

Abstract
The transcription of amtB in Streptomyces coelicolor has been proposed to be counter-regulated by GlnR (a global regulator for nitrogen metabolism) and PhoP (a global regulator for phosphate metabolism). However, the GlnR-protected region, which was deduced to be two 22-bp GlnR binding boxes (gTnAc-n6-GaAAc-n6-GtnAC-n6-GAAAc-n6, abbreviated as a1-b1 and a2-b2), was separated from the PhoP-protected region in the promoter of amtB, leaving the mechanism for this regulation undefined. In this study, another 22-bp GlnR binding box, which consisted of a3-site-n6-b3-site (a3-b3) overlapping with the PhoP-binding sequences, was identified in the promoter region of amtB by a DNase I footprinting assay. An electrophoretic mobility shift assay (EMSA) using purified recombinant GlnR and the synthetic amtB promoter fragments with the three GlnR binding boxes individually mutated demonstrated that every box was involved in GlnR binding in vitro. Further in vivo assays using the egfp reporter gene fused to various kinds of mutated promoter regions of amtB demonstrated that all of the three GlnR binding boxes were required for GlnR-mediated activation of amtB transcription under the nitrogen-limited condition. The results of EMSA using the amtB promoter with mixtures of recombinant His-tagged GlnR and Trx-His-S-tagged PhoP inferred that PhoP might compete against GlnR from binding at the a3-b3 site, attributable to the PhoP/GlnR counter-regulatory function subjected to further experimental proof.
INTRODUCTION
The in vivo ratio of nitrogen versus phosphate affects bacterial growth and metabolism, including the biosynthesis of secondary metabolites (6), and the metabolism of nitrogen and phosphate is coordinated via complex regulatory networks (14). In Streptomyces coelicolor, nitrogen metabolism is globally regulated by an orphan response regulator, GlnR (22), while the expression of phosphate-regulated genes is controlled by the PhoR-PhoP two-component system (18). When phosphate is insufficient, the sensor kinase PhoR is self-phosphorylated and subsequently transfers the high-energy phosphate group to its cognate response regulator PhoP. The phosphorylated PhoP then binds to the PHO boxes, which are comprised of several 11-nucleotide (nt) direct repeat units (DRus) in the promoter regions of its target genes, and regulates their expression (20). Based on microarray data (13), a connection between the phosphate metabolism controlled by PhoP and the nitrogen metabolism regulated by GlnR was proposed. Rodriguez-Garcia et al. (14) subsequently proved that PhoP repressed the expression of both glnR and the GlnR target genes, including glnA, glnII, and the amount operon (amtB-glnK-glnD), through directly binding to the DRus in their promoters.
The proposed cis-element for GlnR binding is complex and is comprised of two GlnR binding boxes, each consisting of one a site and one b site separated by 6 nucleotides (i.e., a1-site-n6-b1-site-n6-a2-site-n6-b2-site-n6 [a1-b1 and a2-b2]) located upstream of most GlnR target genes (including glnA, glnII, and the amount operon) (22). However, for some target genes (e.g., nasA [23] and SCO5163 [8]), only two a sites are characterized in their promoters, separated by variable numbers of nucleotides.
To reveal the mechanisms for the cross talk between the regulations of phosphate and nitrogen coordinated by PhoP and GlnR, Rodriguez-Garcia et al. compared their DNA-binding sequences and found that the PhoP binding DNA sequences overlapped with the predicted GlnR binding boxes upstream of glnA and glnII but not amtB (14). It was therefore proposed that PhoP acted as a competitive repressor by preventing the binding of GlnR to the GlnR binding boxes of glnA and glnII, whereas a “roadblock” model was hypothesized for the regulation of amtB transcription, and PhoP was proposed to block the transcription of amtB initiated from the P2 and P3 sites (14). However, because the transcription from the P1 site, located downstream of the PhoP-binding sequences, contributed the majority of amtB mRNA when S. coelicolor grew under nitrogen-limited conditions (2), the major transcription of amtB was unlikely to be blocked by PhoP binding. Although we cannot exclude the possibility that the different media used in these two separate experiments (2, 14) might influence the transcriptional properties of amtB, the roadblock model apparently may not account for the significant derepression of the amtB promoter in the ΔphoP mutant (>5-fold after 48 h) (14), leaving the question of how PhoP acts to negatively regulate the expression of amount operon open for further investigation.
A DNase I footprinting assay was used here to identify the exact DNA sequences protected by S. coelicolor GlnR in the amtB promoter region and a new stretch of GlnR-binding sequences that not only covered the previously deduced two 22-bp GlnR binding boxes but also revealed a new GlnR binding box consisting of one a3 site and one b3 site (referred to herein as a3-b3). The functions of the three GlnR binding boxes were tested both in vitro and in vivo, and the repressive role of PhoP in regulation of amtB transcription through a plausible competitive binding mechanism was hypothesized.
MATERIALS AND METHODS
Bacterial strains, media, and primers.
All of the bacterial strains and plasmids are listed in Table 1. Escherichia coli DH5α was used for subcloning. S. coelicolor was grown at 30°C in either the nitrogen-rich S medium (11) or nitrogen-limited N-Evans medium (2) with 5 mM nitrate as the sole nitrogen source. When needed, aparamycin (50 μg/ml), kanamycin (50 μg/ml), thiostrepton (50 μg/ml), and ampicillin (100 μg/ml) were added to the media. All of the primers used in the present study are listed in Table 2.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype and/or descriptiona | Source or reference |
|---|---|---|
| Plasmids | ||
| pIJ8660 | Promoter probe vector containing the egfp gene as indicator; oriT RK2 attP aac(3)IV | 20 |
| pIJ8660-hrdBp | 896-bp PCR fragment of hrdB promoter region inserted into the KpnI-BamHI site of pIJ8660 | 16 |
| pIJ8660-amtBpWT | 500-bp PCR fragment of the wild-type amtB promoter region inserted into the BamHI-NdeI site of PIJ8660 | This study |
| pIJ8660-amtBp3 | a3-b3 site mutation within the amtB promoter region from PIJ8660-amtBpWT | This study |
| pIJ8660-amtBp1 | a1-b1 site mutation within the amtB promoter region from PIJ8660-amtBpWT | This study |
| pIJ8660-amtBp2 | a2-b2 site mutation within the amtB promoter region from PIJ8660-amtBpWT | This study |
| pET28a | Expression vector carrying N-terminal His tag; Kmr | Novagen |
| pEXSCR | pET28a carrying the S. coelicolor glnR gene | 23 |
| pET32a | Expression vector carrying the N-terminal Trx-His-S tag; Ampr | Novagen |
| pET32a-PhoP | pET32a carrying the S. coelicolor phoP gene | This study |
| Strains (S. coelicolor) | ||
| M145 | Wild type, SCP1− SCP2− | John Innes Centre |
| M145/pIJ8660 | M145 integrated with pIJ8660 | This study |
| SCamtB-WT | M145 integrated with pIJ8660-amtBpWT | This study |
| SCamtB-3 | M145 integrated with pIJ8660-amtBp3 | This study |
| SCamtB-1 | M145 integrated with pIJ8660-amtBp1 | This study |
| SCamtB-2 | M145 integrated with pIJ8660-amtBp2 | This study |
| M145/pIJ8660-hrdBp | M145 integrated with pIJ8660-hrdBp | 17 |
Ampr, ampicillin resistance; Kmr, kanamycin resistance.
Table 2.
Primers used in this study
| Type of analysis and primer | Sequence (5′–3′) |
|---|---|
| Construction of expression plasmid for PhoP expression in E. coli | |
| SCPhoPEX_F | AATGGATCCGTGCTCGTCGTCGAGGACGA |
| SCPhoPEX_R | AATGAATTCTACGGCTCGAACTTGTAAC |
| DNase I footprinting assay | |
| SCamtBFP(M13F) | GTAAAACGACGGCCAGTCCATGCCAGGTCATTCGGAG |
| SCamtBFP(M13R) | CAGGAAACAGCTATGACGGCGGAGCAGATGAGCATGA |
| M13F-FAM | GTAAAACGACGGCCAGT (5'-FAM labeled) |
| M13R-FAM | CAGGAAACAGCTATGAC (5'-FAM labeled) |
| RT-PCR | |
| hrdB-F | GAGTCCGTCTCTGTCATGGCG |
| hrdB-R | TCGTCCTCGTCGGACAGCACG |
| amtB-F | ATCCTCGTCATCGGCAAGC |
| amtB-R | TTGAAGCCGAACCAGCCGAA |
| egfp-F | GAAGAAGATGGTGCGCTCCT |
| egfp-R | GATGTTGCCGTCCTCCTTGA |
| Introducing mutations in amtB promoter for egfp fusion | |
| For wild-type amtBP | |
| amtBP-1 | CGGGATCCAACCCGAGGAGAGCACCGTG |
| amtBP-2 | GGGAATTCCATATGCGGCGTCTCCTCGTCGT |
| For a3-b3 site mutation | |
| amtBP-3 | CGGCCGGGGCCGGTCGTCGT |
| amtBP-4 | CCTGTCCACGCACGGTACGCACCGTGCCTTCGTCAC |
| For a1-b1 site mutation | |
| amtBP-6 | GAAGGCACGGTGCGTGTTAC |
| amtBP-7 | ACTGTGGCGGCAGGGTAACGAGGGGCTTCCACCGAA |
| For a2-b2 site mutation | |
| amtBP-8 | TCGTTGTTTCGCCGCCGTGA |
| amtBP-9 | GAAATCTCCACCAGGGTCGCGCTGCGTCAATGTCGT |
| Synthesizing mutants of amtB promoter by oligonucleotides for EMSA | |
| For the wild-type oWT | |
| oWTp1 | ACGACGACGACCGGCCCCGGCCGTTCACCCACGCGTAACACGCACCGTGCCTTC |
| oWTp2 | CACGACATTGACGCAGCGCGGTTTCGGTGGAAGCCCCTCGTTGTTTCGCCGCCGTGACGAAGGCACGGTGCGT |
| For oM3, mutagenesis at a3-b3 site | |
| oM3p1 | ACGACGACGACCGGCCCCGGCCGCCTGTCCACGCACGGTACGCACCGTGCCTTC |
| oM3p2 | Same as oWTp2 |
| For oM1, mutagenesis at a1-b1 site | |
| oM1p1 | Same as oWTp1 |
| oM1p2 | CACGACATTGACGCAGCGCGGTTTCGGTGGAAGCCCCTCGTTAGGGTGCCGCCACAGTGAAGGCACGGTGCGT |
| For oM2, mutagenesis at a2-b2 site | |
| oM2p1 | Same as oWTp1 |
| oM2p2 | CACGACATTGACGCAGCGCGACCCTGGTGGAGATTTCTCGTTGTTTCGCCGCCGTGACGAAGGCACGGTGCGT |
Expression and purification of the recombinant GlnR and PhoP.
Expression and purification of recombinant S. coelicolor GlnR were as described previously (23). For PhoP expression, the phoP gene was amplified with the primers SCPhoPEX_F and SCPhoPEX_R using the S. coelicolor chromosome as a template, which was digested with BamHI and EcoRI and then inserted into the same sites of pET32a (Novagen, Darmstadt, Germany) to obtain the expression plasmid pET32a-PhoP. Expression and purification of the recombinant PhoP with Trx, His, and S tags (designated S-PhoP) were performed according to the methods recommended by the manufacturer (Novagen), and the protein concentration was determined using the Bradford method (1). Purified proteins were stored in storage buffer (50 mM Tris-Cl [pH 8.0], 100 mM KCl, 10% glycerol).
DNase I footprinting assay with FAM-labeled primers.
DNase I footprinting assays were performed similar to the method of Zianni et al. (24). A 332-bp promoter region of S. coelicolor amtB was PCR amplified with the primers SCamtBFP(M13F) and SCamtBFP(M13R), and the amplicon was used as the template for further preparation of fluorescent 6-carboxyfluorescein (FAM)-labeled probes with different primer pairs: M13F-FAM and SCamtBFP(M13R) for labeling the coding strand and M13R-FAM and SCamtBFP(M13F) for labeling the noncoding strand. After agarose gel electrophoresis, the FAM-labeled probes were purified by using a QIAquick gel extraction kit (Qiagen, Germany) and quantified with a NanoDrop 2000 (Thermo, USA). For both strand assays, 1 pmol (250 ng) of probes was incubated with 3.5-μg mixtures of recombinant GlnR protein and bovine serum albumin in a total volume of 40 μl in the same buffer as for previously described electrophoretic mobility shift assays (EMSAs) (23). After incubation for 30 min at 25°C, 10 μl of a solution containing 0.015 U of DNase I (Invitrogen) and 100 nmol of freshly prepared CaCl2 was added, followed by further incubation for 1 min at 25°C. The reaction was stopped by adding 140 μl of DNase I stop solution (200 mM unbuffered sodium acetate, 30 mM EDTA, 0.15% sodium dodecyl sulfate) (7). Samples were extracted with phenol-chloroform and precipitated with ethanol, and the pellets were dissolved in 10 μl of Mini-Q water. For preparation of the DNA ladders, the fmol DNA cycle sequencing system (Promega) was used. The volumes of the sequencing reactions were enlarged to 12 μl with 15 ng of amtB promoter region that was amplified with the primers SCamtBFP(M13F) and SCamtBFP(M13R) as the template and 5 pmol of FAM-labeled primer M13F (or primer M13R for the noncoding strand sequencing) as the sequencing primer. The sequencing samples were precipitated with ethanol, dried, and dissolved in 5 μl of Mini-Q water. For both digested DNA fragments and sequencing products, 1 μl of each sample was added to 8.5 μl of HiDi formamide and 0.5 μl of GeneScan-LIZ600 size standards (Applied Biosystems) and was analyzed with 3130xl DNA analyzer and Peak Scanner software v1.0 (Applied Biosystems).
EMSA.
According to the GlnR-protected region in the amtB promoter, probes 101 bp in length representing various types of mutation at each a-b site were synthesized through annealing equal molar amounts of synthetic oligonucleotides (Table 2 and see Fig. 3A), followed by extension using Klenow polymerase fragment (NEB, Massachusetts). All of the probes were labeled with [γ-32P]ATP using T4 polynucleotide kinase (T4 PNK; NEB). EMSA was carried out according to a previously described procedure (23), except that the binding buffer was composed of 50 mM Tris-HCl (pH 8.0), 100 mM KCl, 2.5 mM MgCl2, 1 mM dithiothreitol, and 10% glycerol.
Fig 3.

EMSA using purified recombinant GlnR with various mutants of the amtB promoter. (A) The sequence of the 101-bp DNA fragment covered the amtB promoter region protected by GlnR and PhoP, as revealed by a DNase I footprinting protection assay. The fragments of wild type and mutants used for EMSAs were prepared by Klenow fragment extension after annealing two corresponding synthetic oligonucleotides (see Materials and Methods). For the wild-type oWT sequence, the PhoP-protected region is boxed, the GlnR-protected region is shaded, and the a and b sites are underlined and labeled. Mutated sequences of oM3, oM1, and oM2 are indicated below. (B) The wild-type fragment oWT (0.04 pmol) was used to bind with various amounts of GlnR protein. Two different shifted bands are indicated by arrows. (C) Different amounts of GlnR were incubated with 0.04 pmol of mutated promoter regions of amtB. Three shifted GlnR/DNA complexes are marked by arrows.
egfp fusions and expression.
For egfp fusions, a 500-bp fragment of the upstream region of amtB was PCR amplified with the primers SCamtB-1 and SCamtB-2. The fragment was digested with BamHI and NdeI and introduced into the likewise-digested pIJ8660 (21) to construct pIJ8660-amtBpWT. Site-directed mutations of the ab sites in the amtB promoter were achieved by PCR using paired primers with mutated nucleotides at the 5′ ends (Table 2), yielding pIJ8660-amtBp3, pIJ8660-amtBp1, and pIJ8660-amtBp2, which had mutations in a3-b3 (defined below), a1-b1, and a2-b2, respectively. The mutagenesis procedures resembled the method described in the MutanBEST kit (TaKaRa, Japan), except that the high-fidelity DNA polymerase KOD Plus (ToYoBo, Japan) was used to produce blunt-ended PCR products that allowed direct self-ligation in the presence of T4 PNK. All of the constructed plasmids were introduced into S. coelicolor M145 by conjugation from E. coli ET12567/pUZ8002 (3). Reverse transcription-PCR (RT-PCR) analyses were performed to analyze the expression of the reporter egfp gene in M145.The culture of Streptomyces strains and extraction of total RNA were carried out as described previously (23), and 2.5 μg of total RNA was used for cDNA synthesis with 6-bp random primers using the SuperScript III first-strand synthesis system (Invitrogen). The cDNA (50 ng) was used as the template for the following PCRs, using the hrdB gene as an internal control. Equal amounts of RNA without RT was used as a negative control. The PCR procedures were as follows: heating at 95°C for 5 min, followed by 28 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, and finally incubation for 1 min at 72°C. PCR products were analyzed by agarose gel electrophoresis and visualized by ethidium bromide staining. Two independent samples were used for RT-PCR analysis, and the results were found to be consistent.
RESULTS
DNase I footprinting assay reveals a new section of GlnR-protected sequence in the promoter region of amtB, overlapping with the PhoP-binding sequences.
To identify the exact DNA sequences that GlnR protected in the promoter region of amtB, a DNase I footprinting assay using purified recombinant His-GlnR was performed. Along with the increase of His-GlnR, a clearly protected region of 79 nt extended from positions −177 to −98 relative to the translational start site in the coding strand of the amtB promoter (Fig. 1). Almost the same protected region of 77 nt between positions −173 and −96 was observed in the noncoding strand (see Fig. S1 in the supplemental material). Within the protected region, apart from the previously deduced two 22-bp GlnR binding boxes (a1-b1 and a2-b2), another GlnR binding box consisting of “TTCAC”-n6-“GTAAC” was identified 15 nt before the former predicted a1 site, and these residues were designated the a3 site and the b3 site, respectively (22) (Fig. 2). The results from the DNase I footprinting assay also revealed that GlnR protected the a3-b3 site most efficiently but protected the a2-b2 site with the least efficiency (Fig. 1).
Fig 1.

Identification of the GlnR-protected cis-elements in the S. coelicolor amtB promoter region using a DNase I footprinting assay. The probes used were labeled with FAM dye and are described in Materials and Methods. The whole region of the coding strand DNA is shown in panel A, and the region protected by GlnR from DNase I cleavage was enlarged and is shown separately in panel B. In panel A, the colored lines represent the different concentrations of GlnR used: green, 0 μg; red, 0.2 μg; and blue, 0.4 μg. In panel B, the GlnR binding boxes are underlined, and the DNase I-hypersensitive sites (ACC) are denoted by asterisks. The protected patterns remained the same even when the amount of GlnR used for the binding assay increased up to 2.4 μg (data not shown). The fmol DNA cycle sequencing system was used for DNA sequencing reactions. The sequencing products, together with the digestion products, were analyzed with an ABI 3130xl DNA analyzer and peak scanner software v1.0 (Applied Biosystems). The four sequencing results (G, A, T, and C) are indicated by four different colors separately and are then merged together. The overlapping peaks were probably caused by higher secondary structures within the promoter region. The electropherograms were aligned together with the use of standards. BSA, bovine serum albumin.
Fig 2.
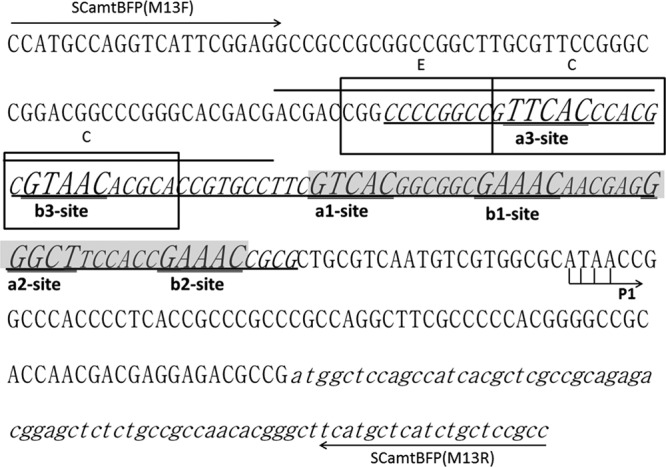
DNA sequence and cis-elements for PhoP and GlnR regulation in the upstream region of amtB. The region protected by GlnR from DNase I digestion is underlined and italicized, while the previously bioinformatically deduced GlnR binding sequences is shaded (22). The a sites and b sites within the GlnR-protected region are enlarged and double underlined. The overlined PhoP binding sequences overlapped with the newly identified a3 site and b3 site. The previously deduced three DRus for PhoP binding are boxed and labeled with an “E” for the extension site and a “C” for the core sites (14). P1, represented by bent arrows, indicates the first and major transcription start site for the amtB operon (2). The primer sequences used for DNase I footprinting assay [SCamtBFP(M13F) and SCamtBFP(M13R)] are labeled by arrows.
Based on both bioinformatics analysis and experimental data, the PHO operators were classified into three groups (19), and the PHO operator upstream of amtB belonged to class III, comprised of two core DRus (C-DRus) and one DRu that extended the protein occupancy beyond the core (E) (19). Interestingly, the a3-b3 site was overlapped by the two previously defined C-DRus of the PhoP-binding sequences (“C” in Fig. 2).
Mutations in a1-b1, a2-b2, or a3-b3 upstream of amtB resulted in different in vitro binding affinities and patterns with GlnR.
A double-stranded wild-type DNA fragment of the region upstream of amtB containing both GlnR- and PhoP-protected regions was synthesized in vitro and designated fragment oWT (see Materials and Methods). EMSA using fragment oWT with purified His-GlnR in a series of titration revealed two GlnR/DNA complexes. The faster-moving complex was designated complex I, whereas the slower one was designated complex II (Fig. 3B).
Considering the physical structures of the nucleotides, the base transition, which interchanges between either two purine nucleotides (A and G) or two pyrimidine nucleotides (C and T), is less likely to introduce large changes in the DNA structure (9). To further study the roles of these three GlnR binding boxes, base transition mutations were introduced into oWT at the a1-b1, a2-b2, and a3-b3 sites to generate fragments oM1, oM2, and oM3, respectively (Fig. 3A). Taking fragment oM3 as an example, only the a3-b3 site was changed from TTCAC-n6-GTAAC to CCTGT-n6-ACGGT, while other DNA sequences remained the same as for oWT. These mutated fragments could still form protein/DNA complexes with GlnR, but the patterns were different from that between GlnR and the wild-type oWT. Specifically, oM3 could only form complex I with GlnR, but the binding affinity was lower than that of the wild-type oWT. When the a1-b1 site was mutated, GlnR could only bind to oM1 with extremely low affinities, with the complexes I and II hardly observed (Fig. 3C). For oM2, only complex I was formed when a lower concentration of GlnR was applied, while complex II between GlnR/oM2 could be successfully formed with increased GlnR concentration, suggesting that complex I was formed prior to the formation of complex II. Under both tested concentrations of GlnR, the binding affinities between GlnR and oM2 seemed unchanged relative to that between GlnR and oWT (Fig. 3C). Therefore, we could infer that the a3-b3 site was required for the complex II formation, the a1-b1 site was necessary for efficient formation of both complex I and complex II, and the a2-b2 site could help the formation of complex II when GlnR was at a lower concentration. In addition, the a3-b3 site could promote the formation of complex I, since mutation in the a3-b3 site led to a lower efficiency in forming complex I.
All three a-b sites are essential for GlnR-mediated activation of amtB transcription in vivo.
Transition mutations were introduced into the three GlnR binding boxes in the amtB promoter region separately (see Materials and Methods), where the mutations at a1-b1, a2-b2, and a3-b3 sites were the same as shown in Fig. 3A. The mutated promoters together with the wild type were coupled to the reporter egfp gene in the promoter-probe plasmid pIJ8660 (21), which were then introduced into S. coelicolor M145 to measure the promoter activities. Plasmid pIJ8660 without a promoter was used as a negative control, and pIJ8660 with the promoter of housekeeping gene hrdB was used as a positive control (17). Strains were shocked with nitrogen-limited conditions before the promoter activities were analyzed with RT-PCR, and the results clearly showed that mutations in each of the a-b sites severely impeded the transcription of egfp gene (Fig. 4). Therefore, all three a-b sites in the amtB promoter were required for GlnR-mediated activation of amtB transcription under nitrogen-limited conditions.
Fig 4.
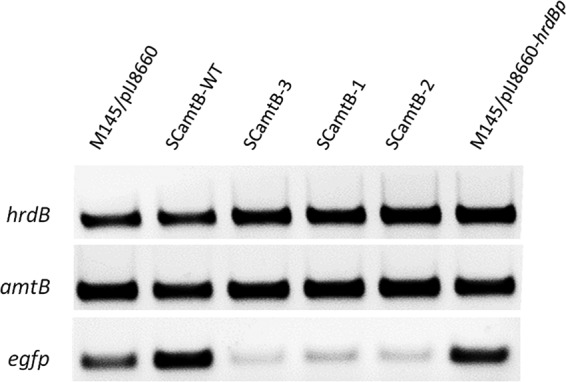
RT-PCR analysis of the activities of wild-type and mutated amtB promoters. Strains used: SCamtB-WT, wild-type amtB promoter; SCamtB-3, a3-b3 site mutation; SCamtB-1, a1-b1 site mutation; SCamtB-2, a2-b2 site mutation.
The EMSA experiment of PhoP/GlnR binding to their overlapping binding sites suggests a competitive binding mechanism for their counter-regulation of amtB transcription. That the a3-b3 site defined in the present study overlapped with two core PhoP-binding DRus (“C”) in the amtB promoter (Fig. 2) suggests that PhoP may compete against GlnR from binding to the a3-b3 site and thus conducts its repressive function.
Recombinant S. coelicolor S-PhoP was expressed and purified from E. coli and was used to conduct the EMSA with both the wild-type and its mutated promoters of amtB. Along with the increase of the concentration of S-PhoP, three major types of S-PhoP/oWT complexes could be observed in the order of complexes I to III, with complex III only visible at the highest concentration of S-PhoP (Fig. 5). When any one of the a-b sites was mutated, only mutation at the a3-b3 site impeded the formation of complex III (Fig. 5).
Fig 5.
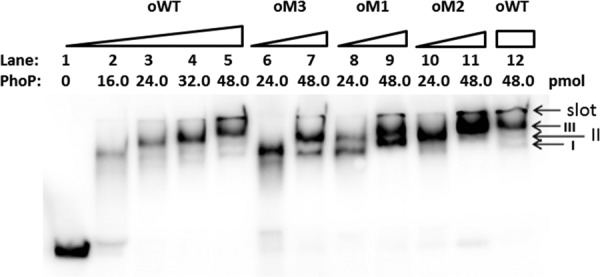
EMSA using purified recombinant S-PhoP with various mutants of the amtB promoter. The probes used are as described for Fig. 3A, and the amounts of PhoP protein used are indicated. Three PhoP/DNA complexes are indicated by arrows. The gel was run in ice-cooled 1× Tris-glycine-EDTA (TGE) buffer at 14 V/cm for about 50 min.
Probe oWT was further incubated with GlnR and PhoP for EMSA (16, 23). After the formation of stable complex II of GlnR/oWT, PhoP was added at incremental concentrations. Along with the addition of PhoP, the GlnR/oWT band disappeared, while more retarded bands were observed with the same migration rates as those of PhoP/oWT complexes I, II, and III (Fig. 6). Because the a3-b3 site was required by PhoP to form the largest complex, PhoP might compete against GlnR in binding to the a3-b3 site in the amtB promoter. However, based on the present data, we cannot prove that the more retarded complexes merely contain PhoP because it cannot be demonstrated that the complexes with the same mobility rates are composed of the same proteins, especially when the formed complexes are extremely large and the mobility rates are analyzed using EMSA. Moreover, although with low affinities, GlnR was able to bind oM3 (Fig. 3C), where the a3-b3 site was mutated, and thus it was possible that PhoP might only replace GlnR in binding to the a3-b3 site while it left GlnR binding to the a1-b1 and a2-b2 sites.
Fig 6.

Competitive EMSA of S. coelicolor amtB promoter with purified recombinant His-GlnR and S-PhoP. The 101-bp DNA fragment of the amtB promoter region (oWT, 0.04 pmol) was used for incubation with various amounts of GlnR and PhoP. The protein-DNA complexes are indicated. The gel was run in ice-cooled 1× TGE buffer at 14 V/cm for about 2 h.
DISCUSSION
Streptomyces spp. have developed complicated mechanisms to adapt their metabolism to the change of the extracellular environments (16). Among these mechanisms, cross talk between different regulators may integrate different signal inputs through fine-tuning the expression of key target genes (10, 15). A regulatory link between nitrogen metabolism and phosphate metabolism was proposed in S. coelicolor, which was coordinated by PhoP through directly binding to the promoters of the nitrogen metabolism-associated genes, including glnA, glnII, and amtB (14). However, the roadblock mechanism suggested for amtB was different from that of the proposed “competitive binding” for glnA and glnII.
Based on the DNase I footprinting data, we defined, in the present study, a new GlnR binding box comprising of an a3-b3 site in addition to the previously well characterized a1-b1 and a2-b2 sites in the promoter region of amtB (22). All of the three GlnR binding boxes were proven essential for GlnR-mediated activation of amtB transcription in vivo. Although the newly characterized a3-b3 site fitted the motif of a-site-n6-b-site, it once again demonstrated the complexities of GlnR binding consensus sequences, e.g., the number of GlnR binding boxes and the distance in between. Through in vitro EMSA analysis, GlnR could form two major complexes with the amtB promoter; however, when the a1-b1 site was mutated, a weak complex III beneath complex I of GlnR/oM1 was detected. Since sheared salmon sperm (100 ng/ml) was added in the binding system, complex III was likely to be the product of GlnR/a3-b3 (or GlnR/a2-b2) rather than a nonspecific binding product between GlnR and oM1. Apart from complex III, which was not observed in GlnR binding of oWT, the transition between complex I and complex II during GlnR binding is still unclear. More work is needed to completely understand the cis-elements and the mechanisms for GlnR binding and regulation.
Since the newly identified a3-b3 site was located within the two C-DRus of PhoP-protected DNA sequences upstream of amtB and the a3-b3 site was essential for GlnR to activate amtB transcription, the repressor role of PhoP in amtB transcription could therefore be inferred, which was the same as in the regulation of glnA and glnII. The mutation of the a3-b3 site in the present study resulted in the failure of GlnR-mediated activation of amtB transcription, which was therefore inappropriate for the study of the in vivo function of a3-b3 for PhoP regulation. Considering the diversities of a-b sequences of GlnR binding boxes, to further confirm the repressor role of PhoP in vivo, future work will focus on screening for partially mutated a3-b3 sites, which could not be recognized by PhoP but could be selectively bound by GlnR and thus the transcriptional activation of amtB by GlnR will not be affected.
In another important industrial strain, Streptomyces venezuelae, the most common motif identified for GlnR binding was found to be comprised of two copies of a sites separated by 6 nt (12). Based on the present data, we compared the amtB promoter sequences of S. venezuelae to that of S. coelicolor using the multiple sequence alignment methodology of CLUSTAL W (4) and found that the two promoters shared high conservation among all three pairs of the a-b sites, with only one base difference at the a2 site (Fig. 7). Although experimental verification is required, the existence of three GlnR binding boxes in the S. venezuelae amtB promoter suggests that the counter-regulation of amtB transcription by PhoP may also exist in S. venezuelae and that the complexities of GlnR binding consensus sequences could be universal.
Fig 7.

Sequence alignment of S. coelicolor (SCO) and S. venezuelae (SVEN) amtB promoters by CLUSTAL W. The GlnR-protected region in S. coelicolor is overlined. Three pairs of “a sites” and “b sites” are underlined and labeled. The sequences below the a3-b3 and a1-b1 sites are the GlnR consensus sites for the S. venezuelae amtB promoter in the study by Pullan et al. (12), and the sections of sequences in boldface and underlined match the proposed GlnR consensus sequence.
The amount operon (amtB-glnK-glnD) encodes an ammonium transporter, a PII protein, and an adenylyltransferase. The S. coelicolor GlnD was proved to function differently from the typical uridylyltransferase, GlnD, in enteric bacteria. Moreover, neither GlnK nor GlnD was required for the rapid adenylation of glutamine synthetase I (GSI) by GlnE in S. coelicolor (5), which was different from that in enteric bacteria, leaving the roles of the GlnK/GlnD system uncharacterized in S. coelicolor. The cross-regulation of the transcription of amount operon by the phosphate-sensing PhoR/PhoP system and nitrogen-sensing GlnR indicates a connection between the overall availabilities of phosphate and nitrogen as well as the function of GlnK/GlnD, opening another window for an in-depth study of the GlnK/GlnD system in S. coelicolor.
Supplementary Material
ACKNOWLEDGMENT
The study was supported by the National Natural Science Foundation of the People's Republic of China (grants 30830002 and 31121001) and the National Basic Research Program of China (grant 2012CB721102).
Footnotes
Published ahead of print 20 July 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 2. Fink D, Weissschuh N, Reuther J, Wohlleben W, Engels A. 2002. Two transcriptional regulators GlnR and GlnRII are involved in regulation of nitrogen metabolism in Streptomyces coelicolor A3(2). Mol. Microbiol. 46:331–347 [DOI] [PubMed] [Google Scholar]
- 3. Gust B, Challis GL, Fowler K, Kieser T, Chater KF. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U. S. A. 100:1541–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98 [Google Scholar]
- 5. Hesketh A, et al. 2002. The GlnD and GlnK homologues of Streptomyces coelicolor A3(2) are functionally dissimilar to their nitrogen regulatory system counterparts from enteric bacteria. Mol. Microbiol. 46:319–330 [DOI] [PubMed] [Google Scholar]
- 6. Hodgson DA. 2000. Primary metabolism and its control in Streptomycetes: a most unusual group of bacteria. Adv. Microb. Physiol. 42:47–238 [DOI] [PubMed] [Google Scholar]
- 7. Le TB, Schumacher MA, Lawson DM, Brennan RG, Buttner MJ. 2011. The crystal structure of the TetR family transcriptional repressor SimR bound to DNA and the role of a flexible N-terminal extension in minor groove binding. Nucleic Acids Res. 39:9433–9447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lewis RA, et al. 2011. Genome-wide transcriptomic analysis of the response to nitrogen limitation in Streptomyces coelicolor A3(2). BMC Res. Notes 4:78 doi:10.1186/1756-0500-4-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Makino K, et al. 1996. DNA binding of PhoB and its interaction with RNA polymerase. J. Mol. Biol. 259:15–26 [DOI] [PubMed] [Google Scholar]
- 10. Martin JF, et al. 2011. Cross-talk of global nutritional regulators in the control of primary and secondary metabolism in Streptomyces. Microb. Biotechnol. 4:165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Okanishi M, Suzuki K, Umezawa H. 1974. Formation and reversion of Streptomycete protoplasts: cultural condition and morphological study. J. Gen. Microbiol. 80:389–400 [DOI] [PubMed] [Google Scholar]
- 12. Pullan ST, Chandra G, Bibb MJ, Merrick M. 2011. Genome-wide analysis of the role of GlnR in Streptomyces venezuelae provides new insights into global nitrogen regulation in actinomycetes. BMC Genomics 12:175 doi:10.1186/1471-2164-12-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rodriguez-Garcia A, Barreiro C, Santos-Beneit F, Sola-Landa A, Martin JF. 2007. Genome-wide transcriptomic and proteomic analysis of the primary response to phosphate limitation in Streptomyces coelicolor M145 and in a ΔphoP mutant. Proteomics 7:2410–2429 [DOI] [PubMed] [Google Scholar]
- 14. Rodriguez-Garcia A, Sola-Landa A, Apel K, Santos-Beneit F, Martin JF. 2009. Phosphate control over nitrogen metabolism in Streptomyces coelicolor: direct and indirect negative control of glnR, glnA, glnII, and amtB expression by the response regulator PhoP. Nucleic Acids Res. 37:3230–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santos-Beneit F, Rodriguez-Garcia A, Martin JF. 2011. Complex transcriptional control of the antibiotic regulator afsS in Streptomyces: PhoP and AfsR are overlapping, competitive activators. J. Bacteriol. 193:2242–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Santos-Beneit F, Rodriguez-Garcia A, Sola-Landa A, Martin JF. 2009. Cross-talk between two global regulators in Streptomyces: PhoP and AfsR interact in the control of afsS, pstS, and phoRP transcription. Mol. Microbiol. 72:53–68 [DOI] [PubMed] [Google Scholar]
- 17. Shu D, et al. 2009. afsQ1-Q2-sigQ is a pleiotropic but conditionally required signal transduction system for both secondary metabolism and morphological development in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 81:1149–1160 [DOI] [PubMed] [Google Scholar]
- 18. Sola-Landa A, Moura RS, Martin JF. 2003. The two-component PhoR-PhoP system controls both primary metabolism and secondary metabolite biosynthesis in Streptomyces lividans. Proc. Natl. Acad. Sci. U. S. A. 100:6133–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sola-Landa A, Rodriguez-Garcia A, Apel AK, Martin JF. 2008. Target genes and structure of the direct repeats in the DNA-binding sequences of the response regulator PhoP in Streptomyces coelicolor. Nucleic Acids Res. 36:1358–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sola-Landa A, Rodriguez-Garcia A, Franco-Dominguez E, Martin JF. 2005. Binding of PhoP to promoters of phosphate-regulated genes in Streptomyces coelicolor: identification of PHO boxes. Mol. Microbiol. 56:1373–1385 [DOI] [PubMed] [Google Scholar]
- 21. Sun J, Kelemen GH, Fernandez-Abalos JM, Bibb MJ. 1999. Green fluorescent protein as a reporter for spatial and temporal gene expression in Streptomyces coelicolor A3(2). Microbiology 145(Pt 9):2221–2227 [DOI] [PubMed] [Google Scholar]
- 22. Tiffert Y, et al. 2008. The Streptomyces coelicolor GlnR regulon: identification of new GlnR targets and evidence for a central role of GlnR in nitrogen metabolism in actinomycetes. Mol. Microbiol. 67:861–880 [DOI] [PubMed] [Google Scholar]
- 23. Wang J, Zhao GP. 2009. GlnR positively regulates nasA transcription in Streptomyces coelicolor. Biochem. Biophys. Res. Commun. 386:77–81 [DOI] [PubMed] [Google Scholar]
- 24. Zianni M, Tessanne K, Merighi M, Laguna R, Tabita FR. 2006. Identification of the DNA bases of a DNase I footprint by the use of dye primer sequencing on an automated capillary DNA analysis instrument. J. Biomol. Tech. 17:103–113 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.