

Abstract
Monocyte-derived macrophages (MDM) are widely distributed in all tissues and organs, including the central nervous system, where they represent the main part of HIV-infected cells. In contrast to activated CD4+ T lymphocytes, MDM are resistant to cytopathic effects and survive HIV infection for a long period of time. The molecular mechanisms of how HIV is able to persist in macrophages are not fully elucidated yet. In this context, we have studied the effect of in vitro HIV-1 infection on telomerase activity (TA), telomere length, and DNA damage. Infection resulted in a significant induction of TA. This increase was directly proportional to the efficacy of HIV infection and was found in both nuclear and cytoplasmic extracts, while neither UV light-inactivated HIV nor exogenous addition of the viral protein Tat or gp120 affected TA. Furthermore, TA was not modified during monocyte-macrophage differentiation, MDM activation, or infection with vaccinia virus. HIV infection did not affect telomere length. However, HIV-infected MDM showed less DNA damage after oxidative stress than noninfected MDM, and this resistance was also increased by overexpressing telomerase alone. Taken together, our results suggest that HIV induces TA in MDM and that this induction might contribute to cellular protection against oxidative stress, which could be considered a viral strategy to make macrophages better suited as longer-lived, more resistant viral reservoirs. In the light of the clinical development of telomerase inhibitors as anticancer therapeutics, inhibition of TA in HIV-infected macrophages might also represent a novel therapeutic target against viral reservoirs.
INTRODUCTION
Infection with human immunodeficiency virus (HIV) causes one of the most devastating illnesses of the human being, AIDS. As a major accomplishment in HIV/AIDS medicine, the introduction of highly active antiretroviral therapy (HAART) has dramatically improved the prognosis of HIV-infected persons. Under therapy, a significant proportion of treated individuals reach an almost normal life expectancy. However, HIV can persist in the body in spite of the presence of drugs that successfully inhibit key steps in the virus life cycle. It is well known that HIV remains detectable in cells of the macrophage-monocyte lineage and in latently infected CD4+ T lymphocytes (2, 21, 30, 54).
Interestingly, while infected activated T CD4+ lymphocytes are killed by the virus, macrophages that are widely distributed in all tissues and organs seem to be used as a potent viral reservoir (48, 74). These cells are resistant to HIV's cytopathic effects and apoptosis and exhibit longer life spans even when they are exposed to oxidative stress (3, 12, 20, 27, 31). Particularly in the central nervous system (CNS), infected macrophages release cytotoxic products like Tat, envelope HIV proteins, and nitric oxide, among others (43). Thereby, they induce the death of neurons and astroglial cells, leading to neurodegenerative diseases especially in long-term-infected patients (61), while infected macrophages survive in this microenvironment (57). Thus, they are able to release virions for weeks after infection and constitute an important source of HIV replication and release in an advanced state of AIDS progression, where the CD4+ T cell count is already very low (2). However, our knowledge of the mechanisms involved in macrophage resistance to HIV cytopathic effects is far from complete.
HIV infection may affect the cellular life span. Recently, it was reported that infection with HIV negatively affects telomerase activity (TA) in CD4+ and CD8+ T lymphocytes (7, 28). In line with this, we have found that HIV infection significantly reduces TA in Jurkat T lymphoblastoid cells (68). Furthermore, it has been demonstrated that HIV-specific CD8+ T cells in HIV controllers have longer telomeres than cells from patients that progress rapidly to AIDS because the HIV controllers exhibit higher levels of constitutive TA (51). Thus, telomerase activity can be controlled by HIV and does play a role in disease progression.
Surprisingly, some of the protective effects in infected macrophages resemble a cellular phenotype that is conferred to cells by noncanonical functions of telomerase. While the canonical function of telomerase is the elongation of the ends of chromosomes that shorten with each round of cell division, the noncanonical functions include inhibition of apoptosis, protection against oxidative stress, and improvement of DNA repair (1, 34) also in normal cells (83). With the refined detection techniques now at hand, it is clear that many normal differentiated human cells display very low telomerase activity, including endothelial cells, fibroblasts, and dendritic cells among others (4, 22, 52, 55, 62). The low level of activity does not allow for the immortalization of cells as observed upon telomerase overexpression (16, 24, 53, 82) and still is not a well-understood function. Furthermore, cells of the immune system are known to be able to reactivate telomerase at least after stimuli that trigger clonal expansion (56, 81).
These hints prompted us to test the hypothesis that noncanonical functions of telomerase are induced by HIV infection and might be part of the protective effects of HIV on macrophages, thus increasing macrophage suitability as a viral reservoir.
Here, we present evidence in favor of this hypothesis as HIV infection is indeed able to induce TA in monocyte-derived macrophages (MDM), and we show that this effect is specific for HIV infection and correlates with p24 antigen production. Furthermore, increased TA by either HIV infection or by overexpression of the catalytic subunit of telomerase (human telomerase reverse transcriptase [hTERT]) leads to higher resistance of MDM against oxidative stress-induced DNA damage. Therefore, we here propose that HIV “hitchhikes” on TA in macrophages to misuse these cells as one of its main reservoirs. In consequence, specific inhibition of telomerase activity in macrophages might represent a therapeutic target against HIV.
MATERIALS AND METHODS
Isolation and culturing of primary monocytes.
Peripheral blood mononuclear cells (PBMCs) were isolated from blood of healthy donors by centrifugation over a density gradient (Ficoll-Hypaque) according to standard procedures (6). Briefly, monocytes were separated from lymphocytes by adherence to plastic flasks. After overnight incubation, nonadherent cells were removed by three washes with warm phosphate-buffered saline (PBS). The purity of monocytes was >90%, as determined by immunofluorescent staining with anti-CD14 monoclonal antibody ([MAb] BD Pharmingen) using a FACSCanto flow cytometer (BD). Monocytes were differentiated into macrophages by culturing in RPMI medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U of penicillin/ml, 100 μg of streptomycin/ml (RPMIc), and 50 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; Sigma-Aldrich, St. Louis, MO) for 6 days prior to infection. The culture medium was replaced every 2 days.
Experiments were approved by the ethics committee of the Huesped Foundation, Buenos Aires, Argentina.
Virus preparation and in vitro HIV-1 infection of MDM.
The HIV strains included R5 (BaL, JRCSF, and SF162), X4 (HXB2 and IIIB), and X4R5 (93BR020; primary viral isolate) variants obtained from the AIDS Research and Reference Reagent Program (AIDS Division, National Institute of Allergy and Infectious Diseases, National Institutes of Health). The HIV BaL stock was prepared by expansion of the inoculum in MDM from healthy HIV-negative donors (47). Clarified supernatant from cell-free culture was aliquoted under different conditions, as follows: (i) ultracentrifugation at 30,000 rpm for 1 h at 4°C in order to pellet the virus but conserve soluble factors such as cytokines in the supernatant, (ii) UV light exposure for 1 h to inactivate virus infectivity, or (iii) storage without modifications. Six days after isolation, MDM cultures were exposed to supernatants treated as described above as well as to infective viral stocks of IIIB, HXB2, 93BR020, JRCSF, and SF162 and to RPMI medium as a negative control for 2 h at 37°C. For virus infection, the inoculum was adjusted to 100 ng of p24 per 1 × 106 MDM.
The efficacy of infection was monitored by p24 antigen enzyme-linked immunosorbent assay (ELISA) in culture supernatants (Coulter HIV-1 p24 antigen assay) and by intracytoplasmic HIV p24 antigen staining using fluorescein isothiocyanate (FITC)-labeled IgG anti-p24 (Coulter Beckman) followed by flow cytometry analysis (FACSCanto; BD). Since MDM are difficult to remove, they were incubated with 0.25% trypsin-EDTA for 20 min, and the cells were finally detached after strong pipetting. MDM were harvested at early time points (2, 5, 7, and 24 h) and at 12 days postinfection.
Vaccinia virus infection.
MDM were infected directly with Western Reserve (WR) and modified vaccinia virus Ankara (MVA) carrying a green fluorescent protein (GFP) reporter gene (MVA-GFP) (kindly provided by M. Gherardi). Briefly, for infection, purified virus was added to the cells at a multiplicity of infection (MOI) of 0.5 or 1 for 90 min at 37°C; a negative control was also performed using RPMIc medium. After incubation, cells were washed three times and cultivated as described above. At 24 h postinfection, cells were harvested, washed with PBS, and stored at −80°C until used or analyzed for GFP expression by flow cytometry (FACSCanto; BD) (see Fig. S1 in the supplemental material).
hTERT gene transduction.
For introduction of hTERT, the retroviral vector pLXSN was used (Clontech Laboratories Inc.). Retroviral particles carrying hTERT were produced, and infection of MDM was performed as described previously (82). Forty-eight hours after infection, MDM infected with pLXSN-hTERT and MDM infected with pLXSN-neo (control) were harvested. To confirm that hTERT transduction indeed resulted in TA, we used the relative TA as normalized to Jurkat cell levels (see Fig. S2A in the supplemental material) (82).
Challenge with Tat and gp120 proteins.
HIV-1 recombinant soluble Tat and gp120 proteins were obtained from the NIH AIDS Research and Reference Reagent Program (www.aidsreagent.org). One million MDM were incubated in 2 ml of RPMIc medium supplemented with either 500 ng of Tat/ml or 500 ng of gp120/ml for 8 and 24 h. The concentrations and time points were chosen in accordance with published data (18, 84). Cells were harvested at the different time points, washed, centrifuged (250 × g for 5 min), and stored at −80°C until use.
Challenge with LPS antigen and IL-4.
MDM in RPMIc medium were stimulated by the addition of 1, 10, or 100 ng/ml lipopolysaccharide ([LPS] Escherichia coli serotype 055:B5; Sigma-Aldrich) (10) and/or 10 ng of human recombinant interleukin-4 (IL-4; BD Biosciences). Cells were harvested 8 and 24 h later, washed with PBS, centrifuged (250 × g for 5 min), and stored at −80°C until used. The concentrations and time points were chosen in accordance with published work (10). Tumor necrosis factor alpha (TNF-α) levels were studied as an activation control (see Fig. S2B in the supplemental material).
Preparation of nuclear and cytoplasmic extract.
Twelve days postinfection, 1 × 107 HIV-1-infected and uninfected MDM were harvested. Nuclear and cytoplasmic extracts were prepared using a Nuclear Extract Kit (Active Motif) according to the manufacturer's instructions. Supernatants were harvested as cytoplasmic fractions. Pellets were resuspended in 50 μl of complete lysis buffer and centrifuged at 14,000 × g for 10 min at 4°C; supernatants were saved as the nuclear fractions.
Measurement of telomerase activity.
MDM (1 × 106) were lysed in 200 μl of CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate) buffer and incubated for 30 min on ice. After incubation, lysates were centrifuged at 14,000 × g for 20 min at 4°C. The protein concentrations of the whole, nuclear, and cytoplasmic extracts were measured using a Bradford assay, followed by dilution to a final concentration of 0.01 μg/μl. Then, TA of extracts was measured by quantitative real-time PCR (qPCR) amplification of telomeric repeat fragments, as described elsewhere (39, 80). The total volume of the reaction mixture was 25 μl per well, containing 12.5 μl of 1× SYBR green Master Mix (Biosystems), 0.1 μg of TS primer (5′-AATCCGTCGAGCAGAGTT-3′), 0.05 μg of ACX primer (5′-GCGCGGCTTACCCTTACCCTTACCCTAACC-3′), 1 μl of RNase-free water, and 10 μl of sample.
The reaction mixture was incubated for 20 min at 25°C. Then, the PCR was started at 95°C for 15 min, followed by 40 cycles of amplification (95°C for 15 s and 60°C for 60 s). The assays were performed in duplicates. RNase-free water and heat-inactivated eluates (85°C for 10 min) were used as negative controls. The threshold cycle values (CT) were determined from semilog amplification plots (log increase in fluorescence versus cycle number) and compared with standard curves generated from serial dilutions of protein extracts from Jurkat E6 cells. TA was quantified and expressed relative to Jurkat cells using the formula described by Herbert et al. (38): TA = 10 (CT (sample) − CT (positive control))/slope.
Telomere length determination.
Telomere length was determined in MDM by a qPCR as described in detail previously (15, 26). In brief, telomere repeat copy numbers (T) and a single-copy gene (S; gene 36B4) were amplified using the following primers: for telomere amplifications, Tel1b (5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3′) and tel2b (5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′); for single-copy gene amplifications, 36B4-sense (5′-CAGCAAGTGGGAAGGTGTAATCC-3′) and 36B4-antisense (5′-CCCATTCTATCATCAACGGGTACAA-3′). Different concentrations of DNA were used for generating a standard curve (2 ng, 20 ng, and 200 ng). All samples, for both the telomere and single-copy gene amplifications, were done in duplicate. The CT values generated were used to calculate the T/S ratio for each sample using the following equation: T/S = 2 − ΔCT, where ΔCT = CT(single-copy gene) − CT(telomere). The T/S ratio is proportional to telomere length (14, 15).
Oxidative stress treatment and DNA damage.
MDM overexpressing hTERT and those transduced with pLXSN-neo were treated with 100 μM paraquat for 3 h. Then, the cells were harvested, and DNA damage levels were evaluated.
A comet assay under alkaline conditions was used for determining DNA damage in both infected and noninfected MDM (79). In brief, 5 × 105 cells/ml were mixed with 1% agarose and cast on microscopic slides. After solidification, the slides were placed in lysis solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Trizma, pH 10, and 1% Triton X-100) for 1 h at 4°C. Thereafter, the slides were sited in a horizontal gel electrophoresis unit filled with fresh electrophoretic buffer (1 mM Na2EDTA and 300 mM NaOH, pH > 13) and left in this buffer for 20 min for DNA unwinding; next, the slides were subjected to electrophoresis for 20 min at 25V at 4°C.
After electrophoresis the slides were rinsed with 0.4 M Tris, pH 7.5 (three times for 5 min each time), and then stained using Trevigen's Comet Assay Silver Staining Kit.
At least 50 comets/slide and 2 slides/sample were quantified, using the software CometScore, version 1.5. (open source software available at http://www.autocomet.com/products_cometscore.php). Thus, based on the percentage of DNA in tails, cells were assigned to classes: class 1 (less than 5% of DNA in tail), class 2 (between 5% and 25%), class 3 (between 25% and 45%), class 4 (between 45% and 65%), and class 5 (major to 65%).
Statistical analysis.
A one-way analysis of variance (ANOVA) test, followed by a Bonferroni test, was used to calculate statistical differences in TA from MDM under different conditions. A Pearson correlation was used to determine the magnitude of association between the levels of p24 antigen in culture supernatant and the TA. A nonparametric Mann-Whitney test was used to determine the significance of differences found in comet distribution in comet assays. P values of <0.05 were considered significant.
RESULTS
HIV infection induces telomerase activity in MDM.
It has been reported that upon HIV infection, the half-life of macrophages is prolonged, in sharp contrast to the cytopathic effects that kill activated CD4+ T lymphocytes. Since telomerase activity is regulated in CD4+ T cells by HIV infection (28, 29) and since the enhanced resistance of macrophages to oxidative stress and apoptosis resembles a phenotype conferred to cells by the noncanonical functions of telomerase, we tested for telomerase activity in HIV-infected MDM. Therefore, monocytes were isolated from human blood from different donors and differentiated in vitro into MDM for 6 days, as outlined in the scheme shown in Fig. 1A. Prior to infection, the purity of the population was confirmed to be above 90% as assayed by staining for the monocyte-macrophage-specific marker CD14 (59) and flow cytometry analysis (Fig. 1B). Then, MDM were infected with HIV-1 BaL, and the infection efficiency was evaluated after 12 days by quantification of intracellular and supernatant levels of p24 antigen. In agreement with previous reports, the susceptibility to HIV infection varied greatly between different donors (9, 25). Table 1 summarizes the levels of p24 antigen in culture supernatants from the 10 donors as measured by ELISA. In accordance, the percentages of MDM expressing intracellular p24 antigen varied largely, depending on the donor, as representative images of the two extremes of 2.97 and 95.21% show (Fig. 1C). As was previously reported (3, 49), even very high p24 levels in MDM did not induce apoptosis (data not shown).
Fig 1.

(A) Time line of HIV experiments. PBMCs were obtained by Ficoll-Hypaque centrifugation at day −7, and monocytes were isolated by adherence after overnight incubation in serum-free medium. At day −6, GM-CSF was added to the cultures, and monocytes were differentiated into macrophages for 6 days. At day 0, MDM were infected with HIV Bal, and after 12 days TA was studied. (B) CD14 expression on monocytes. CD14 density on the monocyte surface was determined by staining with FITC-labeled anti-CD14 antibodies. The purity of the population was more than 90%. (C) Efficiency of infection. HIV infectivity was monitored by intracytoplasmic HIV p24 staining, using FITC-labeled IgG anti-p24 and flow cytometry. Twelve days after infection, the percentage of cells positive for p24 varied between 2.97 and 95.21% depending of the donor. A low efficiency level (left) and high efficiency level (right) of infection are shown as representative figures.
Table 1.
Level of p24 antigen determined in culture supernatant and the intracellular level at day 12 after HIV Bal infection
| Blood donor no. | p24 antigen expression |
|
|---|---|---|
| In culture supernatant (ng/ml) | Intracellular (%)a | |
| 1 | 1026.5 | 95.21 |
| 2 | 87.5 | 2.97 |
| 3 | 192.14 | NA |
| 4 | 256.6 | NA |
| 5 | 406.1 | 11.1 |
| 6 | 192.45 | NA |
| 7 | 870.5 | 36.5 |
| 8 | 607.55 | 15.4 |
| 9 | 252 | 10.56 |
| 10 | 288 | 13.96 |
Values reflect the percentage of MDM expressing intracellular p24 antigen. NA, not available.
In order to test for activation of telomerase, cells were analyzed at 0, 2, 5, 7, and 24 h and at day 12 after infection, which is close to the peak of HIV replication (25, 65). TA was measured by qPCR and is displayed relative to the level in Jurkat cells, which was considered as 100%. During the first hours after infection, a trend toward an increase of TA in infected cells was observed (see Fig. S3A in the supplemental material). On day 12, HIV-infected MDM showed significant (P < 0.05) upregulation of TA in 7 out of 11 cultures (Fig. 2A). In some cases TA mounted to about 30% after HIV infection compared to uninfected Jurkat cells. Of note, a strong correlation between the amount of secreted p24 and TA was observed (R2 = 0.814; Pearson's correlation coefficient, 0.902; P = 0.0004), suggesting that active viral replication is necessary for increasing TA (Fig. 2B). This is in accordance with our experiments using MDM which were incubated with recombinant soluble HIV proteins Tat and gp120 for 8 and 24 h, which did not show differences in TA compared to infected cells (Fig. 2C). These results demonstrate that the increase in TA after HIV-1 infection of MDM is not caused by a bystander effect of Tat or gp120 soluble protein (gp120, P = 0.26; Tat, P = 0.78). Similarly, MDM treated with ultracentrifuged virus-free supernatant and/or inactivated virus did not show induction of TA either (see Fig. S3B).
Fig 2.

(A) TA in MDM. Twelve days after infection, MDM were harvested, lysed with CHAPS buffer, and telomerase activity was studied by quantitative real-time PCR and calculated relative to Jurkat cells. Infection resulted in a significant upregulation of TA in MDM from 7 out of 11 donors. (B). Correlation curve between TA and levels of p24 antigen (Ag) in culture supernatants. When the levels of antigen p24 production were elevated, the increase in TA was greater (R2 = 0.814; Pearson's correlation coefficient, 0.902; P = 0.0004). (C) Challenge with gp120 and Tat soluble proteins. TA was not modified after the treatment with soluble HIV proteins (P > 0.05). Error bars indicate the standard deviation of the mean from the three biological replicates. The horizontal line indicates the ratio value of 1. NI, noninfected; INF, HIV Bal-infected MDM. (D) TA in cytoplasmic and nuclear extracts. TA was detected in both the nucleus and cytoplasm, while being markedly higher in the nucleus. Error bars indicate the standard deviation of the mean from the duplicates of each donor.
It has been shown that telomerase is able to migrate from the nucleus to the cytoplasm, where a fraction of the enzyme is localized to mitochondria (70). Moreover, the subcellular localization of the enzyme has been proposed as an important regulation point of the TA (35, 71). Therefore, TA was differentially determined in nuclear and cytoplasmic extracts. As observed in Fig. 2D, we found an increase of TA in both the nuclear and cytoplasmic compartments of HIV-infected MDM. Accordingly, subcellular telomerase shuttling of MDM could be affected by HIV infection, and telomerase could have functions protecting HIV-infected macrophages both in the nucleus and cytoplasm.
In order to test if telomerase activity is mediated only by the R5-tropic HIV-BaL strain, which exhibits an aggressive replication profile in MDM (69), or if it is a more general phenomenon, we used different HIV strains for infection of MDM, including HIV variants exhibiting X4 tropism (IIIB and HXB2) and R5 tropism (JRCSF and SF162) or X4R5 (93BR020). Among them, all but one were ascribed to subtype B (93BR020, subtype F). As expected, the levels of infection varied greatly between different donors (see Fig. S3C in the supplemental material). Again, telomerase activity was analyzed after 12 days. A trend toward an increase after the infection with all the HIV strains was observed, but due to too large interindividual differences, significance was only reached with the viral strains SF162 and IIIB (Fig. 3A).
Fig 3.

(A) MDM were infected with different HIV strains (93BR020, HXB2, JRCSF, SF162, and IIIB), and after 12 days telomerase activity was studied by quantitative real-time PCR. MDM infected with SF162 and IIIB showed a significant upregulation of TA (P = 0.01 and P = 0.01, respectively). The horizontal bars within boxes represent the median values, and the upper and lower boundaries of boxes represent the 75th and 25th percentiles, respectively. Asterisks represent extreme values. (B) TA during monocyte into macrophage differentiation. Monocytes were differentiated into macrophages by culturing in the presence of GM-CSF for 6 days, and TA was studied at different time points. As observed, the values of the TA are close to zero, indicating no modification of TA during in vitro monocyte-macrophage differentiation. (C) MDM activation and infection with vaccinia virus. MDM were treated with LPS and/or IL-4 in order to produce MDM activation, and TA was studied after 8 and 24 h. As shown, TA was not detected under either condition. In vitro vaccinia virus infection did not affect TA in MDM either. The figure shows TA after infection with WR but is representative of the results found in MDM infected with MVA as well. Error bars indicate the standard deviations of the means from the three biological replicates. The horizontal line indicates the ratio value of 1. RPMI, control; VV, vaccinia virus.
Taken together, these data indicate that in vitro infection of MDM with HIV upregulates TA, and for BaL-1 we observed that such regulation is directly correlated with virus production and might in addition be affecting telomerase subcellular localization.
Telomerase activity is not affected during differentiation of monocytes into macrophages, MDM activation, or modified vaccinia virus Ankara infection.
It has been reported that telomerase is expressed in immature somatic cells and is suppressed in differentiated ones (58). In addition it has been demonstrated that telomerase is upregulated after lymphocyte activation and during dendritic cell differentiation and maturation (62, 67). Moreover, it has been recently published that LPS as a model of bacterial infection is able to augment hTERT gene expression in MDM (32). This prompted us to further investigate whether the upregulation of TA is a specific consequence of HIV infection or could be a nonspecific response to general physiological stimuli. Therefore, we evaluated TA in MDM during differentiation of monocytes into macrophages after LPS and/or IL-4 stimulus, as well as upon vaccinia virus infection. Again, monocytes were differentiated into macrophages by culturing in the presence of GM-CSF for 6 days (13). TA was evaluated at days 0, 1, 4, and 6 and showed that differentiation does not induce TA (Fig. 3B). Similarly, MDM were exposed to LPS and/or IL-4, which again did not induce TA (Fig. 3C). Finally, viral infection per se was tested using vaccinia virus at a multiplicity of infection (MOI) of 0.5 and an MOI of 1. No significant changes were observed (Fig. 3C). Taken together, these data suggest that TA is induced in response to stimuli specifically provided by HIV.
Telomerase increases stress resistance of MDM.
In order to test the consequences of telomerase reactivation in MDM, we analyzed telomere length in noninfected versus infected MDM. Twelve days after infection, cells were harvested, genomic DNA was isolated, and telomere length was studied by qPCR using primers for telomere-specific sequences (T) versus 36B4, a single-copy gene (S). The ratio of T/S is thereby proportional to the telomere length (15). Telomerase activity did not seem to significantly lengthen the telomeres in MDM infected with HIV, with some individual exceptions (Fig. 4A).
Fig 4.

(A) Telomere length. Telomere (T) and single-copy gene (S) were amplified using quantitative real-time PCR, with the T/S ratio proportional to telomere length. Twelve days after infection telomeres from infected MDM were similar to those from noninfected MDM (P > 0.05). The horizontal bars within boxes represent the median values, and the upper and lower boundaries of boxes represent the 75th and 25th percentiles, respectively. The open circles represent the outliers above the end of the top whisker. Asterisks represent extreme values. Telomere length was studied in at least three biological replicates. (B) DNA damage of HIV Bal-infected MDM. DNA was evaluated by comet assay and analyzed using the software CometScore, version 1.5. Based on the percentage of DNA in tails, cells were grouped into five classes, ranging from class 1 (lowest percentage of DNA in tail or undamaged) to class 5 (highest percentage of DNA in tail or severely damaged). MDM from HIV-infected cultures exhibited a lower percentage of damaged cells under basal culture conditions (left panel) and after exposure to oxidative stress (right panel). For more detail, please see the text. Error bars indicate the standard deviations of the means of three biological replicates. (C) MDM were infected with different HIV strains, and DNA damage was studied. Again, MDM infected with HIV showed less DNA damage in both the basal condition (P < 0.001) and after oxidative stress (P < 0.001).
Since MDM are terminally differentiated cells and do not replicate, telomere lengths might not be of prime importance. However, recent studies have demonstrated that telomerase has functions other than telomere synthesis, such as protecting cells from oxidative stress, apoptosis, and necrosis; DNA repair; and stimulating cell proliferation under adverse conditions (11, 73). This prompted us to study resistance to oxidative stress and the DNA damage status of MDM after HIV infection.
Therefore, MDM were infected with HIV, and after 12 days the cells were harvested and treated with 100 μM H2O2 for 10 min, followed by analysis of DNA damage using a comet assay and analyzing 100 cells per condition. The length and shape of the comets classified the cells as undamaged (class 1) to heavily damaged (class 5).
Most of the HIV BaL-infected cells were assigned to the low-DNA-damage classes 1 to 3 (class 1, 46.2%; class 2, 25.2%; class 3, 15.2%; class 4, 8.6%; class 5, 4.8%), whereas noninfected cells showed higher damage (class 1, 32.9%; class 2, 25.2%; class 3, 19.2%; class 4, 12.5%; class 5, 10.1%), as shown in Fig. 4B (P < 0.001). After exposure to oxidative stress, this difference became even more pronounced, as noninfected MDM showed higher percentages of cells in class 4 and 5 than infected ones (class 1, 5.4% versus 14%, noninfected versus infected, respectively; class 2, 11.4% versus 16%; class 3, 21.6% versus 25.6%; class 4, 24.9% versus 20.9%; class 5, 36.3% versus 23.4%; P < 0.001). Notably, we have found similar results when MDM were infected with different HIV strains (Fig. 4C).
Taken together, these data suggest that HIV infection of MDM increases basal DNA repair as well as resistance to oxidative stress-induced DNA damage.
In order to test if, indeed, telomerase is involved in this phenotype, we overexpressed hTERT alone in MDM and tested for DNA damage and stress resistance. Therefore, MDM were transduced with pLXSN-hTERT or with pLXSN-neo, and 48 h later the cells were exposed to oxidative stress. Then, 100 μM paraquat was added to the cell cultures; after 3 h the cells were harvested, and DNA damage and apoptosis levels were studied.
Untreated MDM with and without hTERT overexpression showed similar distributions of DNA-damaged cells (pLXSN-neo class 1, 52.1%; class 2, 26.1%; class 3, 13.2%; class 4, 4.1%; class 5, 4.1%; pLXSN-hTERT class 1, 57.2%; class 2, 21.8%; class 3, 15.1%; class 4, 3.1%; class 5, 2.8%; P > 0.05) (Fig. 5A), suggesting that 48 h after hTERT transduction the effect might not be sufficient for the detection of improved basal repair. However, after exposure to oxidative stress, the DNA damage was significantly higher in empty vector control MDM than in MDM overexpressing TERT (pLXSN-neo class 1, 24.6%; class 2, 18.1%; class 3, 21.5%; class 4, 17.6%; class 5, 18.1%; pLXSN-hTERT class 1, 40.2%; class 2, 26.6%; class 3, 18.8%; class 4, 9.5%; class 5, 4.5%; P < 0.001) (Fig. 5).
Fig 5.
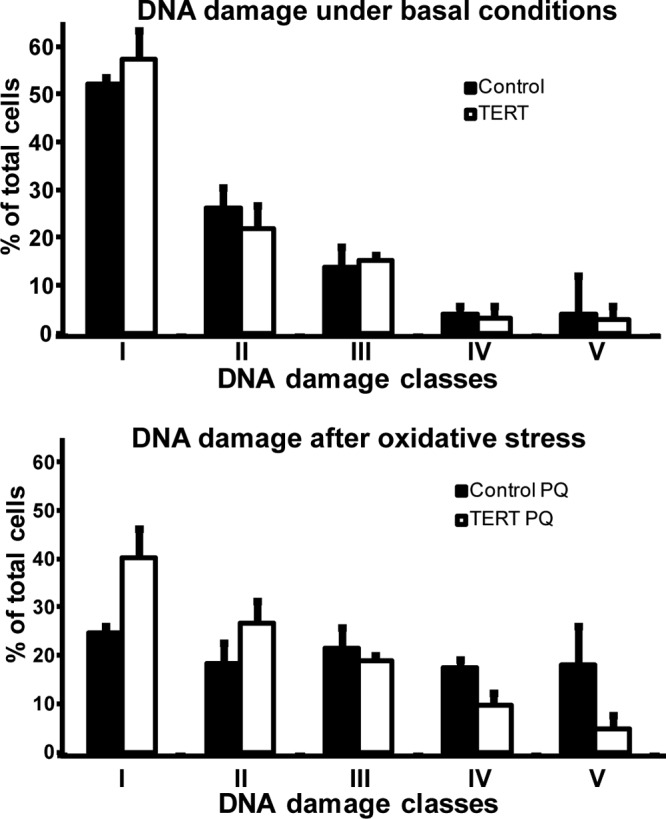
MDM were transduced with pLXSN-hTERT and pLXSN-neo (control) and 48 h later treated with 100 μM paraquat (PQ). DNA damage was analyzed by comet assay. Under basal conditions there were no differences between controls and MDM overexpressing hTERT (upper panel) (P = 0.246). But after oxidative stress treatment, MDM overexpressing hTERT exhibited less DNA damage than control cells (lower panel) (P < 0.001). Error bars indicate the standard deviations of the means from three biological replicates.
Taken together, our data suggest that telomerase reactivation in MDM after HIV infection might contribute to protecting macrophages from oxidative stress and DNA damage and thus might help HIV in establishing a long-lasting viral reservoir.
DISCUSSION
Macrophages are cells crucially involved in HIV pathogenesis. Whereas activated T CD4+ lymphocytes exhibit a half-life between 24 and 48 h after HIV infection, macrophages can survive for several weeks (45). We along with others have shown that TA is downregulated in lymphoid cells infected with HIV and that this mechanism contributes to the depletion of T CD4+ lymphocytes (7, 28, 29, 67, 68). Here, we show that in vitro HIV-1 infection induces TA in MDM. So far, not much is known about low TA activity in somatic cells (40, 50), while work focused on TA in macrophages is limited to one report. There, hTERT expression was found to be upregulated in both U937 cells and MDM, and in the former TA was increased after exposure to LPS or TNF-α as inflammatory stimuli (32). Conversely, in our experiments we did not find any modification in TA after treatment with LPS and/or IL-4. It is known that telomerase has important posttranslational regulations which are necessary for detectable enzymatic activity. Consequently, it is possible that LPS induces hTERT gene expression but not at a level sufficient for positive regulation of TA in MDM. On the other hand, the experimental conditions and the donor characteristics in our work differ from those in the work of Gizard et al. (32). Nonetheless, both studies support the idea that telomerase can be reactivated in macrophages in the presence of specific stimuli that might correspond to immune responses.
MDM have a strong donor-dependent variation of in vitro susceptibility to HIV infection, with differences of up to 3 orders of magnitude in virus production (9, 25). Similarly, we observed a different susceptibility of MDM infection with both R5 and X4 strains. Whether X4-tropic HIV can infect macrophages remains controversial (5, 17, 19, 42). Interestingly, in our model only one X4-tropic strain (IIIB) replicated at high levels, and it was accompanied by an increase in the TA.
Since soluble Tat has been shown to downregulate TA in the nucleus of CD4+ T cells (29), we then tested the possibility that the viral protein Tat or gp120 alone would be sufficient for telomerase reactivation. Tat and gp120 have been implicated in the development of HIV pathogenesis and are known to be cytotoxic and neurotoxic (66), but at the same time they are able to protect HIV-infected macrophages against apoptosis (20, 66, 76). Thus, we found that Tat and gp120 soluble proteins do not per se affect TA in MDM, at least not under the conditions tested. It is yet unclear whether the reactivation of telomerase in MDM could be an indirect effect of HIV infection as a consequence of the activation of cellular proteins, like Sp1 and Akt (60, 77), or if it is directly mediated by viral proteins, like Vpr and/or Nef. On the one hand, Vpr, which is strictly required for HIV replication in MDM, regulates the shuttling of macromolecules between the nucleus and cytoplasm and can act as a transcriptional factor (8, 41, 72). On the other hand, Nef can protect MDM from apoptosis triggered by HIV and contributes to efficient viral replication as well (37, 59). Therefore, more experiments need to be conducted in order to elucidate viral and/or cellular proteins responsible for the TA increase in MDM.
Additionally, we tested if other nononcogenic viral infections might modify TA in MDM. We have used vaccinia virus, which productively infects MDM (46) and has a completely different pathogenesis than HIV. Vaccinia virus infection did not modify TA in MDM, supporting our hypothesis that it is a specific consequence of HIV infection. However, we are conscious that the infection with a different retrovirus would be required for stronger confirmation of our hypothesis.
Furthermore, we observed that infected MDM are more resistant to oxidative stress, similar to MDM overexpressing telomerase. Therefore, we propose here that reactivation of telomerase contributes to the increased survival of HIV-infected macrophages that renders these cells a potent HIV reservoir.
Several lines of evidence suggest that macrophages can indeed act as an important viral reservoir (27, 76). Here, we add telomerase reactivation to the mechanisms that contribute to protecting HIV-infected macrophages. In line with this idea are the findings that telomerase is activated by the phosphatidylinositol 3-kinase (PI3K)/Akt pathway in both somatic and tumor cells (28, 44, 63) and that HIV is able to inhibit the PI3K/Akt pathway in T CD4+ lymphocytes, while it activates this cascade in macrophages (23, 28, 77). It is intriguing to think that the opposed regulation observed in these two cell types could be related to the same opposed regulation of TA, which might help to explain the opposite outcomes for lymphocytes and macrophages after HIV infection.
How might telomerase increase stress resistance in macrophages? We propose that the noncanonical functions of telomerase might be responsible for this. It is known that the positive regulation of telomerase favors cell survival through both canonical and noncanonical functions (33). Interestingly, many functions of telomerase are carried on outside the nucleus. For example, oxidative stress promotes the translocation of telomerase from the nucleus to the cytoplasm (35), which is in keeping with our finding that TA is increased in both nuclear and cytoplasmic extracts. Furthermore, hTERT protects mitochondrial DNA of human fibroblasts against acute and chronic oxidative stress (1). Since HIV infection is characterized by an increase in oxidative stress (75), oxidative damage to brain endothelial cells (36, 78), and oxidative stress-induced telomere shortening in astroglial cells (64), it is intriguing to think that infected macrophages need to be specifically protected against oxidative stress. This is in keeping with our results that HIV-infected MDM have less DNA damage under basal culture conditions and after oxidative stress. Moreover, overexpression of hTERT in MDM was enough to protect the cells against oxidative stress. These results suggest strongly that telomerase can protect HIV-infected macrophages.
In conclusion, the induction of TA together with other HIV-dependent antiapoptotic mechanisms can protect macrophages from the cytotoxic effects and the oxidative stress induced by HIV infection. This phenomenon could contribute to viral persistence and thus to the maintenance of macrophages as an important viral reservoir.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an FWF grant and an International Cooperation grant with the CONICET (Argentina) Research Network, by NRN grant S93-06 of the Austrian Science Fund, GEN-AU Project 820982, Non-coding RNAs, to J.G. as well as by grants from the Herzfelder'sche Familienstiftung and CE.R.I.E.S to J.G. R.R. was supported by EMBO short-term fellowship ASTF 19-2009.
We thank Ana Ceballos and Juan Sabatté for their help with cell culture and Dario Dilernia for his assistance with statistical analysis.
Footnotes
Published ahead of print 11 July 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Ahmed S, et al. 2008. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell Sci. 121:1046–1053 [DOI] [PubMed] [Google Scholar]
- 2. Alexaki A, Liu Y, Wigdahl B. 2008. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 6:388–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aquaro S, et al. 2002. Long-term survival and virus production in human primary macrophages infected by human immunodeficiency virus. J. Med. Virol. 68:479–488 [DOI] [PubMed] [Google Scholar]
- 4. Bachor C, Bachor OA, Boukamp P. 1999. Telomerase is active in normal gastrointestinal mucosa and not up-regulated in precancerous lesions. J. Cancer Res. Clin. Oncol. 125:453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bakri Y, Amzazi S, Mannioui A, Benjouad A. 2001. The susceptibility of macrophages to human immunodeficiency virus type 1 X4 isolates depends on their activation state. Biomed. Pharmacother. 55:32–38 [DOI] [PubMed] [Google Scholar]
- 6. Ball SC, et al. 2003. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes B and C. J. Virol. 77:1021–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ballon G, et al. 2001. Human immunodeficiency virus type 1 modulates telomerase activity in peripheral blood lymphocytes. J. Infect. Dis. 183:417–424 [DOI] [PubMed] [Google Scholar]
- 8. Benko Z, et al. 2007. Antagonistic interaction of HIV-1 Vpr with Hsf-mediated cellular heat shock response and Hsp16 in fission yeast (Schizosaccharomyces pombe). Retrovirology 4:16 doi:10.1186/1742-4690-4-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bol SM, et al. 2009. Donor variation in in vitro HIV-1 susceptibility of monocyte-derived macrophages. Virology 390:205–211 [DOI] [PubMed] [Google Scholar]
- 10. Buechler C, et al. 2000. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and anti-inflammatory stimuli. J. Leukoc. Biol. 67:97–103 [PubMed] [Google Scholar]
- 11. Cao Y, Li H, Deb S, Liu JP. 2002. TERT regulates cell survival independent of telomerase enzymatic activity. Oncogene 21:3130–3138 [DOI] [PubMed] [Google Scholar]
- 12. Carter CA, Ehrlich LS. 2008. Cell biology of HIV-1 infection of macrophages. Annu. Rev. Microbiol. 62:425–443 [DOI] [PubMed] [Google Scholar]
- 13. Cassol E, Alfano M, Biswas P, Poli G. 2006. Monocyte-derived macrophages and myeloid cell lines as targets of HIV-1 replication and persistence. J. Leukoc. Biol. 80:1018–1030 [DOI] [PubMed] [Google Scholar]
- 14. Cawthon RM. 2009. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 37:e21 doi:10.1093/nar/gkn1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cawthon RM. 2002. Telomere measurement by quantitative PCR. Nucleic Acids Res. 30:e47 doi:10.1093/nar/30.10.e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang MW, et al. 2005. Comparison of early passage, senescent and hTERT immortalized endothelial cells. Exp. Cell Res. 309:121–136 [DOI] [PubMed] [Google Scholar]
- 17. Chang TL, et al. 2010. Human peritoneal macrophages from ascitic fluid can be infected by a broad range of HIV-1 isolates. J. Acquir. Immune Defic. Syndr. 53:292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choe W, Volsky DJ, Potash MJ. 2001. Induction of rapid and extensive beta-chemokine synthesis in macrophages by human immunodeficiency virus type 1 and gp120, independently of their coreceptor phenotype. J. Virol. 75:10738–10745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chowdhury IH, Bentsman G, Choe W, Potash MJ, Volsky DJ. 2002. The macrophage response to HIV-1: Intracellular control of X4 virus replication accompanied by activation of chemokine and cytokine synthesis. J. Neurovirol. 8:599–610 [DOI] [PubMed] [Google Scholar]
- 20. Chugh P, et al. 2008. Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy. Retrovirology 5:11 doi:10.1186/1742-4690-5-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coleman CM, Wu L. 2009. HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology 6:51 doi:10.1186/1742-4690-6-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Colitz CM, Barden CA, Lu P, Chandler HL. 2006. Expression and characterization of the catalytic subunit of telomerase in normal and cataractous canine lens epithelial cells. Mol. Vis. 12:1067–1076 [PubMed] [Google Scholar]
- 23. Dabrowska A, Kim N, Aldovini A. 2008. Tat-induced FOXO3a is a key mediator of apoptosis in HIV-1-infected human CD4+ T lymphocytes. J. Immunol. 181:8460–8477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Di Donna S, et al. 2003. Telomerase can extend the proliferative capacity of human myoblasts, but does not lead to their immortalization. Mol. Cancer Res. 1:643–653 [PubMed] [Google Scholar]
- 25. Eisert V, et al. 2001. Analysis of cellular factors influencing the replication of human immunodeficiency virus type I in human macrophages derived from blood of different healthy donors. Virology 286:31–44 [DOI] [PubMed] [Google Scholar]
- 26. Fehrer C, et al. 2006. Techniques in gerontology: cell lines as standards for telomere length and telomerase activity assessment. Exp. Gerontol. 41:648–651 [DOI] [PubMed] [Google Scholar]
- 27. Fernández Larrosa PN, et al. 2008. Apoptosis resistance in HIV-1 persistently-infected cells is independent of active viral replication and involves modulation of the apoptotic mitochondrial pathway. Retrovirology 5:19 doi:10.1186/1742-4690-5-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franzese O, et al. 2007. Telomerase activity, hTERT expression, and phosphorylation are downregulated in CD4+ T lymphocytes infected with human immunodeficiency virus type 1 (HIV-1). J. Med. Virol. 79:639–646 [DOI] [PubMed] [Google Scholar]
- 29. Franzese O, et al. 2004. HIV-Tat down-regulates telomerase activity in the nucleus of human CD4+ T cells. Cell Death Differ. 11:782–784 [DOI] [PubMed] [Google Scholar]
- 30. Gavegnano C, Schinazi RF. 2009. Antiretroviral therapy in macrophages: implication for HIV eradication. Antivir. Chem. Chemother. 20:63–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Giri MS, et al. 2009. Circulating monocytes in HIV-1-infected viremic subjects exhibit an antiapoptosis gene signature and virus- and host-mediated apoptosis resistance. J. Immunol. 182:4459–4470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gizard F, et al. 2011. Telomerase activation in atherosclerosis and induction of telomerase reverse transcriptase expression by inflammatory stimuli in macrophages. Arterioscler. Thromb. Vasc. Biol. 31:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gorbunova V, Seluanov A. 2003. Telomerase as a growth-promoting factor. Cell Cycle 2:534–537 [DOI] [PubMed] [Google Scholar]
- 34. Haendeler J, et al. 2009. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler. Thromb. Vasc. Biol. 29:929–935 [DOI] [PubMed] [Google Scholar]
- 35. Haendeler J, Hoffmann J, Brandes RP, Zeiher AM, Dimmeler S. 2003. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol. Cell Biol. 23:4598–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haorah J, et al. 2007. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 101:566–576 [DOI] [PubMed] [Google Scholar]
- 37. Herbein G, et al. 2008. Nef and TNFα are coplayers that favor HIV-1 replication in monocytic cells and primary macrophages. Curr. HIV Res. 6:117–129 [DOI] [PubMed] [Google Scholar]
- 38. Herbert BS, Hochreiter AE, Wright WE, Shay JW. 2006. Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat. Protoc. 1:1583–1590 [DOI] [PubMed] [Google Scholar]
- 39. Hou M, Xu D, Björkholm M, Gruber A. 2001. Real-time quantitative telomeric repeat amplification protocol assay for the detection of telomerase activity. Clin. Chem. 47:519–524 [PubMed] [Google Scholar]
- 40. Huang W, et al. 2010. Inhibition of telomerase activity alters tight junction protein expression and induces transendothelial migration of HIV-1-infected cells. Am. J. Physiol. Heart Circ. Physiol. 298:H1136–H1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jacquot G, et al. 2007. Localization of HIV-1 Vpr to the nuclear envelope: impact on Vpr functions and virus replication in macrophages. Retrovirology 4:84 doi:10.1186/1742-4690-4-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jayakumar P, et al. 2005. Tissue-resident macrophages are productively infected ex vivo by primary X4 isolates of human immunodeficiency virus type 1. J. Virol. 79:5220–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jones G, Power C. 2006. Regulation of neural cell survival by HIV-1 infection. Neurobiol. Dis. 21:1–17 [DOI] [PubMed] [Google Scholar]
- 44. Kang SS, Kwon T, Kwon DY, Do SI. 1999. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J. Biol. Chem. 274:13085–13090 [DOI] [PubMed] [Google Scholar]
- 45. Kedzierska K, Crowe SM. 2002. The role of monocytes and macrophages in the pathogenesis of HIV-1 infection. Curr. Med. Chem. 9:1893–1903 [DOI] [PubMed] [Google Scholar]
- 46. Kowalczyk A, Guzik K, Slezak K, Dziedzic J, Rokita H. 2005. Heat shock protein and heat shock factor 1 expression and localization in vaccinia virus infected human monocyte derived macrophages. J. Inflamm. (Lond.) 2:12 doi:10.1186/1476-9255-2-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lai JP, et al. 2001. Substance P antagonist (CP-96,345) inhibits HIV-1 replication in human mononuclear phagocytes. Proc. Natl. Acad. Sci. U. S. A. 98:3970–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lambotte O, Deiva K, Tardieu M. 2003. HIV-1 persistence, viral reservoir, and the central nervous system in the HAART era. Brain Pathol. 13:95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Le Douce V, Herbein G, Rohr O, Schwartz C. 2010. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 7:32 doi:10.1186/1742-4690-7-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leri A, et al. 2001. Telomerase expression and activity are coupled with myocyte proliferation and preservation of telomeric length in the failing heart. Proc. Natl. Acad. Sci. U. S. A. 98:8626–8631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lichterfeld M, et al. 2008. Telomerase activity of HIV-1-specific CD8+ T cells: constitutive up-regulation in controllers and selective increase by blockade of PD ligand 1 in progressors. Blood 112:3679–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu J, et al. 2004. Human telomerase reverse transcriptase mRNA is highly expressed in normal breast tissues and down-regulated in ductal carcinoma in situ. Int. J. Oncol. 24:879–884 [PubMed] [Google Scholar]
- 53. MacKenzie KL, Franco S, May C, Sadelain M, Moore MA. 2000. Mass cultured human fibroblasts overexpressing hTERT encounter a growth crisis following an extended period of proliferation. Exp. Cell Res. 259:336–350 [DOI] [PubMed] [Google Scholar]
- 54. Marsden MD, Zack JA. 2009. Eradication of HIV: current challenges and new directions. J. Antimicrob. Chemother. 63:7–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Masutomi K, et al. 2003. Telomerase maintains telomere structure in normal human cells. Cell 114:241–253 [DOI] [PubMed] [Google Scholar]
- 56. Matsumura-Arioka Y, Ohtani K, Hara T, Iwanaga R, Nakamura M. 2005. Identification of two distinct elements mediating activation of telomerase (hTERT) gene expression in association with cell growth in human T cells. Int. Immunol. 17:207–215 [DOI] [PubMed] [Google Scholar]
- 57. Mollace V, et al. 2002. The contribution of oxidative stress in apoptosis of human-cultured astroglial cells induced by supernatants of HIV-1-infected macrophages. J. Leukoc. Biol. 71:65–72 [PubMed] [Google Scholar]
- 58. Nakatake M, Kakiuchi Y, Sasaki N, Murakami-Murofushi K, Yamada O. 2007. STAT3 and PKC differentially regulate telomerase activity during megakaryocytic differentiation of K562 cells. Cell Cycle 6:1496–1501 [PubMed] [Google Scholar]
- 59. Olivetta E, Federico M. 2006. HIV-1 Nef protects human-monocyte-derived macrophages from HIV-1-induced apoptosis. Exp. Cell Res. 312:890–900 [DOI] [PubMed] [Google Scholar]
- 60. Osiecki K, et al. 2005. Identification of granulocyte-macrophage colony-stimulating factor and lipopolysaccharide-induced signal transduction pathways that synergize to stimulate HIV type 1 production by monocytes from HIV type 1 transgenic mice. AIDS Res. Hum. Retroviruses 21:125–139 [DOI] [PubMed] [Google Scholar]
- 61. Ozdener H. 2005. Molecular mechanisms of HIV-1 associated neurodegeneration. J. Biosci. 30:391–405 [DOI] [PubMed] [Google Scholar]
- 62. Ping L, Asai A, Okada A, Isobe K, Nakajima H. 2003. Dramatic increase of telomerase activity during dendritic cell differentiation and maturation. J. Leukoc. Biol. 74:270–276 [DOI] [PubMed] [Google Scholar]
- 63. Plunkett FJ, et al. 2007. The loss of telomerase activity in highly differentiated CD8+ CD28− CD27− T cells is associated with decreased Akt (Ser473) phosphorylation. J. Immunol. 178:7710–7719 [DOI] [PubMed] [Google Scholar]
- 64. Pollicita M, et al. 2009. Apoptosis and telomeres shortening related to HIV-1 induced oxidative stress in an astrocytoma cell line. BMC Neurosci. 10:51 doi:10.1186/1471-2202-10-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Porcheray F, Samah B, Léone C, Dereuddre-Bosquet N, Gras G. 2006. Macrophage activation and human immunodeficiency virus infection: HIV replication directs macrophages towards a pro-inflammatory phenotype while previous activation modulates macrophage susceptibility to infection and viral production. Virology 349:112–120 [DOI] [PubMed] [Google Scholar]
- 66. Price TO, Ercal N, Nakaoke R, Banks WA. 2005. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 1045:57–63 [DOI] [PubMed] [Google Scholar]
- 67. Reynoso R, Laufer N, Bolcic F, Quarleri J. 2010. Telomerase activity in peripheral blood mononuclear cells from HIV and HIV-HCV coinfected patients. Virus Res. 147:284–287 [DOI] [PubMed] [Google Scholar]
- 68. Reynoso R, Minces L, Salomon H, Quarleri J. 2006. HIV-1 infection downregulates nuclear telomerase activity on lymphoblastoic cells without affecting the enzymatic components at the transcriptional level. AIDS Res. Hum. Retroviruses 22:425–429 [DOI] [PubMed] [Google Scholar]
- 69. Rozmyslowicz T, Murphy SL, Conover DO, Gaulton GN. 2010. HIV-1 infection inhibits cytokine production in human thymic macrophages. Exp. Hematol. 38:1157–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Saretzki G. 2009. Telomerase, mitochondria and oxidative stress. Exp. Gerontol. 44:485–492 [DOI] [PubMed] [Google Scholar]
- 71. Sharma NK, et al. 2012. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 40:712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sherman MP, et al. 2003. Nuclear export of Vpr is required for efficient replication of human immunodeficiency virus type 1 in tissue macrophages. J. Virol. 77:7582–7589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Smith LL, Coller HA, Roberts JM. 2003. Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat. Cell Biol. 5:474–479 [DOI] [PubMed] [Google Scholar]
- 74. Smith PD, Meng G, Salazar-Gonzalez JF, Shaw GM. 2003. Macrophage HIV-1 infection and the gastrointestinal tract reservoir. J. Leukoc. Biol. 74:642–649 [DOI] [PubMed] [Google Scholar]
- 75. Steiner J, et al. 2006. Oxidative stress and therapeutic approaches in HIV dementia. Antioxid. Redox Signal. 8:2089–2100 [DOI] [PubMed] [Google Scholar]
- 76. Swingler S, Mann AM, Zhou J, Swingler C, Stevenson M. 2007. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 3:1281–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tachado SD, Li X, Swan K, Patel N, Koziel H. 2008. Constitutive activation of phosphatidylinositol 3-kinase signaling pathway down-regulates TLR4-mediated tumor necrosis factor-alpha release in alveolar macrophages from asymptomatic HIV-positive persons in vitro. J. Biol. Chem. 283:33191–33198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Toborek M, et al. 2003. HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J. Neurochem. 84:169–179 [DOI] [PubMed] [Google Scholar]
- 79. Voglauer R, et al. 2006. SNEV overexpression extends the life span of human endothelial cells. Exp. Cell Res. 312:746–759 [DOI] [PubMed] [Google Scholar]
- 80. Voglauer R, et al. 2005. Establishment of human fibroma cell lines from a MEN1 patient by introduction of either hTERT or SV40 early region. Int. J. Oncol. 26:961–970 [DOI] [PubMed] [Google Scholar]
- 81. Weng NP, Levine BL, June CH, Hodes RJ. 1996. Regulated expression of telomerase activity in human T lymphocyte development and activation. J. Exp. Med. 183:2471–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wieser M, et al. 2008. hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. Am. J. Physiol. Renal Physiol. 295:F1365–F1375 [DOI] [PubMed] [Google Scholar]
- 83. Yasumoto S, et al. 1996. Telomerase activity in normal human epithelial cells. Oncogene 13:433–439 [PubMed] [Google Scholar]
- 84. Zheng L, Yang Y, Guocai L, Pauza CD, Salvato MS. 2007. HIV Tat protein increases Bcl-2 expression in monocytes which inhibits monocyte apoptosis induced by tumor necrosis factor-alpha-related apoptosis-induced ligand. Intervirology 50:224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.