

Abstract
Neurodegenerative diseases are typically associated with an activation of glia and an increased level of cytokines. In our previous studies of prion disease, the cytokine response in the brains of clinically sick scrapie-infected mice was restricted to a small group of cytokines, of which IL-12p40, CCL2, and CXCL10 were present at the highest levels. The goal of our current research was to determine the relationship between cytokine responses, gliosis, and neuropathology during prion disease. Here, in time course studies of C57BL/10 mice intracerebrally inoculated with 22L scrapie, abnormal protease-resistant prion protein (PrPres), astrogliosis, and microgliosis were first detected at 40 days after intracerebral scrapie inoculation. In cytokine studies, IL-12p40 was first elevated by 60 days; CCL3, IL-1β, and CXCL1 were elevated by 80 days; and CCL2 and CCL5 were elevated by 115 days. IL-12p40 showed the most extensive increase throughout disease and was 30-fold above control levels at the terminal stage. Because of the early onset and dramatic elevation of IL-12p40 during scrapie, we investigated whether IL-12p40 contributed to the development of prion disease neuropathogenesis by using three different scrapie strains (22L, RML, 79A) to infect knockout mice in which the gene encoding IL-12p40 was deleted. We also studied knockout mice lacking IL-12p35, which combines with IL-12p40 to form active IL-12 heterodimers. In all instances, knockout mice did not differ from control mice in survival time, clinical tempo, or levels of spongiosis, gliosis, or PrPres in the brain. Thus, neither IL-12p40 nor IL-12p35 molecules were required for prion disease-associated neurodegeneration or neuroinflammation.
INTRODUCTION
Prion diseases, also known as transmissible spongiform encephalopathy (TSE) diseases, are infectious neurodegenerative disorders that affect both humans and animals (24). Prion diseases are characterized by spongiform degeneration of gray matter, gliosis, and accumulation of a misfolded, partially protease resistant form, PrPres (also known as PrPSc), of the normal cellular prion protein, PrPsen (also known as PrPC). Activation and/or proliferation of astroglia and microglia is prominent during prion disease pathogenesis. Microglial and astroglial cells produce inflammatory mediators such as cytokines and chemokines in various neurodegenerative and infectious diseases, and all may contribute to the inflammation in prion disease (5, 7, 9, 18, 25, 27, 35). This neuroinflammatory reaction appears to be a host response to PrPres accumulation and associated brain cell damage. However, this neuroinflammation might also act either to increase or to decrease prion disease pathogenesis, and if so, therapeutic intervention to increase or dampen this reaction might be beneficial.
Our previous study focused on the cytokine response in vitro in glial cells and in vivo in the brains of sick mice after scrapie infection (35). Despite extensive gliosis during prion disease, scrapie-infected mice showed consistent significant elevation of only 6 cytokines (CCL2, CCL5, CXCL1, CXCL10, interleukin 1β [IL-1β], and IL-12p40) (35). The low number of increased cytokines and the low levels attained after scrapie infection differed from the pattern seen in the brain following a typical bacterial or viral infection (3, 13, 31).
In the current experiments, to determine the cause-and-effect relationships of early abnormalities seen in the scrapie pathogenic process, we studied mice at a series of time points starting at 20 days after scrapie infection. PrPres accumulation, astrogliosis, microgliosis, and cytokine levels in the brains of scrapie-infected mice were analyzed. In addition, 2 strains of knockout (KO) mice lacking expression of cytokines of possible importance to disease were infected with scrapie and were monitored for altered pathogenesis.
MATERIALS AND METHODS
Mice.
All mice were housed at the Rocky Mountain Laboratories (RML) in an AAALAC-accredited facility, and experimentation followed NIH RML Animal Care and Use Committee-approved protocols. Female C57BL/10/SnJ mice were used for the initial scrapie pathogenesis experiments (Jackson Laboratories, Bar Harbor, ME). Female B6.129S1-IL12btm1Jm/J (IL-12p40 KO), B6.129S1-IL12atm1Jm/J (IL-12p35 KO), and C57BL/6J (B6) (Jackson Laboratories) mice were used in subsequent scrapie infection studies. In order to reduce genetic differences in background genes, KO mice were backcrossed 11 times to C57BL/6 mice before intercrossing to obtain homozygous KO mice. These three strains should have nearly identical background genes, and differences among them are likely due to changes at the targeted KO loci. Mice were purchased as weanlings and were housed in groups of 4 per box.
Inoculation and observation of mice.
Mice were anesthetized with isoflurane and were inoculated intracerebrally (i.c.) in the left hemisphere at the age of 4 to 6 weeks with 50 μl of a 1% (wt/vol) scrapie brain homogenate stock in phosphate-buffered balanced saline (PBBS) with 2% fetal bovine serum (FBS). C57BL/10/SnJ mice were inoculated with the mouse-adapted scrapie strain 22L (1.0 × 106 50% lethal doses [LD50]) or with normal brain material in PBBS. Mice in this experiment were euthanized at 20-day intervals (4 mice per group) until the onset of scrapie signs. Brains were collected, and the left half was placed in formalin for histologic analysis, while the right half was flash frozen in liquid nitrogen for later biochemical analysis. Mock-infected age-matched controls were processed in a similar manner. IL-12p40 KO, IL-12p35 KO, and B6 mice were inoculated i.c. with either the 22L (1.0 × 106 LD50), RML (4.0 × 104 LD50), or 79A (1.8 × 105 LD50) strain of rodent-adapted scrapie. Following infection, mice were monitored weekly for the onset of scrapie signs. At 100 dpi, mice were monitored biweekly, and mice were euthanized when they displayed consistent signs of ataxia, kyphosis, somnolence, and hind leg weakness. Brains were collected as described above for the C57BL/10/SnJ mice.
Immunohistochemistry.
Mice were euthanized; brains were removed; and the left half of each brain was placed in 3.7% phosphate-buffered formalin for 3 to 5 days before dehydration and embedding in paraffin. Serial 4-μm sections were cut using a standard Leica microtome, placed on positively charged glass slides, and dried overnight at 56°C. Slides were stained with a standard protocol of hematoxylin and eosin (H&E) for observation of overall pathology. For the detection of PrPres, Iba1, and glial fibrillary acidic protein (GFAP), slide processing was completed in a Discovery XT slide stainer (Ventana, Tucson, AZ).
PrPres was detected as described previously (15). In brief, PrPres antigen was retrieved in a Ventana automated Discovery XT stainer by incubation for 188 min at 95°C in CC1 buffer (Ventana) containing Tris-borate-EDTA (pH 8.0), followed by (i) staining of PrPres with the monoclonal human anti-mouse PrP antibody D13 (dilution, 1:500) (In-Pro Biotechnology, South San Francisco, CA) at 37°C for 2 h, (ii) secondary staining with biotinylated anti-human IgG (dilution, 1:250) (Jackson ImmunoResearch, West Grove, PA), and (iii) avidin-horseradish peroxidase (HRP) and chromogen development with a DAB Map kit (Ventana).
Sections were stained for microglia by using the primary antibody Iba1. Slides were exposed to the standard antigen retrieval procedure (44 min at 100°C) and were stained with anti-Iba1 (polyclonal rabbit anti Iba1; kindly provided by John Portis) at 1:2,000 for 40 min at 37°C. The secondary antibody, goat biotinylated anti-rabbit IgG (Biogenex), was applied for 40 min and was followed by streptavidin-alkaline phosphatase. The chromogen used was Fast Red (Red Map kit; Ventana).
Immunostaining of slides for astrocyte detection used a mild antigen retrieval step (22 min at 100°C), followed by (i) staining with polyclonal rabbit anti-GFAP (Dako) at a 1:3,500 dilution for 16 min, (ii) staining with goat biotinylated anti-rabbit IgG as a secondary antibody for 16 min, and (iii) streptavidin-alkaline phosphatase using Fast Red as the chromogen.
All histopathology slides were read either using an Olympus BX51 microscope or as high-resolution scanned slide images visualized on computer monitors (Spectrum and Imagescope software by Aperio). Images were obtained using Microsuite FIVE software on the microscope or Imagescope software on the computer.
Preparation of brain homogenates from scrapie- and mock-infected mice.
Brains were homogenized (20% [wt/vol]) in PBS containing 0.1 mM Pefabloc, using a Mini Bead Beater (BioSpec Products) as described previously (35). Brain homogenates were sonicated for 1 min, vortexed aggressively for 30 s, and frozen in aliquots at −80°C for future use.
Western blotting for PrPres.
For PrPres immunoblotting, tissue samples were analyzed as described previously (21). Briefly, 20 μl of a 20% tissue homogenate was adjusted to 100 mM Tris HCl (pH 8.3), 1% Triton X-100, and 1% sodium deoxycholate in a total volume of 31 μl. Samples were treated with 50 μg/ml of proteinase K (Roche Diagnostics) for 45 min at 37°C. The reaction was stopped by adding 2 μl of 100 mM Pefabloc (Roche Diagnostics), and the reaction product was placed on ice for 5 min. An equal volume of 2× Laemmli sample buffer (Bio-Rad, Hercules, CA) was added, and then tubes were boiled for 5 min. Samples were frozen at −20°C until they were electrophoresed on a 16% Tris-glycine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel (Life Technologies, CA) and blotted to polyvinylidene difluoride (PVDF) membranes using a 7-min transfer, program 3 (P3), on an iBlot (Life Technologies, CA) device. Immunoblots were probed with monoclonal human anti-PrP antibody D13 at a 1:100 dilution (0.66 μg/ml) using a supernatant made in our laboratory from CHO cells expressing the D13 antibody construct (20), kindly provided by R. Anthony Williamson, The Scripps Research Institute, La Jolla, CA (32). The secondary antibody was peroxidase-conjugated anti-human IgG (dilution, 1:5,000; Sigma, St. Louis, MO). Protein bands were visualized using an enhanced chemiluminescence (ECL) detection system (GE Healthcare).
For PrPres quantification during the course of infection, PrPres levels measured in samples from preclinical time points were compared to levels measured in brains obtained at the time of clinical disease. All brain homogenates were treated with proteinase K as described above. Two samples from each time point were analyzed. Fourfold serial dilutions of each brain were tested to facilitate comparisons.
Cytokine quantification.
A multiplex assay kit (Bio-Rad, Hercules, CA) was used to determine the concentration of cytokines in brain homogenates as described previously (34, 35). Nine cytokines (IL-12p40, CCL2, CCL3, CCL5, CXCL1, IL-1β IL-9, IL-13, gamma interferon) were analyzed. Samples were thawed and diluted to the final concentration in Dulbecco's modified Eagle medium (DMEM) with 10% FBS. Samples were usually assayed at a 1% concentration, but in one experiment, CCL2 was analyzed at 4% in order to obtain values in the midrange of the standard curve for this cytokine. All samples were assayed in duplicate. Data were obtained using the Bio-Plex Manager software program (version 4.1.1; Bio-Rad) for standardization and standard curve acquisition and were exported to Microsoft Excel (Microsoft Corporation, Seattle, WA) for further analysis. Statistical analysis was done for each cytokine using one-way analysis of variance (ANOVA) on GraphPad software to compare normal control mice with mice for whom data were obtained at various times after scrapie infection. For these experiments, 3 to 4 uninoculated control B6 mice were studied. In addition, some control B6 mice were inoculated with normal brain homogenate material at the same strength used for the scrapie stocks, and mice were euthanized between 20 and 135 days postinoculation (dpi). The results from all these mice appeared to be similar, and all control mice were grouped for comparison with infected mice for statistical analysis and graphic presentation.
RESULTS
Early PrPres accumulation and gliosis in prion disease.
To determine which pathogenic abnormalities were the earliest to occur after scrapie infection, we followed two hallmark features of prion disease, PrPres and gliosis, and compared these parameters to levels of various cytokines in scrapie-infected brain homogenates at different times after scrapie inoculation. For these experiments, C57BL/10 mice were infected intracerebrally with 1 × 106 50% infective doses (ID50) of the 22L strain of mouse scrapie, and groups of mice were euthanized at approximately 20-day intervals beginning at 20 days postinoculation (dpi). PrPres was first detected by immunoblotting at 40 dpi. The level was 0.1% of the level seen at terminal stage (Fig. 1A and B). Subsequently, PrPres accumulated slowly to about 10% of the terminal level by 100 dpi and increased rapidly thereafter to reach a maximum at the clinical stage at around 135 dpi (Fig. 1B).
Fig 1.

Early detection of PrPres in scrapie-infected brains. (A) Immunoblot detection of PrPres in brain at various times after i.c. inoculation with scrapie strain 22L. The upper panel was exposed for 10 s, and the lower panel was exposed for 30 min to increase sensitivity. Exposure for 120 min did not reveal any bands in the 20-dpi samples. Results similar to those in the lower panel were also seen when samples were analyzed using the phosphotungstic acid precipitation method (28). (B) PrPres was quantitated by density scanning of the gels shown in panel A. The average amount of PrPres detected at each time point is shown as a percentage of the amount detected at the terminal stage.
Infected brain tissue was also analyzed for PrPres by immunohistochemistry, because small localized foci of PrPres deposition might escape detection by immunoblotting of whole-brain homogenates. At 20 dpi, PrPres was not detected in any brain region (Fig. 2A), but at 40 dpi, PrPres was seen in several brain areas of all 4 mice studied, including the hypothalamus, thalamus, and interpeduncular nuclei (Fig. 2B to D). At 80 dpi (Fig. 2E) and at later times up to the clinical endpoint (data not shown), PrPres was found in a progressively wider distribution. Gliosis involving proliferation and activation of microglia and astroglia is a prominent feature of prion disease infection in brain.
Fig 2.

Early detection of PrPres and microglia by immunohistochemistry following scrapie infection. (A to E) Staining with the monoclonal anti-PrP antibody D13 for detection of PrPres (brown) in the thalamus and forebrain at various days postinoculation. (F to -J) Staining of microglia with anti-Iba1 in brain sections parallel to those studied in panels A to E. (A) At 20 dpi, no PrPres was detected; however, weak background staining of PrPsen was seen. Similar staining was seen in uninfected mice but was not seen in PrP-null mice, which confirmed its identity as PrPsen. (B) At 40 dpi, two patchy areas of PrPres were detected, one in the thalamus on the right and several small groups of staining in the forebrain on the left. (C and D) Enlargements of panel B. (E) At 80 dpi, PrPres was found over a more extensive area. (F) Anti-Iba1 staining at 20 dpi detected an even distribution of microglia in the same area. (Inset) Thin processes of cells characteristic of nonactivated microglia. (G) At 40 dpi, increased staining of Iba1 was seen near the same areas where PrPres was found (compare panel B). (H and I) Microglia with short thick processes and enlarged somata typical of activated microglia. (J) At 80 dpi, microglia with larger cell bodies were seen over most of the field shown, and the inset confirmed the morphology of activated cells. Bars, 250 μm (A, B, E, F, G, J), 100 μm (C and D), and 50 μm (H, I, and insets to panels F and J).
Therefore, brains were analyzed by immunohistochemistry using anti-Iba1 and anti-GFAP to detect microglia and astroglia, respectively. At 20 dpi, Iba1-positive cells had normal-appearing somata with thin and delicate projecting processes (Fig. 2F) and were not distinguishable from microglia seen in uninfected mice (not shown). At 20 dpi, the morphology and distribution of astroglia also appeared similar to those seen in uninfected mice (not shown). However, at 40 dpi, in the same brain regions with detectable PrPres, there was notable glial activation. Microglia were hypertrophic with an enlarged, darkened soma and shorter, thicker, less branched processes (Fig. 2G, H, And I), and astroglia exhibited increased GFAP staining with a swollen hypertrophic appearance indicating an activated phenotype (not shown). Gliosis was more extensive at 80 dpi (Fig. 2J) and increased steadily until the clinical endpoint at 135 dpi.
Early cytokine expression in prion disease.
In our previous studies, levels of several cytokines were found to be elevated in scrapie-infected brains at the time of clinical disease (35). Furthermore, in vitro cultures of microglia and astroglia stimulated with scrapie-infected brain homogenates were found to release cytokines. Since gliosis was detected as early as 40 dpi in the brains of mice infected with scrapie (Fig. 2), we investigated whether cytokines were also elevated in the brain at early times after scrapie inoculation. Levels of 9 cytokines in brain homogenates from the current experiment were quantitated using a multiplex assay. This analysis revealed a detectable increase above background for several cytokines. Relative to expression in control mice, IL-12p40 was first detected in scrapie-infected mice at 60 dpi and showed a progressive increase throughout the disease, with a 30-fold increase above the level in control mice at the clinical endpoint (Fig. 3A). CCL3, IL-1β, and CXCL1 were also elevated by 80 dpi, and CCL2 and CCL5 were increased between 115 and 120 dpi (Fig. 3B to F). Thus, there was a reproducible elevation of the levels of several cytokines, most prominently IL-12p40, starting during the early preclinical phase of scrapie infection. However, it was not clear whether these cytokines were actively contributing to the progressive brain damage observed or whether these cytokine responses were part of the host response attempting to repair the scrapie-induced brain damage.
Fig 3.

Detection of cytokines in brain tissue at various times after scrapie infection. Protein levels of 9 cytokines in the brains of scrapie 22L-infected mice were measured by multiplex assays at various days postinoculation. The six cytokines for which results are shown had values significantly elevated over those for normal control mice (N) at various days postinoculation. Data for CCL2 were obtained from samples assayed at a 4% brain concentration, and other cytokines were assayed at a 1% brain concentration. IL-9, IL-13, and gamma interferon were also analyzed, but no values above those for normal controls were detected. Two to four mice per group were studied. Statistical analysis was carried out using one-way ANOVA with GraphPad Prism software. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
Scrapie infection in IL-12p40 and IL-12p35 knockout mice.
Since IL-12p40 was found in the brain near the time of the earliest detectable brain alterations and also persisted throughout the course of the disease, we examined whether IL-12p40 was required for scrapie-induced disease by infection of IL-12p40 KO mice. Since IL-12p40 forms a heterodimer with IL-12p35 in the active IL-12 cytokine, we also examined IL-12p35 KO mice (for a review, see reference 6). C57BL/6 (B6) mice were used as controls for both types of KO mice.
In these experiments, we tested three different scrapie strains (22L, RML, and 79A) because of potential strain-specific differences in the pathogenic processes. In these experiments, each KO mouse type studied developed terminal scrapie disease at mean times similar to those for B6 control mice inoculated with the same scrapie strain (Fig. 4A and B). Disease progressed slightly faster in mice inoculated with strain 22L, likely because the titer of this strain was approximately 10-fold higher than those of the other two strains. Thus, neither IL-12p40 nor IL-12p35 influenced the tempo or incidence of prion-induced disease in mice.
Fig 4.

Scrapie incubation times and PrPres detection in infected knockout mice. (A and B) The periods of incubation until clinical disease in scrapie-infected IL-12p40 KO (A) and IL-12p35 KO (B) mice were compared to those in non-knockout B6 controls. Mice were inoculated intracerebrally with scrapie strain 22L, RML, or 79A. Because experiments were carried out at separate times, each KO mouse strain had a separate set of B6 controls, which were inoculated with the same dilution of the three scrapie strains. Each dot represents one mouse. Horizontal bars show the mean incubation period. Values represent the day postinoculation when the animal was euthanized due to advanced signs of clinical scrapie as described in Materials and Methods. (C and D) Immunoblot detection of PrPres in the brains of representative IL-12p40 KO (C) and IL-12p35 KO (D) mice and their respective control B6 mice at the clinical endpoint after infection by scrapie strain 22L, RML, or 79A. All samples were treated with proteinase K as described in Materials and Methods. Each lane was loaded with 0.5-mg tissue equivalents. The blot was probed using anti-PrP antibody D13 and was developed using enhanced chemiluminescence detection. Blots C and D were exposed for 2.5 and 1.0 min, respectively.
To see whether the disease induced in these KO mice differed from that seen in B6 control mice, we also tested several other parameters of scrapie disease pathogenesis in these experiments. At the time of the clinical endpoint, PrPres immunoblots from KO mice and B6 mice had similar intensities and similar glycoform banding patterns for each scrapie strain tested (Fig. 4C and D). Histopathology of the brain showed microgliosis and astrogliosis, as well as vacuolation of the gray matter by H&E staining, all of which were similar in the two types of KO mice and the B6 control mice (Fig. 5). In addition, at the clinical endpoint after scrapie infection, cytokine levels in the brain were similar to those reported previously (35) (data not shown). Thus, these results gave no evidence that lack of expression of IL-12p40 or IL-12p35 altered the basic parameters of brain disease with any of the three scrapie strains tested.
Fig 5.
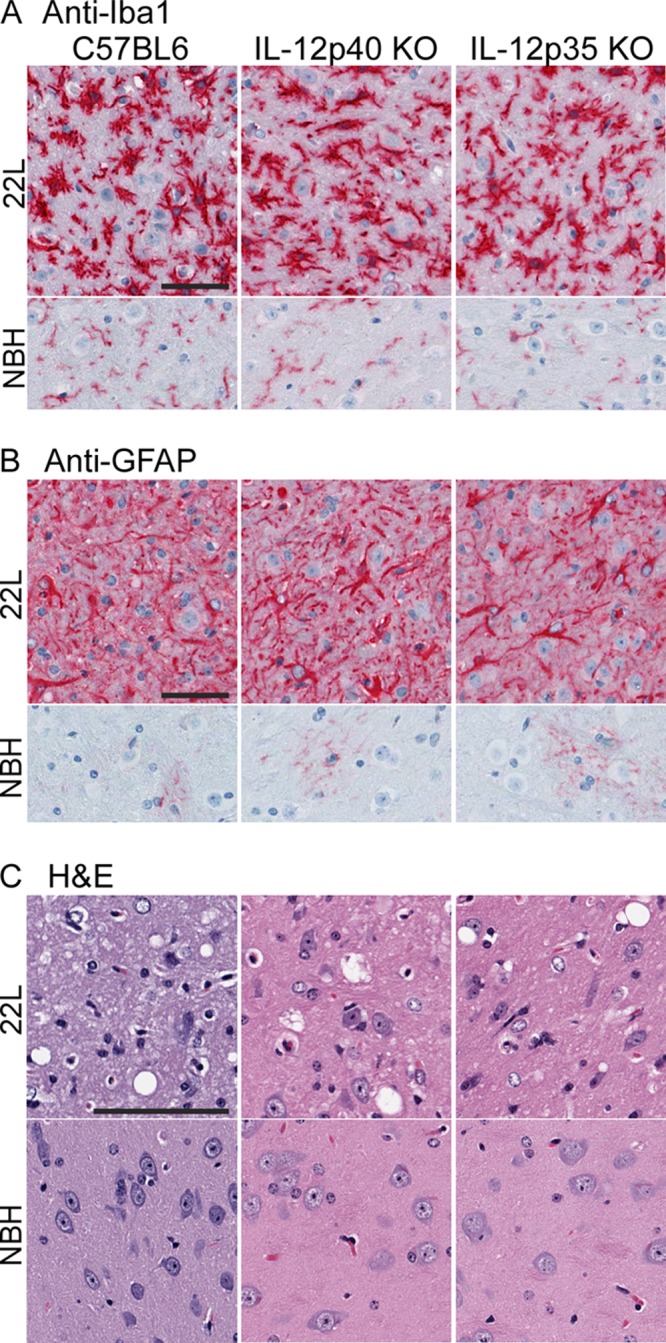
Detection of microgliosis, astrogliosis, and vacuolation in scrapie-infected knockout and control B6 mice. Mice were euthanized at the clinical endpoint (approximately 135 dpi) after infection with scrapie strain 22L. Control mice of each strain were inoculated with normal brain homogenate (NBH) and were euthanized at the same time point. (A and B) Immunohistochemical analysis using anti-Iba1 (A) or anti-GFAP (B) shows that the density and morphology of activated microglia and astroglia in infected mice were similar and differed from those of normal glia seen in NBH-infected controls. (C) H&E staining shows vacuolation of gray matter in all infected mice but not in NBH-infected controls. Images are from the thalamus, but similar observations were made in other gray-matter regions. Bars, 200 μm. All panels within each subgroup are at the same magnification.
DISCUSSION
In the present experiments, we found evidence for PrPres deposition and both astrogliosis and microgliosis in the brain at 40 days after intracerebral scrapie infection. In contrast, at 20 dpi, none of these findings were detectable. Thus, these three aspects of prion disease pathology appeared nearly simultaneously between 20 and 40 dpi. PrPres detection at 40 dpi in this system was 20 to 30 days earlier than in several previous studies (1, 2, 4, 12, 17, 30). This might be due to the rapid tempo of the disease induced by our stock of strain 22L, which is known to induce clinical disease at 135 to 140 dpi and is about 15 days faster than our stocks of other scrapie strains, such as RML, ME7, and 79A. This early time of onset of the pathological process in the brain should provide an opportunity for future detection of key early events in scrapie pathogenesis.
Using quantitative multiplex cytokine protein analyses with the same group of mice, we found elevated levels of several cytokines, including IL-12p40, CCL2, CCL3, CCL5, IL-1β, and CXCL1, starting at 60 to 80 dpi. These cytokines were previously shown to be present in scrapie-infected brains at the end stage of disease and were also produced by microglia and/or astroglia after in vitro stimulation with scrapie-infected brain extracts (35). These cytokines appeared slightly later than the initial PrPres deposits and gliosis, suggesting that cytokine elevation might be induced by these earlier pathological events. Alternatively, our biochemical cytokine assay based on analysis of whole-brain homogenates might not be sensitive enough to detect small foci of early cytokine production simultaneous with, or even preceding, PrPres deposition and gliosis. Because of these caveats, we cannot distinguish whether these cytokines contribute to the ongoing prion infection-induced brain damage and/or to the host response attempting to repair this damage.
The study of mice in which individual cytokine genes have been deleted is a powerful tool for testing the requirement for individual genes in disease pathogenesis or recovery. In previous studies of cytokine or cytokine receptor knockout mice using i.c. scrapie infection, a variety of outcomes have been seen. Depending on the individual gene tested and the scrapie strain utilized, survival times either were unchanged (IL-6 [19], IL-4 and/or IL-13 [33], MyD88 [23]), were slightly increased (CCL2 with strain ME7 [8], CXCR3 [26], or IL-1R [29]), or were slightly decreased (CCL2 with strain RML/Chandler [22], IL-10 [33], or CCR1 [16]). Thus, although most of these cytokines, cytokine receptors, and cytokine regulators would be expected to influence neuroinflammation and neurotoxicity after scrapie infection, no single cytokine or receptor has so far been proven to be required for scrapie disease.
In the current experiments, we attempted to determine whether prion disease brain pathogenesis was influenced by deletion of IL-12p40, the cytokine with the earliest onset and highest fold increase during disease. Knockout mice in which expression of IL-12p35 was deleted were also studied, because a heterodimer of IL-12p40 and IL-12p35 polypeptides forms the active IL-12 cytokine. In these experiments, scrapie was inoculated by the i.c. route in order to focus the study on pathogenesis within the central nervous system (CNS) rather than on aspects of extracerebral infection and neuroinvasion (14, 19). After scrapie infection, these two KO mouse strains showed no differences in survival time, PrPres occurrence, astrogliosis, microgliosis, or neutrophil vacuolation from non-KO C57BL/6 control mice (Fig. 4 and 5). Thus, none of the three cytokines containing IL-12p40 or IL-12p35—i.e., IL-12, IL-23, and the IL-12p40 dimer (see reference 6 for a review)—were required for these aspects of scrapie pathogenesis, nor were they required for fatal scrapie disease. Since many of these cytokines act on T lymphocytes and dendritic cells not found in the CNS during scrapie disease, it may not be surprising that no effect on scrapie was seen by deletion of these genes. However, in microglia, IL-12p70, the IL-12p40 monomer, and the IL-12p40 dimer all stimulate mitogen-activated protein (MAP) kinase and extracellular signal-regulated kinase (ERK) pathways, as well as inducing the production of tumor necrosis factor (TNF) and nitric oxide (10, 11). Nevertheless, based on our current experiments, these effects on microglial function do not influence scrapie disease tempo or survival after inoculation of any of the three scrapie strains tested in our studies.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Allergy and Infectious Diseases, Division of Intramural Research.
We thank Kimberly Meade-White for technical assistance with the mouse experiments, Jeffrey Severson for assistance with animal husbandry, Dan Long, Lori Lubke, and Nancy Kurtz for technical assistance with immunohistochemistry, and Suzette Priola and Karin Peterson for critical reading of the manuscript.
Footnotes
Published ahead of print 11 July 2012
REFERENCES
- 1. Andreoletti O, et al. 2002. Astrocytes accumulate 4-hydroxynonenal adducts in murine scrapie and human Creutzfeldt-Jakob disease. Neurobiol. Dis. 11:386–393 [DOI] [PubMed] [Google Scholar]
- 2. Bencsik AA, Leclere E, Perron H, Moussa A. 2008. New insights into early sequential PrPsc accumulation in scrapie infected mouse brain evidenced by the use of streptomycin sulfate. Histochem. Cell Biol. 129:643–650 [DOI] [PubMed] [Google Scholar]
- 3. Brown AR, et al. 2003. Inducible cytokine gene expression in the brain in the ME7/CV mouse model of scrapie is highly restricted, is at a strikingly low level relative to the degree of gliosis and occurs only late in disease. J. Gen. Virol. 84:2605–2611 [DOI] [PubMed] [Google Scholar]
- 4. Bueler H, et al. 1994. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol. Med. 1:19–30 [PMC free article] [PubMed] [Google Scholar]
- 5. Chen Y, Swanson RA. 2003. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 23:137–149 [DOI] [PubMed] [Google Scholar]
- 6. Cooper AM, Khader SA. 2007. IL-12p40: an inherently agonistic cytokine. Trends Immunol. 28:33–38 [DOI] [PubMed] [Google Scholar]
- 7. Dheen ST, Kaur C, Ling EA. 2007. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 14:1189–1197 [DOI] [PubMed] [Google Scholar]
- 8. Felton LM, et al. 2005. MCP-1 and murine prion disease: separation of early behavioural dysfunction from overt clinical disease. Neurobiol. Dis. 20:283–295 [DOI] [PubMed] [Google Scholar]
- 9. Hanisch UK. 2002. Microglia as a source and target of cytokines. Glia 40:140–155 [DOI] [PubMed] [Google Scholar]
- 10. Jana M, Dasgupta S, Saha RN, Liu X, Pahan K. 2003. Induction of tumor necrosis factor-alpha (TNF-α) by interleukin-12 p40 monomer and homodimer in microglia and macrophages. J. Neurochem. 86:519–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jana M, Pahan K. 2009. IL-12 p40 homodimer, but not IL-12 p70, induces the expression of IL-16 in microglia and macrophages. Mol. Immunol. 46:773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jeffrey M, Martin S, Barr J, Chong A, Fraser JR. 2001. Onset of accumulation of PrPres in murine ME7 scrapie in relation to pathological and PrP immunohistochemical changes. J. Comp. Pathol. 124:20–28 [DOI] [PubMed] [Google Scholar]
- 13. Kielian T. 2004. Microglia and chemokines in infectious diseases of the nervous system: views and reviews. Front. Biosci. 9:732–750 [DOI] [PubMed] [Google Scholar]
- 14. Klein MA, et al. 1997. A crucial role for B cells in neuroinvasive scrapie. Nature 390:687–690 [DOI] [PubMed] [Google Scholar]
- 15. Klingeborn M, et al. 2011. Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J. Virol. 85:1484–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. LaCasse RA, Striebel JF, Favara C, Kercher L, Chesebro B. 2008. Role of Erk1/2 activation in prion disease pathogenesis: absence of CCR1 leads to increased Erk1/2 activation and accelerated disease progression. J. Neuroimmunol. 196:16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lasmezas CI, et al. 1996. Strain specific and common pathogenic events in murine models of scrapie and bovine spongiform encephalopathy. J. Gen. Virol. 77(Pt 7):1601–1609 [DOI] [PubMed] [Google Scholar]
- 18. Lefkowitz DL, Lefkowitz SS. 2008. Microglia and myeloperoxidase: a deadly partnership in neurodegenerative disease. Free Radic. Biol. Med. 45:726–731 [DOI] [PubMed] [Google Scholar]
- 19. Mabbott NA, et al. 2000. Tumor necrosis factor alpha-deficient, but not interleukin-6-deficient, mice resist peripheral infection with scrapie. J. Virol. 74:3338–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsunaga Y, et al. 2001. Cryptic epitopes in N-terminally truncated prion protein are exposed in the full-length molecule: dependence of conformation on pH. Proteins 44:110–118 [DOI] [PubMed] [Google Scholar]
- 21. Meade-White K, et al. 2007. Resistance to chronic wasting disease in transgenic mice expressing a naturally occurring allelic variant of deer prion protein. J. Virol. 81:4533–4539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Shea M, et al. 2008. Investigation of Mcp1 as a quantitative trait gene for prion disease incubation time in mouse. Genetics 180:559–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prinz M, Hausler KG, Kettenmann H, Hanisch U. 2001. β-Adrenergic receptor stimulation selectively inhibits IL-12p40 release in microglia. Brain Res. 899:264–270 [DOI] [PubMed] [Google Scholar]
- 24. Prusiner SB. 1991. Molecular biology of prion diseases. Science 252:1515–1522 [DOI] [PubMed] [Google Scholar]
- 25. Riemer C, et al. 2004. Gene expression profiling of scrapie-infected brain tissue. Biochem. Biophys. Res. Commun. 323:556–564 [DOI] [PubMed] [Google Scholar]
- 26. Riemer C, et al. 2008. Accelerated prion replication in, but prolonged survival times of, prion-infected CXCR3−/− mice. J. Virol. 82:12464–12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rock RB, et al. 2004. Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 17:942–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Safar J, et al. 1998. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 4:1157–1165 [DOI] [PubMed] [Google Scholar]
- 29. Schultz J, et al. 2004. Role of interleukin-1 in prion disease-associated astrocyte activation. Am. J. Pathol. 165:671–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song PJ, et al. 2010. Evaluation of prion deposits and microglial activation in scrapie-infected mice using molecular imaging probes. Mol. Imaging Biol. 12:576–582 [DOI] [PubMed] [Google Scholar]
- 31. Stanton JB, Knowles DP, Call DR, Mathison BA, Baszler TV. 2009. Limited transcriptional response of ovine microglia to prion accumulation. Biochem. Biophys. Res. Commun. 386:345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Striebel JF, Race B, Meade-White KD, LaCasse R, Chesebro B. 2011. Strain specific resistance to murine scrapie associated with a naturally occurring human prion protein polymorphism at residue 171. PLoS Pathog. 7:e1002275 doi:10.1371/journal.ppat.1002275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thackray AM, McKenzie AN, Klein MA, Lauder A, Bujdoso R. 2004. Accelerated prion disease in the absence of interleukin-10. J. Virol. 78:13697–13707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tribouillard-Tanvier D, Carroll JA, Moore RA, Striebel JF, Chesebro B. 2012. Role of cyclophilin A from brains of prion-infected mice in stimulation of cytokine release by microglia and astroglia in vitro. J. Biol. Chem. 287:4628–4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tribouillard-Tanvier D, Striebel JF, Peterson KE, Chesebro B. 2009. Analysis of protein levels of 24 cytokines in scrapie agent-infected brain and glial cell cultures from mice differing in prion protein expression levels. J. Virol. 83:11244–11253 [DOI] [PMC free article] [PubMed] [Google Scholar]