

Abstract
Solvent-producing clostridia are capable of utilizing pentose sugars, including xylose and arabinose; however, little is known about how pentose sugars are catabolized through the metabolic pathways in clostridia. In this study, we identified the xylose catabolic pathways and quantified their fluxes in Clostridium acetobutylicum based on [1-13C]xylose labeling experiments. The phosphoketolase pathway was found to be active, which contributed up to 40% of the xylose catabolic flux in C. acetobutylicum. The split ratio of the phosphoketolase pathway to the pentose phosphate pathway was markedly increased when the xylose concentration in the culture medium was increased from 10 to 20 g liter−1. To our knowledge, this is the first time that the in vivo activity of the phosphoketolase pathway in clostridia has been revealed. A phosphoketolase from C. acetobutylicum was purified and characterized, and its activity with xylulose-5-P was verified. The phosphoketolase was overexpressed in C. acetobutylicum, which resulted in slightly increased xylose consumption rates during the exponential growth phase and a high level of acetate accumulation.
INTRODUCTION
Substrate cost is a major factor impacting the economics of fermentative solvent production by clostridia (7, 21). To reduce the substrate cost, abundant and inexpensive lignocellulosic materials could be used, and one of their major components is pentose-rich hemicellulose (15). Solventogenic clostridia, including Clostridium acetobutylicum and Clostridium beijerinckii, are capable of utilizing the hemicellulosic pentoses xylose and arabinose (25). However, knowledge of clostridial pentose metabolism is very limited.
Recently, our group has characterized two key enzymes in the xylose utilization pathway of C. acetobutylicum, xylose isomerase and xylulokinase, which convert xylose to xylulose-5-P (14). In many bacteria such as Escherichia coli, xylulose-5-P is further catabolized to form the central intermediate glyceraldehyde-3-P by the pentose phosphate pathway enzymes, including transketolase and transaldolase. Another xylose catabolic pathway through the phosphoketolase is found to be present in heterofermentative and facultative homofermentative lactic acid bacteria (19, 37). The thiamine diphosphate-dependent phosphoketolases (EC 4.1.2.9) cleave xylulose-5-P into acetyl-P and glyceraldehyde-3-P. In bifidobacteria, phosphoketolases are key enzymes of the fructose-6-P shunt to convert fructose-6-P to acetyl-P and erythrose-4-P (12, 34).
Little is known about how xylose is metabolized through the metabolic pathways in solventogenic clostridia. Recent studies have shown that genes of the pentose phosphate pathway and a putative phosphoketolase-encoding gene in C. acetobutylicum are induced by pentose sugars (13, 33). However, the contribution of individual pathways to clostridial xylose catabolism has not been analyzed. To manipulate clostridia for efficient xylose utilization, it is important to gain insight into the responses of xylose catabolic pathways to environmental and genetic manipulations.
Metabolic flux analysis can be used to assess the operation of metabolic networks by quantifying intracellular fluxes through individual pathways. However, if parallel or cyclic pathways are present in bioreaction networks, their fluxes cannot be reliably estimated based on only mass balances of intracellular metabolites. A powerful approach for accurately quantifying the fluxes in a complex metabolic network is based on 13C tracer experiments (30, 47). In this approach, the 13C labeling patterns in products of metabolism, which reflect the in vivo activity of metabolic pathways and enzymes, are analyzed by nuclear magnetic resonance or mass spectrometry (MS). Based on the balances of metabolites and isotopomers, a mathematical model relating the intracellular fluxes to the 13C labeling data is constructed (32, 41). Exchange fluxes in reversible reactions are also quantitatively considered in the model to account for their influence on 13C labeling patterns. The intracellular flux distribution is then estimated by finding a best fit for the 13C labeling data within the constructed model. In addition to quantification of fluxes through the known biochemical pathways, the 13C metabolic flux analysis has also demonstrated its value for identification of novel or unexpected metabolic pathways (9, 17). This approach has recently been used to elucidate the glucose metabolic pathways in C. acetobutylicum (2, 6).
The aim of this study was to quantitatively analyze xylose metabolism in C. acetobutylicum. We grew C. acetobutylicum in batch cultures with various initial concentrations of xylose and observed different conversion yields of xylose into solvents. The 13C-based metabolic flux analysis was used to identify xylose catabolic pathways and quantify their fluxes. Our results elucidated the use of the phosphoketolase pathway for xylose catabolism in C. acetobutylicum, and its flux was changed when cells were grown at different concentrations of xylose. Moreover, a phosphoketolase from C. acetobutylicum was purified and characterized, and the effect of phosphoketolase overexpression on xylose fermentation of C. acetobutylicum was investigated.
MATERIALS AND METHODS
Strains and growth conditions.
The C. acetobutylicum strains and plasmids used in this study are given in Table 1. C. acetobutylicum strains were precultured anaerobically on clostridial growth medium (CGM) (43) to late exponential growth phase and washed twice using the P2 minimal medium (4), which contains (per liter) 0.5 g of K2HPO4, 0.5 g of KH2PO4, 2.2 g of CH3COONH4, 0.2 g of MgSO4 · 7H2O, 0.01 g of MnSO4 · H2O, 0.01 g of NaCl, 0.01 g of FeSO4 · 7H2O, 1 mg of p-aminobenzoic acid, 1 mg of vitamin B1, and 0.01 mg of biotin. The cultures were started with the same optical density at 600 nm (OD600; ∼0.04) and performed at 37°C in triplicate in 100-ml sealed glass flasks with 60 ml of P2 minimal medium. d-Xylose was supplied at concentrations of 10, 20, or 60 g liter−1 as a carbon source. For 13C labeling experiments, xylose was added in the form of a mixture of 76% (wt/wt) 1-13C-labeled xylose (99% pure; Sigma) and 24% (wt/wt) natural xylose.
Table 1.
C. acetobutylicum strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Description or sequencea (5′→3′) | Source or reference |
|---|---|---|
| C. acetobutylicum strains | ||
| ATCC 824 | Wild type | ATCC |
| xfp::intron | xfp-inactivated mutant, Emr | This study |
| 824-pIMP1 | Strain carrying pIMP1 | |
| 824-pIMP1XFP | xfp-overexpressed strain, Emr | This study |
| Plasmids | ||
| pWJ1 | Group II intron | 44 |
| pWJ1-xfp | pWJ1 derivative for inserting intron into xfp | This study |
| pIMP1 | Ampr Emr | 29 |
| pIMP1-xfp | pIMP1 derivative carrying CAC1343 gene | This study |
| pET28a | Novagen | |
| pET28a-xfp | pET28a derivative carrying CAC1343 gene | This study |
| Primers | ||
| xfp-IBS | AAAACTCGAGATAATTATCCTTACACTACAGGTTCGTGCGCCCAGATAGGGTG | |
| xfp-EBS1d | CAGATTGTACAAATGTGGTGATAACAGATAAGTCAGGTTCCATAACTTACCTTTCTTTGT | |
| xfp-EBS2 | TGAACGCAAGTTTCTAATTTCGGTTTAGTGTCGATAGAGGAAAGTGTCT | |
| xfp-UNI | CGAAATTAGAAACTTGCGTTCAGTAAAC | |
| pIMP1-xfp_F | CGGGATCCATGCAAAGTATAATAGGAAAAC | |
| pIMP1-xfp_R | TCCCCCCGGGTTATACATGCCACTGCCAATTAG | |
| pET28a-xfp_F | AAGCTTATGCAAAGTATAATAGGAAAACATAAGG | |
| pET28a-xfp_R | GGATCCTTATACATGCCACTGCCAATTAGTT |
The introduced restriction sites are underlined.
Analytical methods.
Cell growth was monitored spectrophotometrically at 600 nm (OD600). For analysis of extracellular metabolites, culture samples were harvested by centrifugation at 15,000 × g for 10 min at 4°C. Xylose concentrations were determined by high-pressure liquid chromatography (HPLC) using an Agilent model 1200 instrument equipped with a Waters Sugar Pak I column (6.5 by 300 mm) and a refractive index detector (Agilent). Double-distilled water was used as the mobile phase at a flow rate of 0.6 ml min−1, and the column was operated at 60°C. Acetone, ethanol, butanol, acetate, and butyrate in culture supernatants were detected by a gas chromatograph (GC) (Agilent model 7890A) equipped with a capillary column (Alltech EC-Wax; 30 m by 0.32 mm) and a flame ionization detector (Agilent). The specific xylose uptake rate was determined during the exponential growth phase as the coefficient of a linear regression of the rate of change in xylose concentration versus the average OD600.
Gene disruption in C. acetobutylicum.
Gene disruption in C. acetobutylicum ATCC 824 was performed by using group II intron-based targetron technology as described previously (35). Briefly, the 350-bp fragment for retargeting the intron to insert within the xfp (CAC1343) gene was generated by one-step assembly PCR using the primers xfp-IBS, xfp-EBS1d, xfp-EBS2, and xfp-UNI (Table 1), according to the protocol of the TargeTron gene knockout system (Sigma). The PCR products were then digested and ligated to targetron plasmid pWJ1 (44). The plasmid pWJ1-xfp was methylated in vivo in E. coli ER2275(pAN1) (23) and electroporated into C. acetobutylicum ATCC 824 according to a previously published method (24) by using a MicroPulser (Bio-Rad). The transformants were selected on CGM plates supplemented with erythromycin. The resulting mutant with an intron insertion in the xfp gene was confirmed by PCR.
Gene overexpression in C. acetobutylicum.
The xfp gene from C. acetobutylicum was PCR amplified using the primers pIMP1-xfp_F and pIMP1-xfp_R (Table 1). The PCR fragment was cloned into the pIMP1 vector (23), under the control of the promoter of the phosphotransbutyrylase gene (ptb), which is an early-growth-associated promoter and drives the gene expression throughout the exponential growth phase (40). The obtained plasmid was electroporated into C. acetobutylicum, generating the strain 824-pIMP1XFP. The pIMP1 empty vector was also expressed in C. acetobutylicum as a negative control.
Protein overexpression and purification.
The xfp (CAC1343) gene was PCR amplified from C. acetobutylicum ATCC 824 genomic DNA using the primers pET28a-xfp_F and pET28a-xfp_R (Table 1). The PCR fragment was ligated into the expression vector pET28a cleaved by BamHI and HindIII. Selected clones were confirmed by DNA sequence analysis. E. coli Rosetta(DE3) (Novagen) harboring the expression plasmid for the N-terminal His6-tagged protein was grown on LB medium to an OD600 of 0.8 at 37°C, induced by 0.2 mM isopropyl-β-d-thiogalactopyranoside, and harvested after 12 h of shaking at 16°C. Protein purification was performed using a rapid nickel-nitrilotriacetic acid (Ni-NTA) agarose minicolumn protocol as described previously (45). Briefly, harvested cells were resuspended in 20 mM HEPES buffer (pH 7) containing 100 mM NaCl, 0.03% Brij 35, 2 mM β-mercaptoethanol, and 2 mM phenylmethylsulfonyl fluoride. Lysozyme was added to a concentration of 1 mg ml−1, and the cells were lysed by freezing-thawing followed by sonication. After centrifugation, the supernatant was loaded onto a Ni-NTA agarose column (0.2 ml). After the column was washed with the 50 mM Tris-HCl buffer (pH 8) containing 1 M NaCl, 0.3% Brij 35, and 2 mM β-mercaptoethanol, bound proteins were eluted with 0.3 ml of the same buffer supplemented with 250 mM imidazole. The buffer was then changed to 10 mM Tris-HCl (pH 7.4) containing 0.3 mM dithiothreitol (DTT), 1 mM EDTA, and 10% glycerol by using Bio-Spin columns (Bio-Rad). The purified protein was run on a sodium dodecyl sulfate-polyacrylamide gel to monitor its size and purity.
Enzyme assays.
The activity of phosphoketolase was assayed in crude cell extracts of C. acetobutylicum or using the purified recombinant protein. The crude cell extracts were prepared from 10-ml aliquots of exponentially growing cultures (OD600, ∼1.5) in minimal medium with 60 g liter−1 of xylose. Cell pellets were harvested by centrifugation, washed twice, and resuspended in 20 mM HEPES buffer (pH 7.0) containing 100 mM NaCl and 2 mM β-mercaptoethanol. Cells were disrupted by two passes through a French press at a pressure of 30 klb/in2 before centrifugation for 15 min at 4°C and 20,000 × g. The supernatant was used for determination of the enzyme activity and protein concentration. Phosphoketolase activity of the purified protein was assayed using a previously published colorimetric method based on formation of ferric acetyl hydroxamate from acetyl-P (22, 46). Briefly, the enzyme sample was added to 75 μl of 150 mM potassium phosphate buffer (pH 6.5) containing 1.9 mM l-cysteine hydrochloride, 23 mM sodium fluoride, 8 mM sodium iodoacetate, 1 mM thiamine pyrophosphate, 5 mM magnesium chloride, and either d-xylulose-5-P (Sigma) or d-fructose-6-P as a substrate (each at a 25 mM concentration). After incubation at 37°C for 30 min, 75 μl of hydroxylamine hydrochloride (2 M, pH 6.5) was added to stop the reaction, and the reaction mixture was incubated at room temperature for 10 min. Then, 50 μl of 15% (wt/vol) trichloroacetic acid, 50 μl of 4 M HCl, and 50 μl of FeCl3 · 6H2O (5% [wt/vol] in 0.1 M HCl) were added to the reaction mixture. The formation of the ferric hydroxamate was monitored at 505 nm with a Beckman DU800 spectrophotometer by using a series of acetyl-P standards for calibration. Phosphoketolase activity of the crude cell extracts was assayed by measuring the acetyl-P formed after addition of xylulose-5-P according to the method of Sonderegger et al. (36) with minor modification. Briefly, acetyl-P was converted to acetate by adding 1 μl of 1 M MgCl2, 1 μl of 30 mM ADP, and 0.2 U of acetate kinase (Sigma) to 75 μl of the assay mixture, followed by incubation at 30°C for 30 min. The acetate produced was then determined by using an enzymatic test kit (r-Biopharm, Darmstadt, Germany) and subtracting the acetate that was formed in an assay mixture without xylulose-5-P from that in the assay mixture containing xylulose-5-P. Specific phosphoketolase activities were expressed as units per milligram of protein, where 1 U is defined as formation of 1 μmol of acetyl-P per min. Protein concentrations were measured by using the Bradford reagent (Sangong Corp., Shanghai, China) with bovine serum albumin (BSA) as a standard.
Determination of intracellular acetyl-P concentration.
The intracellular acetyl-P was extracted and assayed using the method of Zhao et al. (49). Briefly, cell pellets were harvested by centrifuging 10-ml aliquots of exponentially growing cultures (OD600, ∼1.5) in minimal medium with 60 g liter−1 of xylose, treated with ice-cold 3 M HClO4, and incubated for 30 min on ice. After neutralization with saturated KHCO3 and centrifugation, the extract was incubated with activated charcoal (50 mg ml−1) for 15 min on ice to remove ATP and other small adenylated molecules. Acetyl-P was converted to ATP by adding 1 μl of 1 M MgCl2, 1 μl of 30 mM ADP, and 0.2 U of acetate kinase (Sigma) to 1 ml of the extract, followed by incubation at 30°C for 30 min. The concentration of the formed ATP was then determined by using the ATP bioluminescent assay kit (Sigma) and the Varioskan flash multimode reader (Thermo Scientific). The sample without addition of acetate kinase was used as a negative control. A series of acetyl-P standards were used to obtain a calibration curve for determining acetyl phosphate concentration in the cell extracts. The intracellular acetyl-P concentrations were then calculated by normalization to cell density using a predetermined correlation factor of 0.26 g (dry weight) of cells per OD600.
Sample preparation and GC-MS analysis.
From the 13C labeling experiments, cell aliquots were harvested during late exponential growth phase (OD600, ∼1.5) by centrifuging 3 ml of culture broth at 9,000 × g and 4°C for 10 min. The pellet was washed with 1 ml 0.9% (wt/vol) NaCl, resuspended in 200 μl of 6 M HCl, and hydrolyzed at 105°C for 24 h in sealed 2-ml glass vials. The hydrolysate was dried in a vacuum centrifuge at room temperature and derivatized at 85°C for 1 h in 120 μl pyridine and 30 μl N-methyl-N-[tert-butyldimethylsilyl]trifluoroacetamide (Sigma). After filtration with an 0.2-μm-pore-size filter, 4 μl of derivatized sample was injected into an Agilent 6890-5973 GC-MS system with an HP-5MS column (30 m by 0.25 mm by 0.25 μm). GC oven temperature was programmed from 60°C to 180°C at 5°C per min and from 180°C to 260°C at 10°C per min. The flow rate of carrier gas (helium) was set at 1 ml min−1. The mass spectrometer was operated in the electron impact (EI) mode at 70 eV.
Metabolic flux analysis.
The GC-MS data were analyzed as described previously (26). Briefly, a mass isotopomer distribution vector (MDV) of each fragment of alanine, glycine, valine, serine, phenylalanine, histidine, and tyrosine was determined from the respective mass spectra and was corrected for the natural abundance of all stable isotopes, including 13C, 29Si, 30Si, 15N, and 18O. From the MDV of the amino acids, the MDVs of their respective precursor intermediates, including 3-P-glycerate, phosphoenolpyruvate (PEP), pyruvate, ribose-5-P, and erythrose-4-P, could be easily derived. It should be noted that an MDV could be determined for each metabolite fragment containing specific combinations of carbon atoms of the considered metabolite. For example, an MDV was determined for each of the three 3-P-glycerate fragments, including the 3PG1–3 fragment containing all three carbon atoms of 3-P-glycerate, the 3PG2-3 fragment containing C-2 and C-3 atoms, and the 3PG1-2 fragment containing C-1 and C-2 atoms. The summed fractional labeling (SFL) of a metabolite fragment was calculated from the MDV according to , in which n represents the number of carbon atoms in the fragment and mi represents the relative abundance of different mass isotopomers (11). The SFL data of intermediate metabolites were then used for quantification of intracellular fluxes.
For intracellular flux analysis, a bioreaction network was constructed based on the C. acetobutylicum ATCC 824 genome sequence and is shown in Fig. 1. It included xylose uptake and transformation of xylose to xylulose-5-P (v1), the reactions of nonoxidative pentose phosphate pathway (v3, v4, and v5), and the glycolytic reactions converting fructose-6-P to 3-P-glycerate (v6). The reaction catalyzed by phosphoketolase (v2) was also included (see Results). Since the isomerase and epimerase reactions between ribose-5-P, ribulose-5-P, and xylulose-5-P are highly reversible, they were assumed to be in rapid equilibrium; hence, the three pentose-5-P metabolites were treated as one pool. The reactions catalyzed by transketolase (v3 and v5) and transaldolase (v4) were considered reversible, while the phosphoketolase reaction was assumed to be physiologically irreversible, based on the thermodynamic properties of these reactions (38). For a reversible reaction, its net flux and extent of reaction reversibility were represented by vnet and exch, respectively, which can be easily transformed into the forward and backward fluxes of the reaction (42). The intracellular metabolites pentose-5-P, sedoheptulose-7-P, erythrose-4-P, fructose-6-P, and glyceraldehyde-3-P were assumed to be in isotopomeric steady state during the labeling experiments (see Results). Balances on the isotopomers of each metabolite were constructed by using the atom mapping matrices that describe the transfer of carbon atoms from reactants to products in biochemical reactions (42). Based on the balances of metabolites and isotopomers, the SFL measurements of intermediate metabolites were simulated as a function of intracellular flux distributions. The best-fit intracellular fluxes were then estimated by minimizing the deviation between experimentally determined and simulated SFL data, using a least-squares parameter-fitting approach (41). The flux calculation was achieved by developing a computer algorithm using MATLAB 6.0 (Mathworks).
Fig 1.

Pathways of xylose metabolism in C. acetobutylicum. Metabolites in boxes are extracellular substrates or products. Gray arrows indicate precursor withdrawal for the amino acids analyzed by GC-MS. Double-headed arrows indicate reactions assumed to be reversible. TCA, tricarboxylic acid.
RESULTS
Fermentation of C. acetobutylicum at various xylose concentrations.
To study the xylose metabolism in C. acetobutylicum, we grew C. acetobutylicum ATCC 824 with xylose as a carbon source in batch cultures. The initial xylose concentration in the culture medium was 10, 20, and 60 g liter−1, respectively. Cell growth, xylose consumption, and formation of acetone, butanol, ethanol, acetate, and butyrate were measured during the fermentation. As shown in Fig. 2, cultivation at different concentrations of xylose resulted in a remarkable change in product profiles. At a low xylose concentration (i.e., 10 g liter−1), the primary products were acetate and butyrate throughout the cultivation, whereas butanol and acetone were the predominant final products at a high xylose concentration (i.e., 60 g liter−1). Compared to the culture on 10 g liter−1 xylose, the solvents (acetone and butanol) produced per gram xylose consumed were increased about 7-fold, while formation of acids (acetate and butyrate) was reduced substantially, during the fermentation on 60 g liter−1 xylose. The results suggest a shift in xylose metabolism according to xylose concentration in the medium. This prompted us to perform a quantitative analysis of xylose metabolism in C. acetobutylicum.
Fig 2.

C. acetobutylicum ATCC 824 fermentation at initial xylose concentrations of 10 (A), 20 (B), and 60 (C) g liter−1. Cell growth (⧫), xylose consumption (●), and formation of acetone (▶), butanol (★), ethanol (*), acetate (♢), and butyrate (□) were measured during the cultivation. The data points represent the averages of three independent cultures.
Quantification of intracellular fluxes in xylose metabolism.
The pathways of C. acetobutylicum xylose metabolism were studied based on the C. acetobutylicum ATCC 824 genome sequence (27), which showed the presence of the genes encoding all the enzymes of the nonoxidative pentose phosphate pathway, including transketolase and transaldolase (Fig. 1). In addition, the presence of a phosphoketolase (i.e., CAC1343) was inferred according to genome annotation and the encoding gene was shown to be induced by xylose (13, 33). Using an in vitro enzyme assay, we verified the xylulose-5-P phosphoketolase activity (8.3 mU mg−1) in C. acetobutylicum during growth on xylose (Table 2), which is higher than the value reported for the phosphoketolase from Saccharomyces cerevisiae (2.5 mU mg−1) (36). Thus, the bioreaction network used for intracellular flux analysis included the reactions of the nonoxidative pentose phosphate pathway, the glycolytic reactions converting fructose-6-P to 3-P-glycerate, and the reaction catalyzed by phosphoketolase (Fig. 1).
Table 2.
Specific xylulose-5-P phosphoketolase activity in crude cell extracts of C. acetobutylicum strainsa
| Strain | Sp act (mU [mg protein−1])b |
|---|---|
| ATCC 824 | 8.3 ± 1.5 |
| xfp::intron | 5.5 ± 0.8 |
| 824-pIMP1 | 9.4 ± 2.4 |
| 824-pIMP1XFP | 120 ± 10 |
Samples were harvested during exponential growth on xylose.
The average and the standard deviation were determined from three independent experiments.
We performed 13C-based metabolic flux analysis that relies on the [1-13C]xylose tracer experiments and GC-MS analysis of mass isotopomer patterns in cellular amino acids. Because C. acetobutylicum batch cultures are typically biphasic and acids formed during exponential growth are reassimilated at the stationary growth phase, cell aliquots were harvested from 13C-labeled experimental cells at exponential growth phase during which xylose was the primary carbon source for cells. To assess if the isotopic steady state was achieved, samples were taken at different time points during the exponential growth phase. The determined mass isotopomer distributions of key amino acids were almost unchanged with the time of harvest, which is consistent with previous reports that have showed that a (quasi-)steady state can be reached during the exponential growth phase in batch cultures (8, 31).
From the GC-MS data of amino acids, the 13C labeling patterns of the precursor metabolites, including 3-P-glycerate, PEP, pyruvate, ribose-5-P, and erythrose-4-P, were identified, which reflected the in vivo activities of various pathways and enzymes in xylose metabolism. The labeling data of 3-P-glycerate, PEP, and pyruvate were found to be identical, indicating the full equilibration between these metabolite pools. As shown in Fig. 3A, the pentose phosphate pathway generates 3-P-glycerate that is 13C labeled at the C-1 and C-3 positions, while the use of phosphoketolase for xylose catabolism results in dilution of 13C label in 3-P-glycerate. Therefore, the relative contributions of the phosphoketolase and pentose phosphate pathways to xylose catabolic flux could be determined using [1-13C]xylose as the input substrate and the labeling data of 3-P-glycerate at least in the situation where all fluxes are unidirectional.
Fig 3.

Simulations of fractional 13C labeling of intermediate metabolites resulting from [1-13C]xylose input. (A) Principles of analysis of the phosphoketolase flux with [1-13C]xylose. All steps were assumed to be unidirectional. Labeled carbon atoms are marked by a solid circle. (B) SFL of 3-P-glycerate, pentose-5-P, and erythrose-4-P fragments as a function of the phosphoketolase flux. The subscript numbers of the fragments indicate the carbon atoms included in each fragment. The extents of reversibility of transketolase and transaldolase reactions are set at the same values (i.e., 0, 0.2, 0.4, 0.6, and 0.8). The input substrate is a mixture of 76% [1-13C]xylose and 24% natural xylose. The measured SFL values of 3PG1–3 and 3PG2-3 fragments for the cultures on 10 or 20 g liter−1 xylose are indicated.
The 13C label distribution through the pentose phosphate pathway is very complicated due to the presence of highly reversible reactions catalyzed by transketolase and transaldolase (10). To investigate the flux distribution between the pentose phosphate pathway and the phosphoketolase pathway and its sensitivity with respect to measured fractional labeling data, a simulation study was conducted. By using the balances of metabolites and isotopomers in the bioreaction network, a mathematical framework relating the intracellular fluxes to the available labeling data was constructed. Figure 3B illustrates the simulation results of fractional labeling of 3-P-glycerate, pentose-5-P, and erythrose-4-P as a function of the phosphoketolase flux when the extents of reversibility are the same for transketolase and transaldolase reactions. The SFL of the 3-P-glycerate fragments 3PG1–3 and 3PG2-3 is monotonically reduced with the increased flux through phosphoketolase and does not depend on the extent of reversibility of transketolase and transaldolase reactions. On the other hand, the SFL values of the pentose-5-P fragment (P5P1–5), erythrose-4-P fragment (E4P1–4), and 3-P-glycerate fragment (3PG1-2) are noticeably affected by the extent of reaction reversibility. When the reversibility extent is low, these SFL values are insensitive to the phosphoketolase flux. Therefore, the simulation results indicated that the flux through phosphoketolase can be accurately determined from the SFL data of 3PG1–3 and 3PG2-3 fragments.
The 13C-based metabolic flux analysis was performed for C. acetobutylicum grown in the medium containing 10 or 20 g liter−1 xylose. The use of the phosphoketolase pathway for xylose catabolism was revealed, because the measured fractional labeling of 3-P-glycerate was much lower than expected when the phosphoketolase was inactive (Fig. 3B). Furthermore, the measured SFL values of 3PG1–3 and 3PG2-3 were higher at a low xylose concentration (i.e., 10 g liter−1) than at a high xylose concentration (i.e., 20 g liter−1). The intracellular carbon flux distribution was determined as the best fit to the SFL data by repeating a parameter-fitting procedure from different random starting values of the phosphoketolase flux and of reversibility extents of transketolase and transaldolase reactions. As shown in Fig. 4, during growth at a low xylose concentration, 85% of the xylose molecules were routed through the pentose phosphate pathway and 15% entered the phosphoketolase pathway. When cells were grown on 20 g liter−1 xylose, the specific xylose uptake rate was 2.3-fold higher and the split ratio of the phosphoketolase pathway to the pentose phosphate pathway was increased to 40%. Thus, the flux through phosphoketolase was increased from 0.02 to 0.12 mM−1 OD−1 h−1 when the xylose concentration in the medium was changed from 10 to 20 g liter−1.
Fig 4.
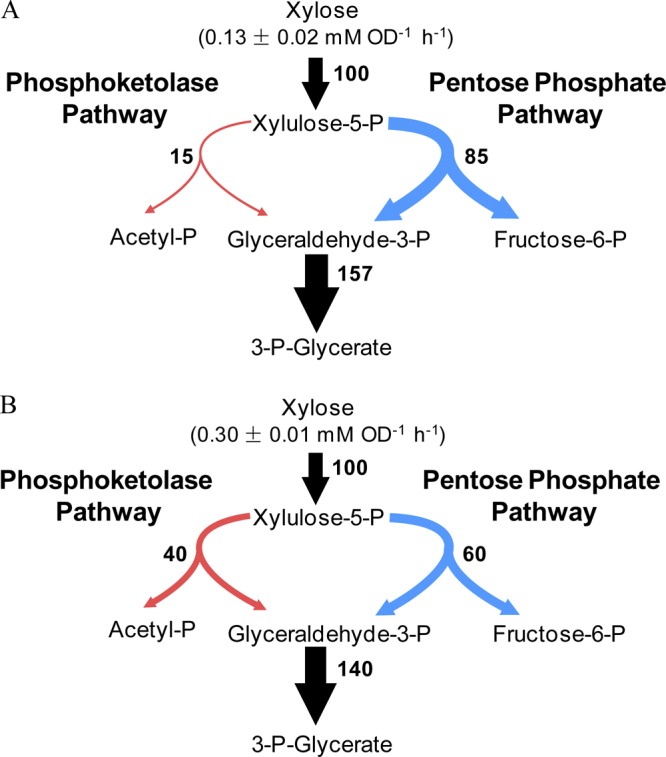
In vivo carbon flux distribution in C. acetobutylicum ATCC 824 during exponential growth on 10 g liter−1 (A) and 20 g liter−1 (B) of xylose. The net flux values are expressed relative to the specific xylose uptake rate that is given above the uptake arrow. Arrows indicate the directions of the net fluxes, and their widths are scaled to the flux values. The 95% confidence intervals were less than 10% for all the fluxes.
Characterization and overexpression of phosphoketolase in C. acetobutylicum.
We then characterized a phosphoketolase from C. acetobutylicum. The CAC1343 gene product had a close homology with the xylulose-5-P/fructose-6-P phosphoketolase (XFP) from Bifidobacterium lactis (46% identity [22]). This putative XFP protein of C. acetobutylicum was overexpressed in E. coli cells with the N-terminal His6 tag and purified with a nickel-chelating affinity column. The purified recombinant protein displayed a xylulose-5-P phosphoketolase activity, and the specific activity value (2.01 U mg−1) was comparable with those reported for the enzymes from B. lactis (22) and Lactobacillus plantarum (46). The fructose-6-P phosphoketolase activity (0.09 U mg−1) was also detected at 25 mM fructose-6-P.
Since the phosphoketolase played an important role in xylose catabolism of C. acetobutylicum, the effect of phosphoketolase overexpression on xylose fermentation was investigated. We constructed a recombinant C. acetobutylicum strain, in which the xfp gene from C. acetobutylicum (i.e., CAC1343) was expressed in a plasmid by the promoter of phosphotransbutyrylase (ptb). Overexpression of phosphoketolase in the recombinant strain was verified using an in vitro enzyme assay. The specific activity of xylulose-5-P phosphoketolase in the recombinant strain was about 12-fold higher than that of the control strain carrying an empty-vector plasmid (Table 2). We then compared the fermentation performances of the phosphoketolase-overexpressing strain and the control strain in a batch culture with 60 g liter−1 xylose. As shown in Fig. 5, during the exponential growth phase, slightly increased rates of cell growth and xylose consumption were observed for the phosphoketolase-overexpressing strain compared to the control strain. However, during the subsequent solventogenic phase, the phosphoketolase-overexpressing strain exhibited a strongly reduced xylose uptake rate and solvent yields. The most prominent effect of phosphoketolase overexpression was the high level of accumulation of acetate (75 mM). Unlike the control strain, the phosphoketolase-overexpressing strain did not reassimilate acetate at the solventogenic phase. Nevertheless, no significant decrease in the culture pH (i.e., 4.3) was observed for the phosphoketolase-overexpressing strain compared to the control strain (pH 4.5) throughout the solventogenic phase. In addition, we determined the intracellular concentration of acetyl-P in exponentially growing cells using a previously published method. The acetyl-P concentration in the phosphoketolase-overexpressing strain was 685 pmol g (dry weight) of cells−1, which was increased over 7-fold compared to that in the control strain (91 pmol g [dry weight] of cells−1). This result indicated that the acetate accumulation of the phosphoketolase-overexpressing strain mostly likely originated from dephosphorylation of acetyl-P formed through the phosphoketolase pathway.
Fig 5.

Effect of phosphoketolase overexpression (filled symbols) on C. acetobutylicum cell growth and xylose consumption (A), solvent production (B), and acid formation (C). The strain carrying an empty-vector plasmid was used as a control (open symbols). Cells were grown in P2 minimal medium containing 60 g liter−1 of xylose as a carbon source. The data points represent the averages of three independent cultures.
To investigate the effect of xfp gene inactivation on xylose fermentation of C. acetobutylicum, we disrupted the CAC1343 gene by inserting an intron and confirmed it by PCR. The resulting xfp-inactivated mutant still displayed a phosphoketolase activity, although it was lower than that in the wild type, during growth on xylose (Table 2). This result revealed that in addition to CAC1343, C. acetobutylicum may have another, hitherto-unknown phosphoketolase for xylose catabolism. Moreover, the influence of CAC1343 gene inactivation on xylose fermentation was assessed by cultivating both wild-type and mutant strains on 20 g liter−1 xylose. The CAC1343-inactivated mutant exhibited a xylose fermentation rate and product yields similar to those of the wild type (Table 3).
Table 3.
Product yield in batch fermentation of wild-type C. acetobutylicum and xfp-inactivated mutanta
| Strain | Yield (mmol product formed per mol xylose consumed) |
||||
|---|---|---|---|---|---|
| Acetone | Butanol | Ethanol | Acetate | Butyrate | |
| Wild type | 149 ± 13 | 271 ± 13 | 61 ± 12 | 110 ± 4 | 188 ± 30 |
| xfp-inactivated mutant | 170 ± 17 | 278 ± 12 | 69 ± 5 | 94 ± 19 | 171 ± 12 |
Fermentation was performed at an initial xylose concentration of 20 g liter−1. Samples were harvested during stationary growth phase (∼149 h). Data shown are means ± standard deviations calculated from three independent experiments.
DISCUSSION
By using 13C-based metabolic flux analysis technique, this study presented evidence that the phosphoketolase pathway played an important role in xylose metabolism in C. acetobutylicum. Up to 40% of the xylose catabolic flux was contributed by the phosphoketolase pathway. To the best of our knowledge, this is the first time that the in vivo activity of the phosphoketolase pathway in clostridia has been revealed. So far, the existence of the phosphoketolase pathway has been described only for the lactic acid bacteria (3), bifidobacteria (22), and yeast and filamentous fungi (28, 36).
The two xylose catabolic routes, the phosphoketolase pathway and the pentose phosphate pathway, exhibit different stoichiometries. Through 13 enzymatic reactions in the pentose phosphate pathway, three xylulose-5-P molecules are converted to five acetyl coenzyme A (CoA) molecules and eight ATP molecules are produced concomitantly. On the other hand, six ATP molecules are formed in the conversion of three xylulose-5-P molecules to six acetyl-CoA molecules through eight enzymatic reactions in the phosphoketolase pathway. Thus, the phosphoketolase pathway could be taken as a shortcut route despite its poor energetic yields. One explanation for the use of the phosphoketolase pathway for xylose catabolism in C. acetobutylicum is the presence of rate-limiting steps in the pentose phosphate pathway. This is supported by a previous study which showed that overexpression of the transaldolase from E. coli in C. acetobutylicum led to a potent increase in the xylose consumption rate (16). The limited capacity of the pentose phosphate pathway may also explain the increase in the fraction of xylose molecules catabolized through the phosphoketolase pathway when the xylose uptake rate was increased during growth at a high xylose concentration. Our study provided the initial insight into the xylose metabolism in C. acetobutylicum. To elucidate the regulatory mechanisms of increased solvent yields at high xylose concentrations compared to low xylose concentrations, further studies are required, including dynamic 13C labeling experiments and systematic analysis of intracellular metabolite concentrations for the solventogenic phase (1).
The purified phosphoketolase from C. acetobutylicum displayed a much higher activity with xylulose-5-P than with fructose-6-P, which is similar to the previously reported dual-substrate phosphoketolase from B. lactis or L. plantarum (22, 46). Orthologs of the C. acetobutylicum phosphoketolase are present in other clostridia, including Clostridium carboxidivorans and Clostridium butyricum. However, no orthologs could be found in another solvent-producing strain, C. beijerinckii, which is capable of utilizing xylose. In C. acetobutylicum, the xfp gene (CAC1343) clusters on the chromosome with arabinose utilization genes. A DNA microarray analysis has shown that this gene was strongly induced by arabinose (33). We have found that expression of this xfp gene was regulated by the transcriptional factor AraR, which controls arabinose utilization in C. acetobutylicum (48). Moreover, we have detected the xylulose-5-P phosphoketolase activity in C. acetobutylicum grown on arabinose by using the in vitro enzyme assay (data not shown). Therefore, besides xylose catabolism, the phosphoketolase may participate in arabinose catabolism of C. acetobutylicum. In addition to the CAC1343 gene product, C. acetobutylicum may have another unknown phosphoketolase for xylose catabolism, as shown by the phosphoketolase activity detected in the CAC1343-inactivated mutant. Similarly, the gene encoding xylulose-5-P phosphoketolase in S. cerevisiae has not been identified, although this enzyme activity was detected (36). It was speculated that transketolase could be responsible for the phosphoketolase activity found in S. cerevisiae (39).
Overexpression of the phosphoketolase in C. acetobutylicum resulted in a high level of acetate accumulation. Extracellular acetate may diffuse passively across the cell membrane in its undissociated form (5) and cause a marked drop in the intracellular pH (18). This may explain the strongly decreased xylose fermentation rate in the phosphoketolase-overexpressing strain during the solventogenic phase. Acetate accumulation and a negative effect of acetate on xylose fermentation were also observed for Saccharomyces cerevisiae into which a phosphoketolase pathway was introduced (36). We found a remarkable increase in the intracellular concentration of acetyl-P in the phosphoketolase-overexpressing strain, indicating that the acetate accumulation mostly likely arises from dephosphorylation of acetyl-P. To prevent phosphoketolase pathway-based acetate formation in C. acetobutylicum, overexpression of phosphotransacetylase may be necessary to convert acetyl-P to acetyl-CoA. In addition, inactivation of acetate kinase is also expected to reduce the production of acetate from acetyl-P (20) and thus increase the carbon flow from acetyl-P to acetyl-CoA that could be used for solvent formation.
ACKNOWLEDGMENTS
This work was supported in part by the Natural Science Foundation of China (31070033 and 31121001), the National Basic Research Program of China (973: 2012CB721101), the Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-EW-G-5 and KSCX1-YW-11C3), and the Open Funding Project of the State Key Laboratory of Bioreactor Engineering.
Footnotes
Published ahead of print 3 August 2012
REFERENCES
- 1. Amador-Noguez D, Brasg IA, Feng XJ, Roquet N, Rabinowitz JD. 2011. Metabolome remodeling during the acidogenic-solventogenic transition in Clostridium acetobutylicum. Appl. Environ. Microbiol. 77:7984–7997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amador-Noguez D, et al. 2010. Systems-level metabolic flux profiling elucidates a complete, bifurcated tricarboxylic acid cycle in Clostridium acetobutylicum. J. Bacteriol. 192:4452–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arskold E, et al. 2008. Phosphoketolase pathway dominates in Lactobacillus reuteri ATCC 55730 containing dual pathways for glycolysis. J. Bacteriol. 190:206–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baer SH, Blaschek HP, Smith TL. 1987. Effect of butanol challenge and temperature on lipid composition and membrane fluidity of butanol-tolerant Clostridium acetobutylicum. Appl. Environ. Microbiol. 53:2854–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baronofsky JJ, Schreurs WJ, Kashket ER. 1984. Uncoupling by acetic acid limits growth of and acetogenesis by Clostridium thermoaceticum. Appl. Environ. Microbiol. 48:1134–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crown SB, et al. 2011. Resolving the TCA cycle and pentose-phosphate pathway of Clostridium acetobutylicum ATCC 824: isotopomer analysis, in vitro activities and expression analysis. J. Biotechnol. 6:300–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Durre P. 2007. Biobutanol: an attractive biofuel. J. Biotechnol. 2:1525–1534 [DOI] [PubMed] [Google Scholar]
- 8. Fischer E, Sauer U. 2003. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 270:880–891 [DOI] [PubMed] [Google Scholar]
- 9. Fischer E, Sauer U. 2003. A novel metabolic cycle catalyzes glucose oxidation and anaplerosis in hungry Escherichia coli. J. Biol. Chem. 278:46446–46451 [DOI] [PubMed] [Google Scholar]
- 10. Follstad BD, Stephanopoulos G. 1998. Effect of reversible reactions on isotope label redistribution: analysis of the pentose phosphate pathway. Eur. J. Biochem. 252:360–371 [DOI] [PubMed] [Google Scholar]
- 11. Gombert AK, Santos MM, Christensen B, Nielsen J. 2001. Network identification and flux quantification in the central metabolism of Saccharomyces cerevisiae under different conditions of glucose repression. J. Bacteriol. 183:1441–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grill JP, Crociani J, Ballongue J. 1995. Characterization of fructose 6 phosphate phosphoketolases purified from Bifidobacterium species. Curr. Microbiol. 31:49–54 [DOI] [PubMed] [Google Scholar]
- 13. Grimmler C, Held C, Liebl W, Ehrenreich A. 2010. Transcriptional analysis of catabolite repression in Clostridium acetobutylicum growing on mixtures of D-glucose and D-xylose. J. Biotechnol. 150:315–323 [DOI] [PubMed] [Google Scholar]
- 14. Gu Y, et al. 2010. Reconstruction of xylose utilization pathway and regulons in Firmicutes. BMC Genomics 11:255 doi:10.1186/1471-2164-11-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gu Y, et al. 2011. Economical challenges to microbial producers of butanol: feedstock, butanol ratio and titer. J. Biotechnol. 6:1348–1357 [DOI] [PubMed] [Google Scholar]
- 16. Gu Y, et al. 2009. Improvement of xylose utilization in Clostridium acetobutylicum via expression of the talA gene encoding transaldolase from Escherichia coli. J. Biotechnol. 143:284–287 [DOI] [PubMed] [Google Scholar]
- 17. Hua Q, Yang C, Baba T, Mori H, Shimizu K. 2003. Responses of the central metabolism in Escherichia coli to phosphoglucose isomerase and glucose-6-phosphate dehydrogenase knockouts. J. Bacteriol. 185:7053–7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang L, Forsberg CW, Gibbins LN. 1986. Influence of external pH and fermentation products on Clostridium acetobutylicum intracellular pH and cellular distribution of fermentation products. Appl. Environ. Microbiol. 51:1230–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kandler O. 1983. Carbohydrate metabolism in lactic acid bacteria. Antonie Van Leeuwenhoek 49:209–224 [DOI] [PubMed] [Google Scholar]
- 20. Kuit W, Minton NP, Lopez-Contreras AM, Eggink G. 2012. Disruption of the acetate kinase (ack) gene of Clostridium acetobutylicum results in delayed acetate production. Appl. Microbiol. Biotechnol. 94:729–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee SY, et al. 2008. Fermentative butanol production by clostridia. Biotechnol. Bioeng. 101:209–228 [DOI] [PubMed] [Google Scholar]
- 22. Meile L, Rohr LM, Geissmann TA, Herensperger M, Teuber M. 2001. Characterization of the D-xylulose 5-phosphate/D-fructose 6-phosphate phosphoketolase gene (xfp) from Bifidobacterium lactis. J. Bacteriol. 183:2929–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mermelstein LD, Papoutsakis ET. 1993. In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol. 59:1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mermelstein LD, Welker NE, Bennett GN, Papoutsakis ET. 1992. Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824. Biotechnology 10:190–195 [DOI] [PubMed] [Google Scholar]
- 25. Mitchell WJ. 1998. Physiology of carbohydrate to solvent conversion by clostridia. Adv. Microb. Physiol. 39:31–130 [DOI] [PubMed] [Google Scholar]
- 26. Nanchen A, Fuhrer T, Sauer U. 2007. Determination of metabolic flux ratios from 13C-experiments and gas chromatography-mass spectrometry data: protocol and principles. Methods Mol. Biol. 358:177–197 [DOI] [PubMed] [Google Scholar]
- 27. Nolling J, et al. 2001. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J. Bacteriol. 183:4823–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Panagiotou G, et al. 2008. Systems analysis unfolds the relationship between the phosphoketolase pathway and growth in Aspergillus nidulans. PLoS One 3:e3847 doi:10.1371/journal.pone.0003847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ren C, et al. 2010. Identification and inactivation of pleiotropic regulator CcpA to eliminate glucose repression of xylose utilization in Clostridium acetobutylicum. Metab. Eng. 12:446–454 [DOI] [PubMed] [Google Scholar]
- 30. Sauer U. 2006. Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2:62 doi:10.1038/msb4100109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sauer U, et al. 1999. Metabolic flux ratio analysis of genetic and environmental modulations of Escherichia coli central carbon metabolism. J. Bacteriol. 181:6679–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmidt K, Carlsen M, Nielsen J, Villadsen J. 1997. Modeling isotopomer distributions in biochemical networks using isotopomer mapping matrices. Biotechnol. Bioeng. 55:831–840 [DOI] [PubMed] [Google Scholar]
- 33. Servinsky MD, Kiel JT, Dupuy NF, Sund CJ. 2010. Transcriptional analysis of differential carbohydrate utilization by Clostridium acetobutylicum. Microbiology 156:3478–3491 [DOI] [PubMed] [Google Scholar]
- 34. Sgorbati B, Lenaz G, Casalicchio F. 1976. Purification and properties of two fructose-6-phosphate phosphoketolases in Bifidobacterium. Antonie Van Leeuwenhoek 42:49–57 [DOI] [PubMed] [Google Scholar]
- 35. Shao L, et al. 2007. Targeted gene disruption by use of a group II intron (targetron) vector in Clostridium acetobutylicum. Cell Res. 17:963–965 [DOI] [PubMed] [Google Scholar]
- 36. Sonderegger M, Schumperli M, Sauer U. 2004. Metabolic engineering of a phosphoketolase pathway for pentose catabolism in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 70:2892–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanaka K, et al. 2002. Two different pathways for D-xylose metabolism and the effect of xylose concentration on the yield coefficient of L-lactate in mixed-acid fermentation by the lactic acid bacterium Lactococcus lactis IO-1. Appl. Microbiol. Biotechnol. 60:160–167 [DOI] [PubMed] [Google Scholar]
- 38. Thauer RK, Jungermann K, Decker K. 1977. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41:100–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thykaer J, Nielsen J. 2007. Evidence, through C13-labelling analysis, of phosphoketolase activity in fungi. Process Biochem. 42:1050–1055 [Google Scholar]
- 40. Tummala SB, Welker NE, Papoutsakis ET. 1999. Development and characterization of a gene expression reporter system for Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol. 65:3793–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wiechert W. 2001. 13C metabolic flux analysis. Metab. Eng. 3:195–206 [DOI] [PubMed] [Google Scholar]
- 42. Wiechert W, de Graaf AA. 1997. Bidirectional reaction steps in metabolic networks: I. Modeling and simulation of carbon isotope labeling experiments. Biotechnol. Bioeng. 55:101–117 [DOI] [PubMed] [Google Scholar]
- 43. Wiesenborn DP, Rudolph FB, Papoutsakis ET. 1988. Thiolase from Clostridium acetobutylicum ATCC 824 and its role in the synthesis of acids and solvents. Appl. Environ. Microbiol. 54:2717–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xiao H, et al. 2011. Confirmation and elimination of xylose metabolism bottlenecks in glucose phosphoenolpyruvate-dependent phosphotransferase system-deficient Clostridium acetobutylicum for simultaneous utilization of glucose, xylose, and arabinose. Appl. Environ. Microbiol. 77:7886–7895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang C, Rodionov DA, Rodionova IA, Li X, Osterman AL. 2008. Glycerate 2-kinase of Thermotoga maritima and genomic reconstruction of related metabolic pathways. J. Bacteriol. 190:1773–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yevenes A, Frey PA. 2008. Cloning, expression, purification, cofactor requirements, and steady state kinetics of phosphoketolase-2 from Lactobacillus plantarum. Bioorg. Chem. 36:121–127 [DOI] [PubMed] [Google Scholar]
- 47. Zamboni N, Fendt SM, Ruhl M, Sauer U. 2009. 13C-based metabolic flux analysis. Nat. Protoc. 4:878–892 [DOI] [PubMed] [Google Scholar]
- 48. Zhang L, et al. 2012. Ribulokinase and transcriptional regulation of arabinose metabolism in Clostridium acetobutylicum. J. Bacteriol. 194:1055–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao Y, Tomas CA, Rudolph FB, Papoutsakis ET, Bennett GN. 2005. Intracellular butyryl phosphate and acetyl phosphate concentrations in Clostridium acetobutylicum and their implications for solvent formation. Appl. Environ. Microbiol. 71:530–537 [DOI] [PMC free article] [PubMed] [Google Scholar]