress SOS expression in log-phase cells of Escherichia coli are identified. These are radA, the concentration of RecA in the cell, and recX. The first two limit SOS expression in wild-type log-phase cells in the absence of external DNA damage, and the third is needed additionally in xthA mutants, possibly because there are more RecA-loading events. It is hypothesized that these factors may change the character of the RecA filament and/or reduce the half-life of RecA filaments in the cell, limiting the time RecA is available to interact with LexA and induce SOS expression. It is additionally shown that the ability of RadA to repress SOS expression is situation dependent, as overproduction of RadA represses SOS constitutive expression in a recA4142 mutant but has no effect on SOS expression after UV irradiation in a wild-type cell.
MATERIALS AND METHODS
Strains and media.
All bacterial strains are derivatives of E. coli K-12 and are described in Table S1 in the supplemental material. The protocol for P1 transduction has been described previously (62). All P1 transductions were selected on 2% agar plates made with either Luria broth or 56/2 minimal media (62) supplemented with 0.2% glucose, 0.001% thiamine, and amino acids. Selection with antibiotics used either 50 μg/ml kanamycin, 25 μg/ml chloramphenicol, or 10 μg/ml tetracycline. All transductants were grown at 37°C and purified on the same type of media on which they were selected.
Preparation and analysis of cells for microscopy.
The cells for SOS expression were prepared as follows. The cells were grown in minimal medium to mid-log phase, and then 3 μl of cells was placed on a 1% agarose pad. A coverslip was then applied on top of the agarose pad. Cells were then imaged under identical settings. Images (phase-contrast and fluorescent) were taken on 3 different days, and 3 different images were taken for each strain each day. The cells were imaged using a 750-ms exposure and a 100× objective. This differed from previous work where a 100-ms exposure and a 60× objective were used. These images were analyzed by a combination of MicrobeTracker software (52) and Matlab R2011a software (Mathworks, Inc.). The relative fluorescence intensity (RFI) for each cell was normalized to the average fluorescence intensity of the JC13509 strain (no gfp). Typically, between 1,000 and 3,000 cells are counted for each strain. Statistical analysis of the data was performed using Student's t test.
The full genotype for the recA-gfp translational fusion used here is recAo1403 recA4155,4136::gfp-901 (45). This is abbreviated to recA4155,4136 in Table S1 in the supplemental material. recAo1403 is an operator mutant that increases the basal or non-SOS-induced level of transcription 2- to 3-fold (60). gfp-901 refers to mut-2 (7) with the additional “monomeric” mutation A206T (65). recA4155 is a mutant allele of recA encoding an arginine-to-alanine change at codon 28. It does not make storage structures in vivo (45). recA4136 refers to the specific fusion of recA to gfp (45).
The recA4155,4136::gfp-901 strains were prepared as in previous publications. Z-stacks of cells were imaged using a 750-ms exposure with an ND4 filter and a 100× objective. This differed from previous work where a 100-ms exposure with no ND4 filter and a 60× objective were used. The Z-stacks of the fluorescent images were processed by deconvolution using Volocity version 5.1 software (Improvision, Inc.). Deconvolved images were then flattened and foci were determined by a special thresholding program written by Q. Wang (personal communication). The phase-contrast images were then converted to binary images using MicrobeTracker (52) software. Programs written in Matlab R2011a (Mathworks, Inc.) were used to analyze the binary and fluorescent images to produce the data in Table 4. Statistical analysis of the data was performed using the chi-square test of homogeneity for an r × c contingency table (37).
Table 4.
Effects of radA, recX, and recAo1403 mutations on SOS expression in log-phase cells
| Strain | Versiona of: |
RFI | % >9-fold (18-fold) | Cells counted | ||
|---|---|---|---|---|---|---|
| recAo | radA | recX | ||||
| SS996 | + | + | + | 1.9 | 1.6 (0.7) | 2,140 |
| SS6088 | 1403 | + | + | 2.2 | 2.4 (0.6) | 1,534 |
| SS7102 | + | del | + | 2.9 | 4.5 (1.0) | 1,254 |
| SS6080 | + | + | cat | 2.9 | 4.4 (0.7) | 995 |
| SS7152 | + | del | cat | 2.4 | 3.7 (0.3) | 1,532 |
| SS7136 | 1403 | del | + | 6.6 | 23.3 (5.3) | 1,399 |
| SS7155 | 1403 | + | cat | 1.2 | 0.3 (0.0) | 1,590 |
| SS5841 | 1403 | del | cat | 5.8 | 24.2 (7.9) | 1,704 |
RESULTS
In this work, SOS expression is measured in individual cells containing a sulAp-gfp transcriptional fusion reporter. This transcriptional fusion has been previously described (34, 39). The sulAp-gfp is inserted in the attλ site on the chromosome. The sulA promoter has been shown to be an early SOS promoter (8). All strains used in this study also have sulB103 (33). sulB103 is an allele of ftsZ that makes the cells insensitive to the action of the sulA SOS cell division inhibitor (3). In all cases, the strains were grown in minimal media at 37°C to log phase and then assayed for the amount of fluorescence in individual cells. New methods for counting cells, detecting foci (when using RecA-GFP as a marker for RecA structures), and measuring the levels of GFP fluorescence have been employed in this work. See Materials and Methods for more details.
In this work, the level of SOS expression is reported in two ways: the average relative fluorescence intensity (RFI) and the percentage of the population having 9-fold (or 18-fold) or greater levels of expression than the average cell having no gfp (see Materials and Methods). The average RFI for a strain is the normalized pixel intensity that has been averaged for each pixel in a cell and then for all cells in the population. The RFI is similar to a bulk measurement of a culture. The 9-fold level was chosen due to results obtained previously from single-cell analysis (33). There it was shown that all individual cells in a recA-deleted strain had levels of fluorescence less than 6-fold above background. It was decided in that work that any cell having fluorescence 6-fold or greater above background would be considered to be induced for SOS expression (33). In this work, a more conservative 9-fold cutoff is used (and the extremely conservative 18-fold cutoff is also reported for comparison).
radA limits SOS expression in a recA4142 mutant.
Constitutive SOS expression in the recA4142 (F217Y) mutant is dependent upon ruvAB (28, 29). Since radA is partially redundant with ruvAB for UV survival and recombination (1), it is possible that radA may also be required for SOS expression in a recA4142 mutant and that the deletion of radA should lower SOS expression. To test this, a strain with recA4142 and a deletion of radA was produced and the resulting double mutant was measured for SOS expression. Surprisingly, the radA recA4142 double mutant showed a large increase in SOS expression relative to either single mutant (compare SS9024 [RFI of 120.8] with SS7102 [RFI of 2.9] and SS9023 [RFI of 15.9] in Table 1; the P values for both are <10−5). This result suggested that, instead of RadA being required for SOS expression in the recA4142 mutant, it may limit or repress SOS constitutive expression.
Table 1.
Effects of radA mutations on SOS expression in a recA4142 mutantc
| Strain | Versiond of: |
RFIa | % >9-fold (18-fold)b | Cells counted | |
|---|---|---|---|---|---|
| recA | radA | ||||
| SS996 | + | + | 1.9 | 1.6 (0.7) | 2,140 |
| SS7102 | + | del | 2.9 | 4.5 (1.0) | 1,254 |
| SS9023 | 4142 | + | 15.9 | 24.7 (9.0) | 1,737 |
| SS9024 | 4142 | del | 120.8 | 82.9 (74.2) | 1,571 |
| SS8253 | + | op | 2.8 | 3.4 (0.2) | 1,761 |
| SS8254 | 4142 | op | 4.6 | 12.4 (1.3) | 2,041 |
| SS6156 | o1403 4142 | + | 102.4 | 100 (99.4) | 787 |
| SS8272 | o1403 4142 | op | 27.6 | 99.9 (17.9) | 918 |
RFI, average relative fluorescence intensity (similar to bulk measurement).
% >9-fold (18-fold), percentage of cells with SOS expression 9-fold or 18-fold above background.
The statistical measure of significance for the data was determined by the Student t test. P values are given in the text, and values of 0.05 or lower are considered significant.
+, wild type; 4142, recA4142 mutant; o1403 4142, recA4142 augmented by a recAo1403 mutation; del, deletion; op, overproducer.
Overproduction of RadA decreases SOS expression in a recA4142 mutant but has no effect on SOS induction after UV treatment.
Given that radA is limiting for SOS expression in a recA4142 mutant, overexpression of radA could decrease SOS expression in this background. To test this, an overexpression mutant of radA was constructed on the chromosome by placing a strong constitutive promoter and optimized ribosome binding site in front of the radA gene (Fig. 1) (47, 56, 63). The construct changes the GTG start codon to an ATG start codon. This overexpression (radAop) mutation was combined with the recA4142 mutation, and a significant decrease in SOS expression was observed relative to that for the recA4142 mutant alone (compare SS9023 [RFI of 15.9] with SS8254 [RFI of 4.6] in Table 1; P value was <10−5). RadA overproduction also decreased SOS constitutive expression 4-fold when recA4142 was augmented by a recAo1403 mutation (compare SS6156 [RFI of 102.4] with SS8272 [RFI of 27.6] in Table 1; P value was <10−5).
Fig 1.

Sequence of DNA that has been added in front of the radA gene to increase its level of transcription and translation. Spaces in the sequence are placed there to separate functional sequences of DNA that are described below or above the sequence. The only omitted sequence is that of the cat gene and is denoted by the multiple dots. The promoter was modeled on the sequence of the recA promoter and 5′ untranslated region. Deviations from the recA sequence to remove SOS regulation and to improve the ribosome binding site are denoted in lowercase letters. The allele numbers of the operator mutations that remove LexA regulation are given below the line. The sequences for −10 and −35 boxes are underlined, and the transcriptional start site is denoted by an asterisk. The construction was verified by DNA sequencing.
Certain recA alleles [i.e., recA4162 (I298V)] can suppress SOS constitutive expression caused by recA4142 in cis or in trans (30). However, this suppression is very specific. While recA4142,4162 double mutants show low levels of SOS constitutive expression, they show UV-induced SOS like the wild type (30). It was therefore of interest to test if RadA overproduction inhibited all SOS expression or just SOS constitutive expression in log-phase cells. To test this, SS8254 was treated with UV irradiation and measured for SOS expression. Table 2 shows that this strain induced SOS expression 6-fold after UV treatment (compare SS8254 [RFI of 4.6] in Table 1 to SS8254 in Table 2 [RFI of 32.6]; P value was <10−5), and this strain behaved similarly to the wild type.
Table 2.
Summary of phenotypes in the radA overexpression strain relative to a wild-type straina
| Strain | Versionc of: |
UV survival | Amt of SOS expression |
Rel. recb | ||
|---|---|---|---|---|---|---|
| recA | radA | RFI | % >9-fold (18-fold) | |||
| SS996 | + | + | 0.91 ± 0.3 | 32.8 | 98.2 (36.4) | 1.00 ± 0.2 |
| SS8253 | + | op | 0.92 ± 0.4 | 31.2 | 97.2 (32.3) | 1.12 ± 0.3 |
| SS9023 | 4142 | + | NDd | 34.8 | 99.3 (41.3) | ND |
| SS8254 | 4142 | op | ND | 32.6 | 98.9 (31.3) | ND |
Data are the averages of three experiments or the counting of 800 to 1,000 cells. For all three experiments, the cells were grown in minimal media at 37°C into log phase before the treatment. The UV survival reported is survival after 20 J/m2 of irradiation. For the SOS test, cells were irradiated at 10 J/m2, imaged after 1.5 h of incubation at 37°C in the dark, and quantified as described in Materials and Methods.
Rel. rec., relative recombination. Recombination was measured by inheritance of a marker after P1 transduction. An equal titer of P1 lysate (multiplicity of infection of 0.1) was used for each strain.
Abbreviations are as for Table 1.
ND, not determined.
From these experiments it is concluded that radA can suppress SOS constitutive expression in a recA4142 mutant. This ability is proportional to the amount of RadA in the cell, is specific for SOS constitutive expression in log-phase cells, and has no detectable effect on SOS expression after UV treatment.
radA, recX, and the amount of RecA each contribute to limiting SOS expression in an xthA mutant.
The goal of this work was to test the idea that there may be a mechanism(s) repressing SOS expression at RecA filaments in wild-type log-phase cells. The experiment above shows that removal of radA can increase SOS constitutive expression in a recA4142 mutant. It is also known that in a recA4142 mutant, recX (deletion) and recAo1403 mutations increase SOS constitutive expression 3-fold and 10-fold, respectively (29). Thus, using recA4142 as a guide, it is possible that radA, recX, and the concentration of monomeric RecA in the cell could repress SOS expression in wild-type log-phase cells at the level of filament stability. A priori, these could function either in the same pathway or in different pathways. It is also known that xthA (exonuclease III) mutants have about 3-fold-higher SOS expression than a recA4142 mutant. This, however, is thought to occur because they have 3-fold more RecA loading events (6, 29), not because XthA somehow affects the RecA filament. To test if recX, radA, and/or the level of recA transcription was limiting SOS expression in an xthA mutant (where wild-type RecA formed the filament and not RecA4142), recX, radA, and recAo1403 mutations were combined with an xthA mutation.
The RFI values in Table 3 show that the deletion of radA from or the addition of recAo1403 to the xthA mutant led to small, but not significant, increases relative to the value for xthA alone (compare SS4857 [RFI of 3.6] with SS9040 [RFI of 3.9] and SS7118 [RFI of 4.7] in Table 3; P values were 0.9 and 0.06, respectively). Deletion of recX in an xthA mutant, however, led to a slightly larger and significant increase in SOS expression (compare SS4857 [RFI of 3.6] with SS9041 [RFI of 4.8] in Table 3; P values are <0.001). It is concluded that, as a single mutation, only the recX mutation (and not the radA or recAo1403 mutation) leads to a small increase in SOS expression in the xthA strain.
Table 3.
Effects of radA, recX, and recAo1403 mutations on SOS expression in log-phase xthA cellsa
| Strain | Version of: |
RFI | % >9-fold (18-fold) | Cells counted | |||
|---|---|---|---|---|---|---|---|
| recAo | radA | recX | xthA | ||||
| SS996 | + | + | + | + | 1.9 | 1.6 (0.7) | 2,140 |
| SS4857 | + | + | + | del | 3.6 | 9.5 (2.0) | 1,582 |
| SS9040 | 1403 | + | + | del | 3.9 | 10.2 (2.4) | 945 |
| SS7118 | + | del | + | del | 4.7 | 19.0 (3.4) | 1,343 |
| SS9041 | + | + | cat | del | 4.8 | 16.7 (3.3) | 1,291 |
| SS7132 | + | del | cat | del | 10.7 | 71.0 (31.4) | 2,409 |
| SS7128 | 1403 | del | + | del | 6.7 | 32.4 (6.9) | 1,286 |
| SS9045 | 1403 | + | cat | del | 5.1 | 23.5 (5.5) | 779 |
| SS7129 | 1403 | del | cat | del | 19.8 | 91.3 (42.6) | 1,136 |
cat, chloramphenicol. Other abbreviations are as defined for Table 1.
Since the single mutations had very small, if any, effects on SOS expression, it was tested whether combinations of these three mutations (doubles or triples) could increase expression in an xthA mutant. Table 3 shows that the deletion of both radA and recX led to a significant increase (2-fold) relative to that for either single mutant (compare SS7132 [RFI of 10.7] with SS7118 [RFI of 4.7] and SS9041 [RFI of 4.8] in Table 3; P values are both <10−5). The combination of recAo1403 and deletion of radA also increased expression significantly from that for both single mutants (compare SS7128 [RFI of 6.7] with SS7118 [RFI of 4.7] and SS9040 [RFI of 3.9] in Table 3; P values are <10−3 and <10−5, respectively). The last double-mutant combination of recAo1403 and deletion of recX yielded the smallest significant increase relative to either single mutant (compare SS9045 [RFI of 5.1] with SS9041 [RFI of 4.8] and SS9040 [RFI of 3.9] in Table 3; P values are 0.03 and <10−5, respectively).
Finally, the combination of all three mutations was tested by the construction of the recAo1403 radA recX triple mutant in the xthA background. This yielded a large and significant increase (2- to 5-fold) relative to any of the three double mutants (compare SS7129 [RFI of 19.8] with SS7132 [RFI of 10.7], SS7128 [RFI of 6.7], and SS9045 [RFI of 5.1] in Table 3; P values were of <10−5 for all three).
It is concluded that radA, recX, and the level of recA transcription form three independent pathways for the repression of SOS expression in log-phase cells. However, the quantitative contributions of these pathways may be not equal or simply additive.
radA and the level of recA transcription (but not recX) each contribute to limiting SOS expression in wild-type cells.
In the above-described experiments, it is seen that radA, recX, and the level of recA transcription serve to repress SOS expression in an xthA mutant. It is possible that these three mechanisms also repress SOS expression in a wild-type cell. To test this idea, the same strategy as above was used. All single-, double-, and triple-mutant combinations were made in a wild-type strain (SS996). Addition of any single mutation to the wild-type strain led to a small, but not significant, increase in SOS expression (compare SS996 [RFI of 1.9] with SS6088 [RFI of 2.2], SS7102 [RFI of 2.9], or SS6080 [RFI of 2.9] in Table 4; P values are 0.2, 0.6, and 0.8, respectively). It is possible that to see a significant increase one has to make at least double-mutant combinations. When these combinations are made, it is seen that only the combination of recAo1403 and radA led to a significant (2- to 3-fold) increase relative to either single mutant (compare SS7136 [RFI of 6.6] with SS6088 [RFI of 2.2] or SS7102 [RFI of 2.9] in Table 4; P values were <10−5 for both). The radA recX double mutation led to an unexpected small, significant decrease relative to the radA single mutation (compare SS7152 [RFI of 2.4] with SS7102 [RFI of 2.9] in Table 3; P value was 10−3). A small and not significant decrease was seen relative to the recX single mutation (compare SS7152 [RFI of 2.4] with SS6080 [RFI of 2.9] in Table 4; P value was 0.2). The last double mutation, recAo1403 recX, led to a significant 2-fold decrease relative to either the recAo1403 or recX single mutation (compare SS7155 [RFI of 1.2] with SS6088 [RFI of 2.2] or SS6080 [RFI of 2.9] in Table 4; P values were <10−5 for both). Lastly, the recAo1403 recX radA triple mutation led to a significant increase relative to two of the double mutations (compare SS5841 [RFI of 5.8] with SS7152 [RFI of 1.2] or SS7155 [RFI of 1.2] in Table 3; both P values are <10−5). Comparison of the triple mutant with the recAo1403 radA mutant (the one showing the greatest effect on expression) revealed a small but significant decrease (compare SS5841 [RFI of 5.8] with SS7136 [RFI of 6.6] in Table 4; P value is <10−3).
These results suggest that RadA and the level of recA transcription provide two independent and additive pathways for repressing SOS expression in wild-type cells. RecX does not play a significant role in repression of SOS expression in wild-type cells, as it does in the xthA mutant. However, RecX may play a role in helping to provide SOS expression in some situations in wild-type cells.
radA alone and the radA recX double mutation do not affect the number of RecA-GFP foci.
Since the goal of this work is to test the hypothesis that there are factors that repress the ability of RecA filaments to induce the SOS response in log-phase cells, it is necessary to test if these same mutations affect the number of RecA loading events (as measured with RecA-GFP foci). Therefore, whether removing both radA and recX affects the number of RecA-GFP foci in xthA mutant and wild-type strains was tested. Previous work has shown that when wild-type cells are grown in minimal media the removal of recX slightly increases (20%) the number of RecA-GFP foci per unit area of cell. This increase, however, was not significant (46). Note that all recA-gfp strains are additionally recAo1403.
Table 5 shows that in a log-phase population about 25% of the cells have at least one RecA-GFP focus. This number is about 2-fold higher than those in previous reports (45) and is likely due to improved methods of image acquisition and analysis (See Materials and Methods). Table 5 shows that, upon the removal of recX, there is an approximately 30% increase in RecA-GFP foci per unit area of cell. This is barely significantly different from the wild-type value (P = 0.03). Removal of radA alone, however, produced a smaller, approximately 20% increase, in the number of foci per unit area of the cell. This was not significantly different from the wild-type value (P = 0.2). The distribution of RecA-GFP foci in the radA recX double mutant was also not significantly different from that for either of the single mutants or wild type (Table 5). Therefore, it is concluded that removal of radA alone or radA and recX has no significant effect on the number of RecA-GFP foci per unit area of the cell.
Table 5.
Effects of radA, recX, and xthA mutations on RecA-GFP focus formationa
| Strain | Versionb of: |
Foci/unit area | % of cells with: |
Cells counted | Foci counted | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| radA | recX | xthA | 0 foci | 1 focus | 2 foci | 3 or more foci | ||||
| SS3085 | + | + | + | 0.44 | 75.9 | 16.5 | 5.9 | 1.7 | 990 | 257 |
| SS4560 | + | + | del | 1.52 | 33.6 | 40.8 | 26.4 | 5.4 | 979 | 959 |
| SS2647 | + | del | + | 0.63 | 64.2 | 26.4 | 7.0 | 2.4 | 1,266 | 623 |
| SS7261 | del | + | + | 0.57 | 66.4 | 23.5 | 7.1 | 3.0 | 1,221 | 578 |
| SS9043 | del | del | + | 0.51 | 74.6 | 18.6 | 6.0 | 2.7 | 1,061 | 336 |
| SS9048 | del | del | del | 1.93 | 16.7 | 43.8 | 29.6 | 9.9 | 635 | 857 |
The statistical measure of significance for the data was determined by a chi-square test of homogeneity for an r × c contingency table (37). The single mutants were compared to the wild type, and the double and triple mutants were compared to the corresponding single or double mutant. P values of 0.05 or lower are considered significant. P values are given in the text.
Abbreviations are as defined for Table 1.
Table 5 further shows that removal of both radA and recX in an xthA mutant increases the number of foci per unit area of the cell about 20% (compare SS4560 with SS9048; P < 0.001). It is concluded that, in an xthA mutant, removing both radA and recX can increase the number of RecA-GFP foci in a small but significant way.
The percentage of cells in a population of recX radA (with or without xthA) mutants with RecA-GFP foci correlates with the percentage of cells with elevated levels of SOS expression.
The goal of this work was to test the hypothesis that there are specific mechanisms that repress SOS expression when RecA forms a filament to repair housekeeping types of DNA damage. If those mechanisms are removed, then the percentage of cells with a RecA structure (i.e., with RecA-GFP foci) should be approximately equal to the number of cells with high levels of SOS expression. Three tentative mechanisms that involve radA, recX, and the level of recA transcription have been identified above.
Inspection of the data in Tables 3, 4, and 5 allows one to test this hypothesis. Comparison of the wild-type cases reveals that 25% of cells have RecA-GFP foci (SS3085 in Table 5) and about 1.6% of cells have high levels of SOS expression (SS996 in Table 3). Similarly, removing xthA increases the percentage of RecA-GFP foci to 83.3% and increases the percentage of cells with high levels of SOS expression to 9.5%. In each case (xthA+ and xthA mutant) removal of radA and recX and the addition of recAo1403 allow the number of foci to remain unchanged while increasing the percentages of cells with high levels of SOS expression to 24.2% (SS5841 in Table 4) and 91.2% (SS7129 in Table 3). This supports the idea that the increase in SOS expression due to adding recAo1403 and deleting recX and radA is due to a release of repression at the existing RecA filaments.
Overproduction of RadA has no effect on UV survival, recombination, or SOS induction in a wild-type cell.
The data above show that normal levels of RadA and overproduction of RadA could inhibit SOS expression in recA4142 cells but had no effect on SOS expression after UV treatment. The ability of radAop to inhibit SOS expression may be specific to recA4142 cells or may extend to recA+ cells. Since it is also known that overexpression of recX inhibits several of RecA's abilities both in vivo and in vitro (54), it was of interest to test if RadA overproduction would have any negative effect on RecA function in a recA+ strain. Table 1 shows that overproduction of RadA did not significantly change SOS expression in log-phase cells relative to that in the wild type (compare SS996 [RFI of 1.9] with SS8253 [RFI of 2.8] in Table 1; P value was 0.3). Table 2 shows that overproduction of RadA does not significantly change the ability to survive UV irradiation, undergo recombination (as measured by P1 transduction), or induce SOS expression after UV irradiation relative to a wild-type strain (SS996). We conclude that overexpression of radA has no detectable effect on these recA phenotypes.
DISCUSSION
The mechanism of induction of the SOS response is thought to proceed by RecA binding to ssDNA produced as a consequence of DNA damage, which in turn leads to the assembly of a RecA-DNA filament. The LexA protein can then interact with the RecA-DNA filament and increase its rate of autoproteolysis. This reduces the amount of LexA in the cell binding to various promoters such that RNA polymerase can transcribe a set of genes that can help to repair DNA, mutagenize DNA, inhibit cell division, and perform other functions yet to be discovered (there are still many SOS genes of unknown function). SOS mutagenesis has often been thought of as a “last resort” tactic useful to a population of cells trying to survive some external insult. It is likely detrimental to the individual as increases in mutation frequency are more likely to inactivate important genes rather than mutate genes in favorable ways. Thus, it seems prudent that cells would have a mechanism to prohibit SOS induction when RecA is used to repair housekeeping types of DNA damage and would induce SOS only under the most dire of circumstances. This work shows that the absence of radA and the presence of slightly higher levels of recA transcription independently can lead to higher SOS expression in an otherwise wild-type strain. In an xthA mutant, where there are about 3-fold more RecA loading events, maximal levels of SOS expression are achieved if recX is additionally deleted. The effects of radA, recX, and recAo1403 on SOS expression appear independent. Thus, it is hypothesized that SOS expression in log-phase cells where RecA has formed filaments on the DNA is repressed by three mechanisms that work in parallel to minimize the half-life of RecA filaments (Fig. 2). A shorter half-life would lead to higher LexA levels in the cell, whereas a longer half-life would allow more time for RecA to interact with LexA and decrease its level in the cell. It is also possible that some character of the RecA filament changes in some way to allow SOS expression. Possibilities for these character traits may include length of the filament and/or the continuous (or discontinuous) nature of the filament (43, 58). Other characteristics are also possible (35). A more detailed hypothesis for the contributions of each of the three mechanisms follows.
Fig 2.
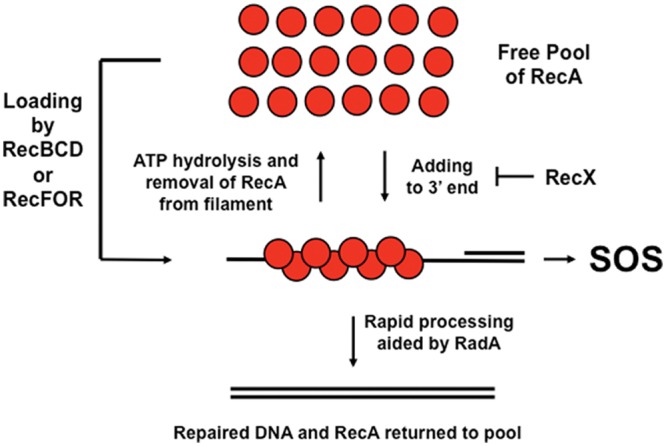
Summary of a model to explain how the three independent mechanisms serve to limit SOS expression in log-phase cells that have RecA-DNA filaments in the absence of external DNA damage. The model proposes that RecA is loaded by either RecBCD or RecFOR depending on the DNA substrate (double-strand end or single-strand gap, respectively). Once loaded, the RecA filament can grow in the 5′-to-3′ direction and lose monomers from the 5′ end through ATP hydrolysis. The half-life of the RecA-DNA filament can be prolonged by increasing the concentration of RecA in the cell, thus increasing the rate at which RecA adds to the 3′ end. RecX can decrease the half-life of the RecA filament by inhibiting 3′ addition. The filament will shorten since ATP hydrolysis will still remove RecA from the 5′ end. Lastly, RadA can decrease the half-life of the filament by processing it toward repaired DNA. It should be noted that other proteins that can also affect the half-life of RecA filaments, such as DinI and UvrD, are not depicted here. The red circles are RecA, and the solid lines are indicative of DNA.
Given that previous studies on RadA revealed a synergistic activity with RuvAB and RecG in processing postsynaptic recombination structures, it is possible that the role of RadA in repression of SOS in log-phase cells is the rapid processing of RecA-bound recombinational intermediates. If the repair of DNA occurs quickly, then the half-life of RecA filaments that can interact with LexA will be short and little if any LexA will be cleaved. Other models for how RadA might repress SOS expression at RecA filaments in log-phase cells are also possible. One suggests that RadA could interact directly with RecA filaments in the groove and compete with LexA binding, preventing the increase in the rate of autoproteolysis (44). Another model suggests that RadA could repress SOS by degrading the RecA protein in the filament with its Lon protease-like domain (1).
The second mechanism is based on the observations that recAo1403 and the deletion of radA increase SOS expression. As mentioned above, RecA filament growth is dynamic, occurring at both ends, with a net increase and decrease to the 3′ and 5′ ends, respectively. The decrease, or RecA dissociation from the filament, occurs when ATP is hydrolyzed (50). Therefore, if the ATP hydrolysis rate is constant, increasing the rate of addition at the 3′ end by increasing the RecA concentration should increase the length and half-life of the RecA filament. Supporting this model is the observation that about 25% of a population of cells containing the recA730,2201 mutant, which cannot hydrolyze ATP but still can bind ATP and ssDNA, have elevated levels of SOS expression (20).
Third, RecX has been shown to inhibit filament extension at the 3′ end, presumably by binding and physically blocking RecA from adding to that end (4, 19, 25; reviewed in reference 9). The overall effect of RecX is then to destabilize the filaments since the RecA-ATP hydrolysis rate remains unchanged. Therefore, removal of RecX could also increase the length and half-life of RecA filaments in cells where the RecA ATP-hydrolysis rate remains constant. It has also been shown that RecX can bind in the middle of the RecA filament and presumably cause discontinuities (43, 58).
A complicating issue with the use of the recAo1403 mutation is that it not only increases RecA expression but also increases recX expression since recA and recX are transcribed from the same promoter. It is known that the amount of transcription and thus expression of RecX is much lower than those of RecA because there is significant Rho-independent termination between the two genes (38). Thus, the balance between RecA and RecX in the cell is of great importance when considering the half-life of RecA-DNA filaments.
Are the contributions of the three factors, radA, recX, and maintaining the amount of RecA below a critical value, completely independent and additive in their abilities to repress SOS expression at RecA filaments? If this were absolutely true, then one would expect to see one-third of the full increase in SOS expression seen in the triple mutant in each of the single mutants and two-thirds of the full amount in the double mutants. This is not seen at all in the single mutants and in two out of three double mutants in the xthA background. Most of the single and double mutants show fairly low, equal levels of expression. This suggests that the mechanisms are independent and that one mechanism is mostly adequate for repression but that the absence of two mechanisms (depending on which ones) is not. There are, however, two notable exceptions: the large increase in the radA recX double mutant (xthA background) (Table 3, SS7132) and the decrease in expression of the recAo1403 recX xthA mutant (Table 4, SS7155). The reasons for these departures are not yet clear.
In the xthA+ strains, there is the question of why recX appears to have no contribution. One idea to explain why removal of recX is needed in the xthA mutant cells for maximal SOS expression (but not in the xthA+ cells) is suggested by the fact that xthA mutants have more RecA loading events. Assuming that the RecA concentrations in xthA+ and xthA mutant cells are equal, then the RecA filaments in xthA mutants are likely to have a shorter half-life or length (because the concentration of RecA remains constant), so the removal of recX is needed to allow for sufficient increase in the half-life of the RecA filament. It is also possible that the three factors may not be completely equal and independent in all types of mutants. Here it is proposed that the abilities of RadA may be more important than those of RecX or maintenance of the concentration of RecA.
If these mechanisms serve to repress SOS expression in log-phase cells, then how does the cell overcome these when SOS is required? For this discussion, two examples will be considered. The first is SOS induction after UV irradiation. It requires DNA replication and RecFOR function (22, 48, 61). The second is a double-stand break caused by I-SceI cleavage at an I-SceI site, mediated by RecBCD (34, 39). In each case, either multiple or repeated RecA-DNA filaments that are more extensive, longer-lived recombinational repair structures that eventually and assuredly lead to SOS expression may be formed. Under conditions of UV irradiation, both forks are likely to encounter lesions in the DNA multiple times before the DNA damage is cleared. The gaps that are produced will lead to daughter strand gap repair, producing many recombination structures. Depending on the efficiency of nucleotide excision repair, the forks may repetitively encounter DNA damage, reloading RecA each time, lengthening the time that the RecA filament can interact with LexA and drive down the LexA concentration in the cell. In the I-SceI case, there is only one site per chromosome. Once the DNA is cut, there are two double-strand ends that RecBCD can then use to load RecA. The only homologous sequences, however, that might be available for repair will be found in the sequestered sister nucleoid (and only if it has not yet been cut by I-SceI). Thus, it is likely that the RecA filaments will spend a long time searching (possibly nonproductively) for homologous sequences and will be available for LexA cleavage.
The data gathered in this work reflect SOS expression and the number of RecA structures in individual cells. It must also be considered that the data are static pictures of dynamic situations. One cannot tell in any one cell if the SOS levels are increasing or decreasing or if RecA structures are being built or taken apart. Presumably across the population both are occurring. One would not expect to see maximal levels of SOS expression (about 200-fold above background for a LexA-defective mutant) because one would expect that the lifetime of the RecA-DNA filament would be short, since RecA would quickly repair the DNA damage so that the cell could resume normal DNA replication and growth as soon as possible. Attaining the maximal level of SOS expression is likely only if RecA filaments were found in all cells, all the time, and the rate of LexA autoproteolysis was greater than its rate of production.
For the multiply mutant strains, there is now reasonable agreement between the percentage of cells with RecA-GFP foci and the percentage of cells with elevated levels of SOS expression if the 9-fold cutoff is used (24% and 91% for the wild type and xthA mutant for SOS, respectively, and 33% and 83% with and without xthA for RecA-GFP foci, respectively). If, however, the extremely conservative 18-fold cutoff is used, the percentage of cells with elevated levels of SOS expression decreases 2- to 3-fold to 8% and 42% of the population of the wild type and xthA mutant, respectively. These numbers are less correlative but show the same trends.
It is possible that the reason why recAo1403 recX radA mutants in either wild-type or xthA mutant strains have higher levels of SOS is the presence of more DNA damage. This seems unlikely for two reasons. First, the single mutants show no significant increase in SOS in either case. This is also true for the recX radA double mutant in the wild-type background. Second, if the presence of RecA-GFP foci is indicative of RecA loading at DNA damage, then one also does not see a significant increase in the number of RecA-GFP foci in the radA and radA recX strains (Table 5).
The work began with the finding that SOS expression in the recA4142 mutant increased when radA was deleted and decreased when radA was overexpressed. It has also been shown that the recA4142 mutant shows an increase in SOS expression with recAo1403 or the deletion of recX or xthA individually (29). This is not seen with RecA+. One way to explain this is to hypothesize that some property of the RecA4142 protein, possibly its high degree of cooperativeness in forming a filament (13), is able to poise the RecA4142 filament for a longer half-life (or length) such that only one of the other mutations is needed for a measurable increase in SOS expression. Since the RecA+ filament has a lower degree of cooperativeness, the release of other repression mechanisms is necessary for a measurable increase in SOS expression.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by AI059027 from the National Institutes of Health.
We thank Kevin Griffith for reading the manuscript before publication and offering suggestions. We especially thank Quanli Wang for assistance with the thresholding program and Oleskii Sliusarenko for assistance with the MicrobeTracker program.
Footnotes
Published ahead of print 27 July 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Beam CE, Saveson CJ, Lovett ST. 2002. Role for radA/sms in recombination intermediate processing in Escherichia coli. J. Bacteriol. 184:6836–6844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bernard R, Marquis KA, Rudner DZ. 2010. Nucleoid occlusion prevents cell division during replication fork arrest in Bacillus subtilis. Mol. Microbiol. 78:866–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bi E, Lutkenhaus J. 1990. Analysis of ftsZ mutations that confer resistance to the cell division inhibitor SulA (SfiA). J. Bacteriol. 172:5602–5609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bork JM, Cox MM, Inman RB. 2001. RecA protein filaments disassemble in the 5′ to 3′ direction on single-stranded DNA. J. Biol. Chem. 276:45740–45743 [DOI] [PubMed] [Google Scholar]
- 5. Brendel V, Brocchieri L, Sandler SJ, Clark AJ, Karlin S. 1997. Evolutionary comparisons of RecA-like proteins across all major kingdoms of living organisms. J. Mol. Evol. 44:528–541 [DOI] [PubMed] [Google Scholar]
- 6. Centore RC, Lestini R, Sandler SJ. 2008. XthA (exonuclease III) regulates loading of RecA onto DNA substrates in log phase Escherichia coli cells. Mol. Microbiol. 67:88–101 [DOI] [PubMed] [Google Scholar]
- 7. Cormack BP, Valdivia RH, Falkow S. 1996. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173:33–38 [DOI] [PubMed] [Google Scholar]
- 8. Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cox MM. 2007. Motoring along with the bacterial RecA protein. Nat. Rev. Mol. Cell Biol. 8:127–138 [DOI] [PubMed] [Google Scholar]
- 10. Cox MM. 2007. Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 42:41–63 [DOI] [PubMed] [Google Scholar]
- 11. Cox MM, et al. 2000. The importance of repairing stalled replication forks. Nature 404:37–41 [DOI] [PubMed] [Google Scholar]
- 12. Cromie GA, Leach DRF. 2000. Control of crossing over. Mol. Cell 6:815–826 [DOI] [PubMed] [Google Scholar]
- 13. De Zutter JK, Forget AL, Logan KM, Knight KL. 2001. Phe217 regulates the transfer of allosteric information across the subunit interface of the RecA protein filament. Structure 9:47–55 [DOI] [PubMed] [Google Scholar]
- 14. Diver WP, Sargentini NJ, Smith KC. 1982. A mutation (radA100) in Escherichia coli that selectively sensitizes cells grown in rich medium to X- or u.v.-radiation, or methyl methanesulphonate. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 42:339–346 [DOI] [PubMed] [Google Scholar]
- 15. Drees JC, Lusetti SL, Chitteni-Pattu S, Inman RB, Cox MM. 2004. A RecA filament capping mechanism for RecX protein. Mol. Cell 15:789–798 [DOI] [PubMed] [Google Scholar]
- 16. Drees JC, Lusetti SL, Cox MM. 2004. Inhibition of RecA protein by the Escherichia coli RecX protein: modulation by the RecA C-terminus and filament functional state. J. Biol. Chem. 279:52991–52997 [DOI] [PubMed] [Google Scholar]
- 17. Erill I, Campoy S, Barbé J. 2007. Aeons of distress: an evolutionary perspective on the bacterial SOS response. FEMS Microbiol. Rev. 31:637–656 [DOI] [PubMed] [Google Scholar]
- 18. Friedberg EC, et al. 2006. DNA repair and mutagenesis. ASM Press, Washington, DC [Google Scholar]
- 19. Galletto R, Amitani I, Baskin RJ, Kowalczykowski SC. 2006. Direct observation of individual RecA filaments assembling on single DNA molecules. Nature 443:875–878 [DOI] [PubMed] [Google Scholar]
- 20. Gruenig MC, et al. 2008. RecA-mediated SOS induction requires an extended filament conformation but no ATP hydrolysis. Mol. Microbiol. 69:1165–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harmon FG, Rehrauer WM, Kowalczykowski SC. 1996. Interaction of Escherichia coli RecA protein with LexA repressor. II. Inhibition of DNA strand exchange by the uncleavable LexA S119A repressor argues that recombination and SOS induction are competitive processes. J. Biol. Chem. 271:23874–23883 [PubMed] [Google Scholar]
- 22. Hegde S, Sandler SJ, Clark AJ, Madiraju MV. 1995. recO and recR mutations delay induction of the SOS response in Escherichia coli. Mol. Gen. Genet. 246:254–258 [DOI] [PubMed] [Google Scholar]
- 23. Kowalczykowski SC. 2000. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 25:156–165 [DOI] [PubMed] [Google Scholar]
- 24. Krejci L, et al. 2004. Role of ATP hydrolysis in the antirecombinase function of Saccharomyces cerevisiae Srs2 protein. J. Biol. Chem. 279:23193–23199 [DOI] [PubMed] [Google Scholar]
- 25. Lindsley JE, Cox MM. 1990. Assembly and disassembly of RecA protein filaments occur at opposite filament ends. Relationship to DNA strand exchange. J. Biol. Chem. 265:9043–9054 [PubMed] [Google Scholar]
- 26. Little JW, Edmiston SH, Pacelli LZ, Mount DW. 1980. Cleavage of the Escherichia coli lexA protein by the recA protease. Proc. Natl. Acad. Sci. U. S. A. 77:3225–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Little JW, Mount DW. 1982. The SOS regulatory system of Escherichia coli. Cell 29:11–22 [DOI] [PubMed] [Google Scholar]
- 28. Long JE, Massoni SC, Sandler SJ. 2010. RecA4142 causes SOS constitutive expression by loading onto reversed replication forks in Escherichia coli K-12. J. Bacteriol. 192:2575–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Long JE, Renzette N, Centore RC, Sandler SJ. 2008. Differential requirements of two recA mutants for constitutive SOS expression in Escherichia coli K-12. PLoS One 3:e4100 doi:10.1371/journal.pone.0004100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Long JE, Renzette N, Sandler SJ. 2009. Suppression of constitutive SOS expression by recA4162 (I298V) and recA4164 (L126V) requires UvrD and RecX in Escherichia coli K-12. Mol. Microbiol. 73:226–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lovett ST. 2006. Replication arrest-stimulated recombination: dependence on the RecA paralog, RadA/Sms and translesion polymerase, DinB. DNA Repair (Amst.) 5:1421–1427 [DOI] [PubMed] [Google Scholar]
- 32. Lusetti SL, Cox MM. 2002. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu. Rev. Biochem. 71:71–100 [DOI] [PubMed] [Google Scholar]
- 33. McCool JD, et al. 2004. Measurement of SOS expression in individual Escherichia coli K-12 cells using fluorescence microscopy. Mol. Microbiol. 53:1343–1357 [DOI] [PubMed] [Google Scholar]
- 34. Meddows TR, Savory AP, Grove JI, Moore T, Lloyd RG. 2005. RecN protein and transcription factor DksA combine to promote faithful recombinational repair of DNA double-strand breaks. Mol. Microbiol. 57:97–110 [DOI] [PubMed] [Google Scholar]
- 35. Menetski JP, Bear DG, Kowalczykowski SC. 1990. Stable DNA heteroduplex formation catalyzed by the Escherichia coli RecA protein in the absence of ATP hydrolysis. Proc. Natl. Acad. Sci. U. S. A. 87:21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mount DW. 1977. A mutant of Escherichia coli showing constitutive expression of the lysogenic induction and error-prone DNA repair pathways. Proc. Natl. Acad. Sci. U. S. A. 74:300–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ott L. 1988. An introduction to statistical methods and data analysis, 3rd ed PWS-Kent, Boston, MA [Google Scholar]
- 38. Pages V, Koffel-Schwartz N, Fuchs RP. 2003. recX, a new SOS gene that is co-transcribed with the recA gene in Escherichia coli. DNA Repair (Amst.) 2:273–284 [DOI] [PubMed] [Google Scholar]
- 39. Pennington JM, Rosenberg SM. 2007. Spontaneous DNA breakage in single living Escherichia coli cells. Nat. Genet. 39:797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Persky NS, Lovett ST. 2008. Mechanisms of recombination: lessons from E. coli. Crit. Rev. Biochem. Mol. Biol. 43:347–370 [DOI] [PubMed] [Google Scholar]
- 41. Petit MA, Ehrlich D. 2002. Essential bacterial helicases that counteract the toxicity of recombination proteins. EMBO J. 21:3137–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Phizicky EM, Roberts JW. 1981. Induction of SOS functions: regulation of proteolytic activity of E. coli RecA protein by interaction with DNA and nucleoside triphosphate. Cell 25:259–267 [DOI] [PubMed] [Google Scholar]
- 43. Ragone S, Maman JD, Furnham N, Pellegrini L. 2008. Structural basis for inhibition of homologous recombination by the RecX protein. EMBO J. 27:2259–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rehrauer WM, Lavery PE, Palmer EL, Singh RN, Kowalczykowski SC. 1996. Interaction of Escherichia coli RecA protein with LexA repressor. I. LexA repressor cleavage is competitive with binding of a secondary DNA molecule. J. Biol. Chem. 271:23865–23873 [PubMed] [Google Scholar]
- 45. Renzette N, et al. 2005. Localization of RecA in Escherichia coli K-12 using RecA-GFP. Mol. Microbiol. 57:1074–1085 [DOI] [PubMed] [Google Scholar]
- 46. Renzette N, Gumlaw N, Sandler SJ. 2007. DinI and RecX modulate RecA-DNA structures in Escherichia coli K-12. Mol. Microbiol. 63:103–115 [DOI] [PubMed] [Google Scholar]
- 47. Sandler SJ, Clark AJ. 1990. Factors affecting expression of the recF gene of Escherichia coli K-12. Gene 86:35–43 [DOI] [PubMed] [Google Scholar]
- 48. Sassanfar M, Roberts JW. 1990. Nature of the SOS-inducing signal in Escherichia coli. The involvement of DNA replication. J. Mol. Biol. 212:79–96 [DOI] [PubMed] [Google Scholar]
- 49. Seitz EM, Brockman JP, Sandler SJ, Clark AJ, Kowalczykowski SC. 1998. RadA protein is an archaeal RecA protein homolog that catalyzes DNA strand exchange. Genes Dev. 12:1248–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shan Q, Bork JM, Webb BL, Inman RB, Cox MM. 1997. RecA protein filaments: end-dependent dissociation from ssDNA and stabilization by RecO and RecR proteins. J. Mol. Biol. 265:519–540 [DOI] [PubMed] [Google Scholar]
- 51. Shinohara A, Ogawa T. 1999. Rad51/RecA protein families and the associated proteins in eukaryotes. Mutat. Res. 435:13–21 [DOI] [PubMed] [Google Scholar]
- 52. Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C. 2011. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol. Microbiol. 80:612–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steiner WW, Kuempel PL. 1998. Sister chromatid exchange frequencies in Escherichia coli analyzed by recombination at the dif resolvase site. J. Bacteriol. 180:6269–6275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stohl EA, et al. 2003. Escherichia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. J. Biol. Chem. 278:2278–2285 [DOI] [PubMed] [Google Scholar]
- 55. Sutera VA, Lovett ST. 2006. The role of replication initiation control in promoting survival of replication fork damage. Mol. Microbiol. 60:229–239 [DOI] [PubMed] [Google Scholar]
- 56. Uhlin BE, Volkert MR, Clark AJ, Sancar A, Rupp WD. 1982. Nucleotide sequence of a recA operator mutation. LexA/operator-repressor binding/inducible repair. Mol. Gen. Genet. 185:251–254 [DOI] [PubMed] [Google Scholar]
- 57. van Gool AJ, Hajibagheri NM, Stasiak A, West SC. 1999. Assembly of the Escherichia coli RuvABC resolvasome directs the orientation of Holliday junction resolution. Genes Dev. 13:1861–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. VanLoock MS, et al. 2003. Complexes of RecA with LexA and RecX differentiate between active and inactive RecA nucleoprotein filaments. J. Mol. Biol. 333:345–354 [DOI] [PubMed] [Google Scholar]
- 59. Walker G. 1987. The SOS response of Escherichia coli, p 1346–1357 In Neidhardt FC, et al. (ed), Escherichia coli and Salmonella typhimurium, vol 2 American Society for Microbiology, Washington, DC [Google Scholar]
- 60. Wertman KF, Mount DW. 1985. Nucleotide sequence binding specificity of the LexA repressor of Escherichia coli K-12. J. Bacteriol. 163:376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Whitby MC, Lloyd RG. 1995. Altered SOS induction associated with mutations in recF, recO and recR. Mol. Gen. Genet. 246:174–179 [DOI] [PubMed] [Google Scholar]
- 62. Willetts NS, Clark AJ, Low B. 1969. Genetic location of certain mutations conferring recombination deficiency in Escherichia coli. J. Bacteriol. 97:244–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu X, Egelman EH. 1993. The LexA repressor binds within the deep helical groove of the activated RecA filament. J. Mol. Biol. 231:29–40 [DOI] [PubMed] [Google Scholar]
- 64. Yu X, Jacobs SA, West SC, Ogawa T, Egelman EH. 2001. Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc. Natl. Acad. Sci. U. S. A. 98:8419–8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zacharias DA, Violin JD, Newton AC, Tsien RY. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.