

Abstract
Although HLA-B*57 (B57) is associated with slow progression to disease following HIV-1 infection, B57 heterozygotes display a wide spectrum of outcomes, including rapid progression, viremic slow progression, and elite control. Efforts to identify differences between B57-positive (B57+) slow progressors and B57+ rapid progressors have largely focused on cytotoxic T lymphocyte (CTL) phenotypes and specificities during chronic stages of infection. Although CTL responses in the early months of infection are likely to be the most important for the long-term rate of HIV-1 disease progression, few data on the early CTL responses of eventual slow progressors have been available. Utilizing the Multicenter AIDS Cohort Study (MACS), we retrospectively examined the early HIV-1-specific CTL responses of 14 B57+ individuals whose time to development of disease ranged from 3.5 years to longer than 25 years after infection. In general, a greater breadth of targeting of epitopes from structural proteins, especially Gag, as well as of highly conserved epitopes from any HIV-1 protein, correlated with longer times until disease. The single elite controller in the cohort was an outlier on several correlations of CTL targeting and time until disease, consistent with reports that elite control is typically not achieved solely by protective HLA-mediated CTLs. When targeting of individual epitopes was analyzed, we found that early CTL responses to the IW9 (ISPRTLNAW) epitope of Gag, while generally subdominant, correlated with delayed progression to disease. This is the first study to identify early CTL responses to IW9 as a correlate of protection in persons with HLA-B*57.
INTRODUCTION
Understanding the mechanisms that underlie rare instances of immunological suppression of HIV-1 infection offers an important opportunity to identify correlates of immune control, which is critical to the rational design of HIV-1 vaccines (18, 54, 60, 69, 71). The overrepresentation of certain HLA class I alleles among slow progressors, those who delay disease onset in the absence of antiretroviral therapy (38, 56, 62), is consistent with the critical role that cytotoxic T lymphocytes (CTLs) play in HIV-1 suppression, both restricting viral propagation (9, 44, 72) and constituting a major force driving viral evolution (2).
HLA-B*57 (B57) is one such protective allele, found in less than 5% of the general population, but enriched to between 39% and 85% among cohorts of slow progressors (14, 25, 29, 38, 45, 56, 62). Genome-wide association analyses further support the contribution of B57 to HIV-1 control (26, 61, 63). However, the B57 haplotype itself is not sufficient for protection: a substantial fraction of B57-positive (B57+) individuals progress normally and even rapidly to disease (8, 29, 56, 58). This spectrum of outcomes has spurred efforts to identify the immunological characteristics that distinguish B57+ slow progressors from B57+ average progressors.
The ability to elicit more effective CTLs is thought to account for the protective effect of B57, and in both slow and rapid progressors, B57-restricted CTLs dominate the immune response throughout the course of HIV-1 infection (3, 5, 56). Functional characterization of the CTLs from B57+ slow progressors revealed preserved expression of multiple cytokines compared to those from B57+ rapid progressors (8), which was followed by some controversy about whether such polyfunctionality was the consequence rather than the cause of sustained low viremia (57, 66, 76). Recent work suggests that superior CTL cytotoxicity, in association with the ability to proliferate in response to antigen, is a more direct correlate of immune control of HIV-1 infection, although causality is impossible to determine in cross-sectional studies of persons with chronic infection (16, 24, 25, 34, 55, 57, 68, 70).
Complicating the effort to identify the correlates of the most effective CTL responses in B57+ individuals is the frequent targeting of three highly conserved B57-restricted epitopes in the Gag-capsid protein: IW9 (ISPRTLNAW, amino acids [aa] 147 to 153), KF11 (KAFSEPVIPMF, aa 162 to 172), and TW10 (TSTLQEQIGW, aa 240 to 249) (33, 56). It is not clear whether the ability to target three conserved CTL epitopes in Gag-capsid is the key to B57-mediated protection or whether targeting of one is more important than targeting of the others. Furthermore, it is unclear whether targeting of these epitopes mediates protection in an additive manner or whether temporal patterns of CTL targeting are important. Efforts to identify patterns of epitope targeting that distinguish B57+ slow progressors from B57+ rapid progressors have focused on chronic stages of infection: overall, CTL targeting during chronic infection appears to be similar between B57+ slow and typical progressors, although slow progressors have CTL responses that are more narrowly focused on TW10, KF11, and IW9 than do rapid progressors (56).
Importantly, the contribution of HIV-specific CTLs to the control of HIV-1 infection is most apparent early in infection, and these early CTL responses are thought to be important for long-term outcomes (9, 31, 47, 53, 77, 79, 81, 82). Consistent with this, B57 exerts its protective effect early in infection (3, 4, 11, 12, 29, 77, 78). Therefore, to identify the correlates of B57-mediated immune control of HIV-1, it is important to compare the early CTL responses of B57+ slow progressors with those of B57+ normal or rapid progressors. However, the long lag between infection and evidence of long-term disease suppression has hampered efforts to correlate specific B57-specific early immunological events with long-term protection.
To address this issue, we have taken advantage of the unique longitudinal resources of the Multicenter AIDS Cohort Study (MACS), which, beginning in 1984, has enrolled at-risk homosexual men, tested for HIV-1, recorded clinical data, and cryopreserved blood samples on a semiannual schedule until the present time. The study has thus captured participants within months of infection, including those who showed no acute symptoms and ultimately controlled infection exceptionally well, and has followed these individuals for up to 27 years. To determine whether early patterns of CTL targeting are associated with differential long-term outcomes, we have conducted CTL epitope mapping studies on samples taken shortly after HIV-1 infection from 14 B57+ MACS participants with different long-term disease progression outcomes.
MATERIALS AND METHODS
Multicenter AIDS Cohort Study.
Established to study the natural history of HIV-1 infection, the MACS is an ongoing prospective cohort study of men who have sex with men that operates at four sites within the United States of America (19, 20, 22, 39). Samples and information are collected from participants semiannually, with samples cryopreserved at each visit. Longitudinal information regarding CD4+ T cell count, viremia, and drug treatment is available for each of the study participants. Decisions to treat are made by the physicians overseeing the care of the participants outside the MACS. Samples and information on participants have been collected under informed consent and institutional review board-approved protocols at each of the four MACS sites, and the current study has been conducted after approval by the UCLA Institutional Review Board.
Study participants.
Fourteen male HLA-B*57+ participants (13 HLA-B*5701+ and 1 HLA-B*5703+ individuals) were identified and selected for study on the basis of HIV-1 seroconversion while enrolled in MACS with subsequent follow-up until either progression to AIDS, initiation of combination antiretroviral therapy (cART), death due to AIDS, or the time of analysis (November 2011) (Table 1). Additional criteria for inclusion included homozygosity for the wild-type allele of CCR5 and availability of cryopreserved early-postseroconversion peripheral blood mononuclear cells (PBMCs). cART was defined as combination therapy with at least two nucleoside reverse transcriptase inhibitors and a protease inhibitor or nonnucleoside reverse transcriptase inhibitor. Viably cryopreserved PBMCs were obtained from the MACS repository for the first available postseroconversion time point for each participant.
Table 1.
Clinical characteristics of 14 HLA-B*57 participants from the MACS cohort
| Participant | HLA genotype | Characteristics at time of sampling |
Long-term outcome |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Time (mo) since infectiona | CD4b | VLc | Event | Time (yr) to eventd | |||||||
| A | A0100 | A3000 | B1801 | B5701 | C0501 | C0602 | 0–12 | 833 | 20,501 | Death | 3.5 |
| B | A0201 | A3301 | B3501 | B5701 | C0401 | C0602 | 5–6 | 789 | 14,811 | Death | 5.5 |
| C | A0101 | A0201 | B4402 | B5701 | C0501 | C0602 | 0–24 | 317 | 30,468 | CD4 < 200 | 6 |
| D | A0101 | A2402 | B4401 | B5701 | C0304 | C0602 | 0–6 | 1098 | 8,868 | CD4 < 200 | 6 |
| E | A0201 | A0201 | B5001 | B5701 | C0602 | C0602 | 10–15 | 800 | 2,927 | CD4 < 200 | 6.25 |
| F | A0101 | A0201 | B0702 | B5701 | C0602 | C0702 | 0–6 | 744 | 11,955 | CD4 < 200 | 9 |
| G | A0101 | A0201 | B5701 | B5701 | C0602 | C0702 | 0–7 | 665 | 4,414 | CD4 < 200 | 11 |
| H | A0101 | A2501 | B1801 | B5701 | C0602 | C1203 | 0–6 | 1637 | 3,151 | CD4 < 200 | 12 |
| I | A0201 | A0201 | B1501 | B5701 | C0304 | C0602 | 3–8 | 958 | 4,989 | cART | 13 |
| J | A0100 | A2400 | B4403 | B5701 | C0303 | C0602 | 4–6 | 801 | 43,400 | Nonee | 13 |
| K | A0101 | A0201 | B5101 | B5701 | C0602 | C0702 | 0–6 | 757 | 109,373 | CD4 < 200 | 13.5 |
| L | A0200 | A3201 | B0702 | B5701 | C0602 | C0700 | 9–15 | 853 | 82,009 | CD4 < 200 | 15 |
| M | A0201 | A6801 | B5703 | B5802 | C0602 | C1801 | 6–12 | 642 | 5,538 | cART | 17 |
| N | A0101 | A0301 | B1518 | B5701 | C0602 | C0704 | 0–9 | 1200 | < 400 | Nonee | 25 |
Number of months that elapsed between seroconversion and donation of the samples used in this study.
Number of CD4+ T lymphocytes/mm3 of blood at the time of sampling.
Number of HIV-1 RNA copies/ml of plasma at the time of sampling.
Number of years elapsed between date of infection and the earliest of the following events: decline of CD4+ T cell counts below 200/mm3, initiation of combination antiretroviral therapy (cART), or death.
Participants J and N still had CD4 counts above 200, as of 2011.
Polyclonal expansion of CD8+ T lymphocytes from PBMCs.
Cryopreserved PBMCs were thawed and subjected to nonspecific polyclonal expansion using a CD3-CD4-bispecific monoclonal antibody as previously described (35). Briefly, 106 PBMCs were incubated at 37°C in complete RPMI medium containing 10% heat-inactivated fetal calf serum (Omega), 1 μg/ml anti-CD3/CD4 antibody (83, 84), and 50 units/ml interleukin-2 (NIH AIDS Research and Reference Reagent Repository). Cells were fed with medium twice per week and kept in culture for 14 days. This procedure produced an average of 15 × 106 cells; cell viability was >85% by trypan blue exclusion; the CD8+ purity was >90% and the CD4+ contamination was <3%, as determined by flow cytometry. This approach has been shown to generate nonspecifically expanded CD8+ T lymphocytes with frequencies of HIV-1-specific CTLs that are similar to those before expansion (35, 37, 74).
Mapping of HIV-1-specific CTL responses.
HIV-1-specific CD8+ T-cell responses were mapped using a standard gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) assay (35). Briefly, 3 × 105 cells were plated per well and exposed to a library of HIV-1 peptides (consecutive 15-mers overlapping by 11 amino acids) spanning all HIV-1 proteins, obtained from the NIH AIDS Research and Reference Reagent Repository (Gag, catalog number 8116; Pol, catalog number 6208; Env, catalog number 9487; Nef, catalog number 5189; Tat, catalog number 5138; Rev, catalog number 6445; Vpr, catalog number 6447; Vpu, catalog number 6444; Vif, catalog number 6446; all were clade B consensus sequences, with the exception of Env, which is clade MN). Peptides were screened in 53 pools of 12 to 16 peptides; each peptide was added to the wells at a final concentration of 5 μg/ml. Responses were mapped to the peptide level using 4-by-4 matrix pools and then single peptides as previously described (86). Each plate included three negative-control wells with expanded CD8+ T cells without peptides and three positive-control wells containing 0.4 μg/ml of anti-CD2/CD2R and anti-CD28 monoclonal antibodies (Becton Dickinson, San Jose, CA). Plates were read on an automated ELISpot counting system (Cellular Technologies Limited, Cleveland, OH). A positive response was defined as being higher than four times the mean of the negative controls or 60 spot-forming cells (SFCs)/106 cells, whichever was greater. All confirmatory single-peptide ELISpot assays were conducted in duplicate, and the average of the two SFC values was used. Because many epitopes are included in two consecutive overlapping 15-mer peptides, only the higher SFC value for positive ELISpot assay responses against 2 sequential peptides was used.
Assigning minimal epitopes within peptides.
For 43 of the 47 targeted peptides identified by IFN-γ ELISpot assay analysis, we were able to identify the likely minimal epitope (9-mer, 10-mer, or 11-mer) within the peptide, guided by (i) the overlap between consecutive recognized 15-mer peptides, (ii) the HLA genotype(s) of the individual(s) being tested, and (iii) the online resources of the Los Alamos National Laboratory (LANL) database of HIV-1 Epitope Maps (http://www.hiv.lanl.gov/content/immunology/maps/maps.html) and the Immune Epitope Database (IEDB) Epitope Predictor (http://tools.immuneepitope.org/analyze/html/mhc_binding.html) (64).
Assessment of epitope sequence variability.
We took two approaches to quantifying the degree of sequence variability of the epitopes identified: Shannon entropy, a measure of variability at each amino acid that takes into account both the number and the frequency of observed amino acids (42), and the Simpson diversity index (Ds), a measure of variability accounting for both the number and the frequency of possible epitope variants (6, 50, 75).
Shannon entropy was calculated using the online tool at the Los Alamos National Laboratory HIV-1 Sequence Database (http://www.hiv.lanl.gov/content/sequence/ENTROPY/entropy_one.html) for each amino acid position in the 2008 alignment of clade B sequences, where low values indicate low variability, as previously described (85). Epitope entropy was calculated as the mean Shannon entropy value of all the amino acid positions in the predicted minimal epitope. For the 4 targeted 15-mer peptides for which we could not identify a specific minimal epitope candidate, we calculated the mean entropy for the entire 15-mer.
Ds was calculated for each epitope by considering the entire sequence as a unit and using all clade B epitope sequences occurring more than once, as identified by the LANL QuickAlign tool (http://www.hiv.lanl.gov/content/sequence/QUICK_ALIGN/QuickAlign.html). Ds was calculated as
where c is the number of epitope variants occurring more than once in the database, ni is the number of occurrences of variant i, and N is the total number of sequences in the database. (1 − Ds) was used as the measure of diversity, so that low numbers indicate low diversity/high conservation, as with the Shannon entropy index.
Statistics.
Relationships between disease-free years (defined in Results) and the breadth or magnitude of the CTL response were assessed using Spearman rank correlations. Tests comparing the number of disease-free years for subjects who did and did not target particular epitopes were performed using the Wilcoxon rank-sum test. We used these nonparametric approaches because of the likelihood of nonnormality with some of these measures (which could not be adequately assessed due to the small sample sizes) and the presence of outliers. However, the results were similar when using the equivalent parametric tests. Due to the variation in time of sampling relative to seroconversion, we also examined whether adjusting for this factor affected the strengths of the observed relationships by fitting linear regressions on the ranks, with sampling time included as a covariate.
RESULTS
Selection of HLA-B*57+ participants with available early postseroconversion samples and known long-term outcomes.
To identify early in HIV-1 infection the CTL targeting characteristics of HLA-B*57 (B57) individuals that correlate with eventual long-term control of infection, we selected 14 participants who had seroconverted while enrolled in the MACS and for whom long-term outcomes were known and early postseroconversion samples were available. Thirteen participants were HLA-B*5701+, including a single B*5701 homozygote. The 14th participant, participant M, was a B*5703-B*5802 heterozygote (Table 1).
In most cases, the time that had elapsed between seroconversion and the first available sample could be defined to within 6 months, on the basis of the dates of the last known seronegative visit and the first known seropositive visit (Table 1), with infection estimated to have occurred at the midpoint between those dates. For several participants, the range could be further narrowed due to additional testing performed at about the time of seroconversion between the usual semiannual study visits. For four participants, the time between the last seronegative visit and the first seropositive visit was greater than 6 months due to missed study visits. In some cases, the earliest cryopreserved postseroconversion sample available was from a time point later than the first seroconversion visit. The time of sampling thus ranged from an estimated 3 to 12.5 months after seroconversion, with a median of 5.25 months.
For each individual, the disease-free interval was calculated as the time from seroconversion to the earliest of the following 3 events: decline of CD4+ T cell counts below 200/mm3, initiation of cART, or death attributed to HIV infection. Participant I had a CD4 count of 622/mm3 and a viral load (VL) which had spiked to 41,465/ml from a set point of about 5,000/ml in his last visit before cART was initiated; 3 years later, his CD4 count declined below 200/mm3. Participant M had a CD4 count of 360/mm3 and a VL which had increased to 8,840/ml from a set point consistently below 5,000/ml in his last visit before cART was initiated (2009); as of November 2011, his CD4 count remained above 200/mm3. The disease-free intervals ranged from 3.5 to more than 25 years, with a median of 11.5 years (Table 1 and Fig. 1). As of November 2011, two of the participants (participant J and N) continued to maintain their CD4+ T cell counts above 450/mm3 without antiretroviral therapy.
Fig 1.
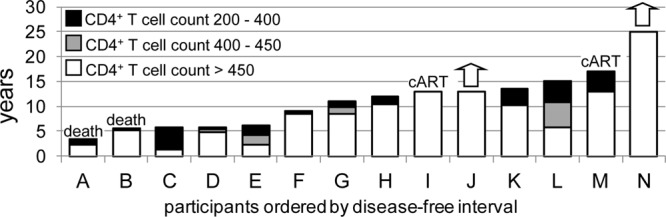
Distribution of disease progression profiles among the 14 HLA-B*57 participants. Column heights indicate number of years from seroconversion until decline of CD4+ T cell counts below 200/mm3 (8 participants) or other threshold disease-defining events, including initiation of cART (2 participants, I and M) and death (2 participants, A and B). White, years that the CD4+ T cell count was sustained above 450/mm3; gray and black, additional years that CD4+ counts were sustained above 400 and 200 cells/mm3, respectively. Two participants (J and N) continue to maintain CD4+ T cell counts above 450 cells/mm3 of blood at this time (arrows).
Early clinical markers vary among HLA-B*57 participants.
Plasma viremia at the time of sampling ranged from <400 to 109,373 RNA copies/ml (Table 1). No correlation was seen between viremia at the time of sampling and eventual time until disease (see Fig. S1 in the supplemental material). For all participants except participant K, these levels were similar to the early-set-point viremia (data not shown). Participant K had a lower level of subsequent viremia (set point, about 3,000 copies/ml; data not shown); the early time of sampling and the high level of viremia suggest that participant K was sampled during early or acute infection. Participant N is an elite controller who has suppressed viremia below the level of detection and maintained CD4+ T cell counts between 800 and 1,200/mm3 for 25 years (data not shown).
Because the time of sampling relative to seroconversion varied among the participants, we tested whether viremia levels were associated with time of sampling but did not find evidence of such a relationship (see Fig. S1 in the supplemental material), further suggesting that these viremia levels reflected early steady state.
CD4+ T cell counts at the time of sampling ranged from 317 to 1,637/mm3 (Table 1). Although declining CD4+ T cell counts are generally an indicator of HIV-1 disease progression, CD4 counts were not significantly associated with lower levels of viremia or longer disease-free intervals (see Fig. S1 in the supplemental material).
B57-restricted responses dominate the CD8+ T cell response early in infection but do not correlate with disease outcomes.
Because CTLs are important effectors of HIV-1 immune containment, particularly during early stages of infection, we sought to determine whether any specific parameters of early CTL responses, including breadth, magnitude, or specific epitope targeting, correlated with effective long-term control of infection. To this end, epitope-specific CTL targeting was quantified by IFN-γ ELISpot assay using overlapping 15-mer peptides spanning the viral proteome.
Overall, CTL responses to 47 peptides were detected among the 14 participants (91 total positive responses; Table 2). Eighty-seven of the 91 CTL responses could be assigned to a likely minimal epitope within the 15-mer. Of the 47 peptides, 19 contained minimal epitopes previously reported to be B57 restricted or to contain anchor motifs consistent with B57 binding and did not contain any epitopes predicted to be restricted by other HLA alleles of the participant (Table 2; see Table S1 in the supplemental material). Another 4 peptides contained both predicted B57-restricted epitopes and epitopes predicted to be restricted by another HLA of the participant (Table 2; see Table S1 in the supplemental material). The fraction of the epitopes targeted that were presumed to be B57 restricted ranged from 25% to 100% (mean, 71%), and B57-restricted CTL responses accounted for 39% to 100% (mean, 75%) of the overall magnitude of the response. These results agree with prior studies showing that B57-restricted CTLs generally dominate the CTL response (3, 4, 56).
Table 2.
CTL epitopes targeted by 14 participantsa

Forty-seven peptides identified by IFN-γ ELISpot analysis. Values shown are number of SFCs/106 cells. The positions and entropy values refer to the boldfaced epitope within the 15-mer peptide. For four 15-mers for which it was not possible to identify the smaller epitope (*), the positions and entropy values refer to the entire 15-mer. Position numbering matches LANL HXB-2 alignment. The 13 lowest entropy values (≤0.1) and diversity values (1 − Ds ≤ 0.5) are indicated in bold in gray boxes. B57 denotes the 19 B57-resticted epitopes; the SFC values for these epitopes are in bold and boxed in dark gray. B57? denotes the four epitopes for which it was not possible to resolve whether they were restricted by B57 or another allele of the participant; the SFC values for these epitopes are in bold and boxed in light gray. The highest-magnitude response for each participant is double boxed. For participants E, G, I, K, L, and M, the 2nd-highest response was within 10% of the highest and is also double boxed.
Neither the breadth nor the magnitude of the total or of the B57-restricted early CTL response was significantly associated with time to disease or with viremia or CD4+ T cell counts.
An elite controller is an outlier on some bivariate analyses relating CTL targeting and disease progression.
During the analyses, it emerged that participant N, the elite controller, was an outlier in terms of the bivariate relationships between some immune parameters and disease progression. While participant N clearly had the slowest progression to disease, maintaining both very low viremia levels and high CD4+ counts, by various measures his CTL responses were quite unremarkable. This suggests that his CTL response may not fully account for his long-term control of HIV, which is consistent with recent reports that elite controllers often benefit from both protective HLA alleles and additional factors, such as infection with weak viral strains or CD4+ T cells with reduced susceptibility to HIV-1 (15, 46, 67). Since our goal was to identify correlates of the early CTL response that correlate with eventual long-term control, we conducted our subsequent analyses both including and excluding data for participant N.
Greater breadth of CTL targeting of conserved epitopes correlates with slower progression to disease, while magnitude is associated with time since infection.
CTL targeting of highly conserved epitopes has been proposed to confer better protection than targeting of variable epitopes because conserved epitope sequences are more constrained in their ability to mutate and evade CTL recognition (87). We took two approaches to quantifying epitope conservation: both the Shannon entropy score and Simpson's diversity index (1 − Ds) were calculated for each of the 47 targeted epitopes. The Shannon entropy index can be understood to be the average of the variabilities of the amino acids in an epitope, whereas Simpson's diversity index quantifies the variability of the epitope as a whole. Cases where amino acids in an epitope covary, commonly seen when secondary mutations compensate for fitness costs of escape mutations, result in a higher entropy score than diversity score.
Plotting the Shannon entropy scores of the 47 targeted epitopes revealed a cluster of 13 epitopes with scores below 0.1 (Fig. 2a); CTL responses to these 13 low-entropy (LE) epitopes were assessed, as were CTL responses to the 13 epitopes with the lowest Simpson diversity scores (<0.5; low diversity [LD]) (Table 2 and Fig. 2a, b, f, and g). Although the breadths of the LE and LD responses were not correlated with the time since seroconversion, a significant association between the magnitudes of the responses to these minimally variable epitopes and the time since seroconversion was observed (R = 0.53 and P = 0.05 for LE response; R = 0.62 and P = 0.02 for LD response; see Fig. S3 in the supplemental material). This led us to adjust for the effect of time of sampling when exploring potential associations between these CTL responses and time until disease.
Fig 2.

Greater targeting of conserved epitopes early in infection correlates with longer times until disease. The 47 epitopes/peptides targeted by participants in this study were plotted by Shannon entropy score (a) and Simpson's diversity index (f). Low scores indicate greater sequence conservation. (a) The 13 epitopes with entropy scores below 0.1 were defined as LE (boxed), and more detailed results are shown in panel b. (f) The 13 epitopes with diversity scores below 0.5 were defined as LD (boxed), and more detailed results are shown in panel g. Numbers in panels b and g indicate the number of participants targeting individual epitopes; unlabeled epitopes were targeted by only one participant. (c and h) Numbers of LE and LD epitopes, respectively, targeted by individual participants versus years to disease. (d and l) Numbers of LE and LD epitopes, respectively, targeted by individual participants versus years to disease data but with data for participant N (circle with a cross) excluded. (e and j) Magnitudes of CTL response against LE and LD epitopes, respectively, versus years until disease. All participants are represented in all graphs with the exception of those in panels d and i, in which data for participant N are excluded from the analysis. The relationships between breadth and magnitude and time until disease were also analyzed by adjusting for the time of sampling; the adjusted (adj.) P values are shown (c to e and h to j). Correlations and P values shown are calculated on the basis of Spearman's rank correlation.
When the entire cohort was analyzed, no relationship between breadth of the LE response and time until disease was observed (R = 0.36; P = 0.21; P = 0.16 when adjusted for the effect of time of sampling; Fig. 2c). However, when data for elite controller N were excluded from the analysis, this relationship was significant (R = 0.57; P = 0.04; adjusted P = 0.03; Fig. 2d). A greater breadth of the LD response was also significantly correlated with slower times until disease, both for the whole cohort (R = 0.61, P = 0.02, adjusted P = 0.006) and when data for elite controller N were excluded (R = 0.79, P = 0.001, adjusted P = 0.00002) (Fig. 2h and i). The magnitudes of the responses to LE and LD epitopes were not significantly correlated with time until disease (Fig. 2e and j). Taken together, these results suggest that CTL targeting of more highly conserved epitopes early in infection is associated with better long-term control of infection.
CTL responses to structural epitopes correlate with delayed progression to disease.
CTL responses against proteins encoded by all 9 genes of HIV-1 were detected (Table 2 and Fig. 3a). All participants demonstrated CD8+ T cell responses to both structural (Gag, Pol, Env) and regulatory (Nef, Tat, Rev) proteins, and six participants targeted at least one of the accessory (Vpr, Vif, Vpu) proteins. Greater breadths of both the total and the B57-restricted CTL response to structural proteins were associated with longer times until disease (total, R = 0.67 and P = 0.009; B57, R = 0.68 and P = 0.008; Fig. 3b and c). Breadth of targeting of regulatory and accessory epitopes did not correlate with time until disease (data not shown).
Fig 3.
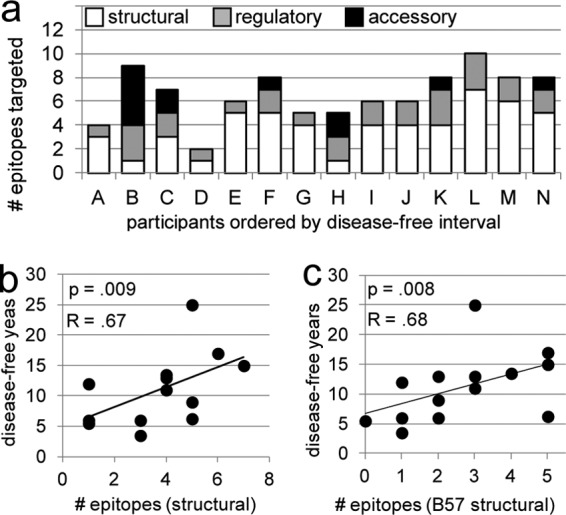
CTL targeting of epitopes from structural proteins early in infection correlates with better long-term outcomes. (a) Column height indicates the number of unique epitopes targeted within each individual. White, gray, and black shading, portion of the response that is specific for epitopes of structural, regulatory, and accessory protein origin, respectively. The greater breadth of the CTL response to both total structural epitopes (b) and B57-restricted structural epitopes (c) correlates similarly with slower progression to disease. The strength and statistical significance of the associations shown in panels b and c were similar when the analysis was adjusted for the effect of the time of sampling on the breadth of targeting and when data for participant N were excluded. Data for all participants are represented in the graphs. Correlations and P values shown are calculated on the basis of Spearman's rank correlation.
In no case was the magnitude of the response to any subset of epitopes associated with time until disease. Previous studies have also found CTL breadth to be a more reliable correlate of protection than magnitude (1). Therefore, for the remainder of the analyses, only the associations between the breadth of the CTL response and time until disease are shown.
B57-restricted targeting of Gag epitopes early in infection correlates with slower disease progression.
CTL targeting of Gag is inversely associated with viremia levels during early and chronic infection (23, 40, 48, 52, 59). Consistent with this was the lack of Gag targeting in participants A and B, who progressed the most rapidly to disease (Table 2; Fig. 4a). While the association between the breadth of the total early Gag-specific CTL response and time until disease for the whole cohort fell short of statistical significance (R = 0.48, P = 0.08; Fig. 4b), a greater breadth of targeting of B57-restricted Gag epitopes early in infection was correlated with slower disease progression (R = 0.62, P = 0.02; Fig. 4c).
Fig 4.

Early targeting of B57-restricted Gag epitopes correlates with better long-term outcomes. (a) Column height indicates the number of unique epitopes targeted within each individual. Black, dark gray, and light gray shading, epitopes derived from Gag, Pol, and Env, respectively; white, responses to epitopes from nonstructural proteins. The association between the breadth of CTL targeting of B57-restricted epitopes of Gag and slow progression to disease (c) was stronger than the association of CTL targeting of all Gag epitopes with slow progression (b). Exclusion of the data for participant N from the analyses in panels b and c yielded similar findings (not shown). The association between breadth of targeting of 3 immunodominant B57-restricted Gag epitopes (IW9, KF11, TW10) and slow disease progression (d) was stronger when the data for participant N were excluded from the analysis (e). The degree to which the CTL response was focused on IW9, KF11, and TW10 (quantified as the fraction of the total breadth that responded to these epitopes represented) did not correlate with time until disease (f). All participants are represented in the graphs, with the exception of that in panel e, in which the data for participant N were excluded from the analysis. The strength and statistical significance of the correlations were similar when adjusted for the effect of time of sampling on CTL targeting. Correlations and P values shown are calculated on the basis of Spearman's rank correlation.
Migueles et al. (56) previously reported that, during chronic infection, B57+ elite controllers demonstrate highly focused CTL responses to four conserved B57-restricted epitopes in Gag, ISPRTLNAW (IW9), KAFSPEVIPMF (KF11), TSTLQEQIGW (TW10), and QASQEVKNW (QW9). While none of the participants from our cohort responded to QW9, the remaining three epitopes were targeted by several individuals. Surprisingly, the breadth of targeting of IW9, KF11, and TW10 correlated less well with outcomes than did the breadth of targeting of all B57-specific Gag epitopes, only trending toward significance (R = 0.51, P = 0.06; Fig. 4d; compare with Fig. 4c). However, when data for elite controller N were excluded from the analysis, the association between early targeting of these three conserved epitopes and longer times to disease reached significance (R = 0.65, P = 0.02; Fig. 4e). There was no correlation between a narrowly focused response to these epitopes early in infection and eventual slow progression (Fig. 4f), even when data for participant N were excluded.
Within the first year of infection, time of sampling did not strongly affect the breadth or magnitude of targeting of total or B57-specific Gag epitopes (see Fig. S3 in the supplemental material).
Early B57-restricted CTL targeting of the IW9 Gag epitope correlates with delayed progression to disease.
Rank-sum analyses comparing the time to disease for targeters versus nontargeters of individual epitopes were possible for all epitopes targeted by more than 5 participants, which included IW9, KF11, and TW10, as well as 2 additional presumed B57-presented epitopes, HW9 from Nef and LY11 from Rev (Table 2 and Fig. 5). Of these 5, the only epitope for which differences in early targeting between eventual slow versus rapid progressors were observed was IW9; individuals who targeted IW9 early had a median time to disease of 13 years, whereas the median time to disease was only 6 years for nontargeters (Fig. 5a and b; P = 0.037 by 1-sided Wilcoxon rank-sum test because the hypothesis was directional; the 2-sided P value is 0.073, also trending toward significance). When the analysis was repeated by excluding the data for elite controller N, the 1-sided P value for the association between early IW9 targeting and longer times until disease strengthens to 0.006 and the 2-sided P value strengthens to 0.012 (Fig. 5b).
Fig 5.

CTL targeting of the IW9 epitope of Gag early in infection is correlated with eventual slow progression to disease. IW9 (ISPRTLNAW), KF11 (KAFSEPVIPMF), and TW10 (TSTLQEQIGWM) from Gag, HW9 (HTQGYFPDW) from Nef, and LY11 (LKTVRLIKFLY) from Rev were all targeted early in infection by at least 5 of 14 participants, permitting analysis of whether early targeting correlated with long-term protection. (a) Hollow circles, nontargeters for each epitope; filled circles, targeters for each epitope; bars, median times until disease. IW9 was the only epitope for which targeters had significantly longer times to disease than nontargeters: median, 13 years versus 6 years. (b) Analysis similar to that for panel a, but with data for participant N excluded. (c) Bars, median times at which samples were taken relative to seroconversion among targeters (filled circles) and nontargeters (hollow circles) for each epitope. P values shown are calculated on the basis of the 1-sided Wilcoxon rank-sum test.
To address the possibility that this finding was an artifact of the particular disease threshold definition chosen, CD4+ T cell counts declining below 200/mm3, we also analyzed the association between early IW9 targeting and the time until CD4 cell counts declined below 400/mm3 and found that those with early IW9 targeting trended toward a delay in reaching this threshold as well (median, 5.3 years for nontargeters versus 10.3 years for targeters, P = 0.10; see Fig. S4 in the supplemental material).
Some epitopes are characteristically targeted early in infection, whereas others elicit stronger responses later in infection (32). We examined whether the patterns of CTL targeting found for individual epitopes were an artifact of the time that had elapsed since infection at the time of sampling but did not find significant differences in time of sampling between targeters and nontargeters for any of the 5 epitopes examined (Fig. 5c). Therefore, it is not likely that the observed association between early targeting of IW9 and eventual slow progression to disease was an artifact of time of sampling.
All 7 participants who targeted IW9 also targeted other Gag epitopes, and 6 of the 7 also targeted TW10 and/or KF11. To quantify the degree to which IW9 targeting contributed to the association between targeting of various subsets of Gag epitopes and slow disease progression, we reanalyzed these relationships with data for IW9 targeting excluded, as well as with data for TW10 and KF11 targeting excluded, for comparison (see Fig. S5 in the supplemental material). For the association between B57-specific Gag epitope targeting and slow progression (R = 0.62, P = 0.02), removing the data for IW9 had a stronger impact on the association (R = 0.42, P = 0.14) than did removing the data for TW10 (R = 0.57, P = 0.03). For the association between targeting of the IW9-TW10-KF11 set (R = 0.51, P = 0.06), removing the data for IW9 had a stronger impact (R = 0.34, P = 0.24) than did removing the data for either TW10 (R = 0.44, P = 0.11) or KF11 (R = 0.40, P = 0.16). This trend was also seen when the data for participant N were excluded from the analyses. These data suggest that IW9 targeting is not protective in isolation and that targeting of other epitopes contributes to protection. However, IW9 targeting early in infection does appear to contribute more strongly to long-term protection than does early targeting of TW10 or KF11 or other B57-specific Gag epitopes.
DISCUSSION
The goal of this study was to determine whether any characteristics of the CTL response early in HIV-1 infection distinguish HLA-B57+ individuals who eventually demonstrate long-term control of infection from those who progress more rapidly to AIDS. Early CTL responses are believed to be more important for control of HIV-1 infection than later CTL responses, both because they limit peak viremia and impose early fitness costs and because they exhibit more effective cellular phenotypes, likely benefiting from more robust CD4+ T cell help and less antigen-induced exhaustion than later responses (13, 79). However, correlating early CTL targeting events with long-term outcomes has been problematic due to the difficulty in reliably identifying those individuals, while still within the early stages of infection, who will eventually prove to be slow progressors. By enrolling thousands of at-risk men, documenting HIV infections and subsequent disease outcomes over decades, as well as archiving cryopreserved samples, the MACS has facilitated this type of analysis. In addition, the prospective enrollment strategy of the MACS ensures better representation of slow progressors, who are less likely to exhibit symptoms of acute retroviral syndrome and are thus often excluded from acute-infection cohorts (3).
The results shown here identify CTL targeting of the IW9 epitope of Gag in the early months following HIV-1 infection to be a novel immune correlate of long-term control among B57+ individuals. Although the cohort is small, many of the observations in this study are consistent with prior reports, suggesting that the cohort is representative and that the correlation between early IW9 targeting and slow progression is real. In no participant was IW9 the sole B57-specific Gag epitope targeted, suggesting that IW9 targeting may not be protective in isolation. Our findings do not distinguish whether early IW9 targeting is directly protective or an indirect correlate of a separate mechanism of protection.
Surprisingly, our study failed to find a correlation between early targeting of two other commonly targeted conserved B57-restricted epitopes, KF11 and TW10, and delayed progression to disease. Although CTL targeting of IW9 and the existence of viral escape from such targeting have been well described, more data exist regarding the dynamics of targeting of TW10 and KF11, which often dominate the early and later CTL responses, respectively (4, 77). However, subdominant CTL responses can be important for the control of HIV-1 (27). It is possible that IW9-specific CTLs, while few in number, can have superior avidity or antiviral activity, as has been seen in isolated IW9-specific CTL lines from a small number of individuals (16, 34). Alternatively, although the fitness costs to HIV-1 of mutating to escape IW9-specific CTL surveillance do not appear to be more severe than the costs of escape from TW10- or KF11-specific CTL surveillance (10), it remains to be determined whether the costs associated with IW9 escape are more sustained, perhaps due to fewer options for compensation by secondary mutations (11).
The timing of particular CTL responses may be an important factor in their influence on disease progression. Most B57+ individuals infected with HIV-1 target IW9 at some point; in one cohort study, only 23% targeted IW9 during acute/early infection, whereas CTL responses to IW9 were detectable in 60% of B57+ individuals during chronic infection (77). The overrepresentation of slow progressors in our cohort may account for the finding of IW9 targeting in 50% of participants in our study. It is possible that IW9 responses are most useful if they are early, in contrast to KF11 targeting, which is effective during chronic infection (40).
It is instructive to compare our findings with those in a recent report which also sought to identify early B57-restricted CTL responses that correlated with slow HIV-1 disease progression (21). Within a cohort of 9 B57+/B58+ individuals, targeting of TW10 within the first 1 to 4 months of infection was significantly correlated with a slower decline of CD4+ T cell numbers in the 8 years after infection (21). This is consistent with our findings: of the 6 participants in the present study whose CD4+ T cell count declined to below 200 within 10 years of infection (which also occurred for all 9 participants in the prior study), the 3 who did not target TW10 advanced to disease much more quickly than those who did (3.5, 5.5, and 6 years for participants A, B, and C, respectively, versus 6, 6.25, and 9 years for participants D, E, and F, respectively). CTL targeting of TW10 and fitness-impacting escape mutations in this epitope have been shown to be associated with control of HIV in the early years of infection (21, 30, 51, 73, 77); however, our study suggests that the protective effect of early TW10 targeting may not extend beyond 10 years. In agreement with this, the fitness costs to HIV-1 of CTL escape mutations in TW10 were shown to wane during chronic infection due to the accumulation of compensatory mutations (11). Importantly, the subjects in the earlier study were recruited from those who presented with acute symptoms of HIV-1 infection and did not include any slow progressors, whereas 50% of our participants maintained CD4+ T cell counts above 500/mm3 for more than 7 years, 0% of the subjects in that study did (21). Finally, the earlier study limited the analysis of B57-restricted targeting to the TW10 and KF11 epitopes (21). In contrast, by conducting proteome-wide ELISpot assay analyses, we made no a priori assumptions as to which early CTL responses would be important and identified early IW9 targeting as an unforeseen correlate of long-term disease suppression.
We also report here that during early infection a greater breadth of targeting of highly conserved epitopes, regardless of HLA restriction or parent protein, correlated with longer times until disease, as did a greater breadth of targeting of B57-restricted epitopes of Gag. The latter findings are consistent with those from a prior report that found that targeting of conserved and Gag-derived epitopes correlated with lower viremia levels during primary infection (48). CTL targeting of Gag epitopes has also been found to correlate with lower viremia levels during chronic infection (23, 40, 52, 59). The present study extends these findings both by focusing on the differences in targeting among B57+ individuals and by linking early CTL targeting events with long-term outcomes. The effectiveness of early responses to conserved and Gag-derived epitopes for not only immediate control but also long-term control of HIV-1 reaffirms the case for the design of vaccines that elicit responses to such epitopes in persons of all HLA genotypes.
Strikingly, the single elite controller in our study was an outlier in many bivariate analyses relating patterns of early CTL targeting and outcomes. This provides additional support for the notion that elite controllers typically have more than simply a protective HLA genotype and associated CTL responses in their favor, but also benefit from additional factors, such as CD4+ T cells resistant to infection or infection with an attenuated virus (15, 46, 49, 67), although we have no evidence for this in the case of participant N. Importantly, however, this model does suggest that efforts to identify CTL-mediated correlates of infection in B57+ individuals should not focus solely on elite controllers; rather, comparisons between viremic slow and rapid progressors are likely to be more fruitful.
The greatest difficulty in interpreting our results is the range of time that elapsed between infection and time of sampling among our participants. The effect of sampling time on the magnitude of the CTL response to highly conserved epitopes was especially evident. This effect is consistent with the increase in the fraction of the CTL response devoted to conserved epitopes that occurs over time, as CTL responses to epitopes that escape progressively decay (7, 36). The length of time that elapsed between infection and time of sampling did not have a strong effect, however, on the pattern of targeting of any specific epitope. CTL targeting is highly dynamic in the early weeks of infection but appears to stabilize somewhat as viremia declines toward the set point and the rate of epitope escape slows (31). Consistent with this, the immunodominance patterns of HIV-specific CD8+ T cells were very similar between acute and early stages of infection for most HLA genotypes, including B57 (4). With the exception of participant K, whose viremia level suggests that he was sampled during primary infection, most of our participants were likely sampled slightly later, during early infection, which could explain the observed lack of impact of time since infection on specific epitopes targeted.
A potential source of error in our work is in the assignment of individual peptides to likely presenting HLA alleles, because epitopes can frequently be presented by more than one HLA allele (28, 65). For most cases where more than one possible HLA allele was predicted for a given peptide, we were able to narrow down to a single HLA allele by eliminating alleles that were not possessed by the targeting participant. However, it is likely that other HLA alleles not predicted by IEDB or LANL algorithms could also present some of the peptides, and it is therefore possible that some of our assignments are incorrect. Because a relatively large fraction of the peptides presented by HLA-B*57 are not presented by any other HLA allele (65), we do not expect that this would significantly impact our findings.
One limitation of our study comes from the use of consensus sequences to detect CTL responses across the HIV-1 proteome. Most likely to be missed with this approach are responses to less conserved viral peptides. CTL responses to such variable epitopes are generally thought to be less important and less protective because the virus can easily mutate to evade targeting; however, determining responses to autologous viral sequences is necessary to fully characterize host CTL responses.
Another caveat is that the polyclonal expansion of CD8+ T cells that we used could potentially have skewed the T cell population, with cells of some specificities expanded more efficiently than others. While we and others have shown that this technique does not generally introduce significant bias at the level of either the epitopes targeted or clonotype populations (35, 37, 74), such bias nonetheless remains possible. However, since any such bias would be due to the loss of some T cell responses rather than to the generation of spurious ones, this possibility does not undermine our key findings.
Finally, the stringent criteria for inclusion in our study limited the size of our cohort and, consequently, the statistical power of the analysis.
While clear correlates of B57-mediated CTL control of HIV-1 infection remain to be firmly defined, two distinct hypotheses compete to explain why the B57 allele is protective. The fortuitous or additive model argues that it is by presenting multiple immunodominant epitopes from highly conserved sections of the Gag p24 protein that B57 uniquely exerts its protective effect (10, 11, 17). The intrinsic model holds that B57-restricted CTLs are superior and are perhaps distinguished by an ability to maintain replication capacity in the face of chronic antigen stimulation (34) or by an ability to cross-react with the epitope sequence variants generated in highly mutable viruses such as HIV-1 and hepatitis C virus (41, 43, 80). Our finding that greater targeting of three conserved B57-restricted epitopes in Gag correlated with better outcomes supports the additive model; however, the finding that targeting of only IW9 early in infection was specifically associated with longer times until disease suggests that the mechanism may be more complex and that the timing of CTL responses may be important. This work, although not powered by a large cohort and necessarily exploratory in nature, does suggest that the role of IW9 targeting in B57-mediated protection merits closer attention. Understanding the detailed mechanisms by which B57 is associated with slow progression to disease will reveal underlying principles of immune control of HIV-1, which is critical for the development of rational vaccine design strategies.
Supplementary Material
ACKNOWLEDGMENTS
We thank the MACS participants for their contributions to this study.
We acknowledge the expert services of the biostatistics core of the UCLA AIDS Institute Center for AIDS Research (CFAR) (NIH/NIAID AI028697). ELISpot assay analysis was conducted in the cytometry core (supported by NIH awards CA-16042 and AI-28697 and by the UCLA Jonsson Comprehensive Cancer Center, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA). This work was supported by the NIH (R21 AG032942 and R21 AI60486), an IDEA award to C.A.B. from the California HIV/AIDS Research Program (ID10-LA-007), and the MACS (AI-35040). The MACS is funded by the National Institute of Allergy and Infectious Diseases, with additional supplemental funding from National Cancer Institute grants UO1-AI-35042, UL1-RR025005 (GCRC), UO1-AI-35043, UO1-AI-35039, UO1-AI-35040, and UO1-AI-35041, with its website located at http://www.statepi.jhsph.edu/macs/macs.html.
Data in this report were collected by MACS, with centers at The Johns Hopkins Bloomberg School of Public Health; Howard Brown Health Center, Feinberg School of Medicine, Northwestern University, and Cook County Bureau of Health Services; University of California, Los Angeles; and University of Pittsburgh.
Footnotes
Published ahead of print 18 July 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Addo MM, et al. 2003. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J. Virol. 77:2081–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Allen TM, et al. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 79:13239–13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Altfeld M, et al. 2003. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 17:2581–2591 [DOI] [PubMed] [Google Scholar]
- 4. Altfeld M, et al. 2006. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T cell response against HIV-1. PLoS Med. 3:e403 doi:10.1371/journal.pmed.0030403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bailey JR, Williams TM, Siliciano RF, Blankson JN. 2006. Maintenance of viral suppression in HIV-1-infected HLA-B*57+ elite suppressors despite CTL escape mutations. J. Exp. Med. 203:1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balamurugan A, Ng HL, Yang OO. 2010. Rapid T cell receptor delineation reveals clonal expansion limitation of the magnitude of the HIV-1-specific CD8+ T cell response. J. Immunol. 185:5935–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bansal A, et al. 2005. CD8 T-cell responses in early HIV-1 infection are skewed towards high entropy peptides. AIDS 19:241–250 [PubMed] [Google Scholar]
- 8. Betts MR, et al. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. 1994. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68:6103–6110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boutwell CL, Rowley CF, Essex M. 2009. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J. Virol. 83:2460–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brockman MA, et al. 2010. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J. Virol. 84:11937–11949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brumme ZL, et al. 2008. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J. Virol. 82:9216–9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cao J, McNevin J, Malhotra U, McElrath MJ. 2003. Evolution of CD8+ T cell immunity and viral escape following acute HIV-1 infection. J. Immunol. 171:3837–3846 [DOI] [PubMed] [Google Scholar]
- 14. Cao K, et al. 2001. Analysis of the frequencies of HLA-A, B, and C alleles and haplotypes in the five major ethnic groups of the United States reveals high levels of diversity in these loci and contrasting distribution patterns in these populations. Hum. Immunol. 62:1009–1030 [DOI] [PubMed] [Google Scholar]
- 15. Chen H, et al. 2011. CD4+ T cells from elite controllers resist HIV-1 infection by selective upregulation of p21. J. Clin. Invest. 121:1549–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen H, et al. 2009. Differential neutralization of human immunodeficiency virus (HIV) replication in autologous CD4 T cells by HIV-specific cytotoxic T lymphocytes. J. Virol. 83:3138–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crawford H, et al. 2009. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J. Exp. Med. 206:909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deeks SG, Walker BD. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27:406–416 [DOI] [PubMed] [Google Scholar]
- 19. Detels R, et al. 2012. The Multicenter AIDS Cohort Study, 1983 to …. Public Health 126:196–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Detels R, et al. 1992. Recent scientific contributions to understanding HIV/AIDS from the Multicenter AIDS Cohort Study. J. Epidemiol. 2:S11–S19 [Google Scholar]
- 21. Dinges WL, et al. 2010. Virus-specific CD8+ T-cell responses better define HIV disease progression than HLA genotype. J. Virol. 84:4461–4468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dudley J, et al. 1995. The Multicenter AIDS Cohort Study: retention after 9 1/2 years. Am. J. Epidemiol. 142:323–330 [DOI] [PubMed] [Google Scholar]
- 23. Edwards BH, et al. 2002. Magnitude of functional CD8+ T-cell responses to the Gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J. Virol. 76:2298–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elahi S, et al. 2011. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat. Med. 17:989–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emu B, et al. 2008. HLA class I-restricted T-cell responses may contribute to the control of human immunodeficiency virus infection, but such responses are not always necessary for long-term virus control. J. Virol. 82:5398–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fellay J, et al. 2007. A whole-genome association study of major determinants for host control of HIV-1. Science 317:944–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frahm N, et al. 2006. Control of human immunodeficiency virus replication by cytotoxic T lymphocytes targeting subdominant epitopes. Nat. Immunol. 7:173–178 [DOI] [PubMed] [Google Scholar]
- 28. Frahm N, et al. 2007. Extensive HLA class I allele promiscuity among viral CTL epitopes. Eur. J. Immunol. 37:2419–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao X, et al. 2005. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat. Med. 11:1290–1292 [DOI] [PubMed] [Google Scholar]
- 30. Goepfert PA, et al. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 205:1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goonetilleke N, et al. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 206:1253–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goulder PJ, et al. 2001. Substantial differences in specificity of HIV-specific cytotoxic T cells in acute and chronic HIV infection. J. Exp. Med. 193:181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goulder PJ, et al. 1996. Novel, cross-restricted, conserved, and immunodominant cytotoxic T lymphocyte epitopes in slow progressors in HIV type 1 infection. AIDS Res. Hum. Retroviruses 12:1691–1698 [DOI] [PubMed] [Google Scholar]
- 34. Horton H, et al. 2006. Preservation of T cell proliferation restricted by protective HLA alleles is critical for immune control of HIV-1 infection. J. Immunol. 177:7406–7415 [DOI] [PubMed] [Google Scholar]
- 35. Ibarrondo FJ, et al. 2005. Parallel human immunodeficiency virus type 1-specific CD8+ T-lymphocyte responses in blood and mucosa during chronic infection. J. Virol. 79:4289–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jamieson BD, et al. 2003. Epitope escape mutation and decay of human immunodeficiency virus type 1-specific CTL responses. J. Immunol. 171:5372–5379 [DOI] [PubMed] [Google Scholar]
- 37. Jones N, et al. 2003. Evaluation of antigen-specific responses using in vitro enriched T cells. J. Immunol. Methods 274:139–147 [DOI] [PubMed] [Google Scholar]
- 38. Kaslow RA, et al. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2:405–411 [DOI] [PubMed] [Google Scholar]
- 39. Kaslow RA, et al. 1987. The Multicenter AIDS Cohort Study: rationale, organization, and selected characteristics of the participants. Am. J. Epidemiol. 126:310–318 [DOI] [PubMed] [Google Scholar]
- 40. Kiepiela P, et al. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53 [DOI] [PubMed] [Google Scholar]
- 41. Kim AY, et al. 2011. Spontaneous control of HCV is associated with expression of HLA-B 57 and preservation of targeted epitopes. Gastroenterology 140:686–696.e1 doi:10.1053/j.gastro.2010.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Korber BT, et al. 1994. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: evidence of conserved elements in the V3 region of the envelope protein of brain-derived sequences. J. Virol. 68:7467–7481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Košmrlj A, et al. 2010. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature 465:350–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koup RA, et al. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68:4650–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lambotte O, et al. 2005. HIV controllers: a homogeneous group of HIV-1-infected patients with spontaneous control of viral replication. Clin. Infect. Dis. 41:1053–1056 [DOI] [PubMed] [Google Scholar]
- 46. Lassen KG, et al. 2009. Elite suppressor-derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and kinetics. PLoS Pathog. 5:e1000377 doi:10.1371/journal.ppat.1000377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lichterfeld M, et al. 2007. Selective depletion of high-avidity human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T cells after early HIV-1 infection. J. Virol. 81:4199–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu Y, et al. 2009. Conserved HIV-1 epitopes continuously elicit subdominant cytotoxic T-lymphocyte responses. J. Infect. Dis. 200:1825–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lobritz MA, Lassen KG, Arts EJ. 2011. HIV-1 replicative fitness in elite controllers. Curr. Opin. HIV AIDS 6:214–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Magurran AE. 2004. Measuring biological diversity. Blackwell, Oxford, United Kingdom [Google Scholar]
- 51. Martinez-Picado J, et al. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80:3617–3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Masemola A, et al. 2004. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: correlation with viral load. J. Virol. 78:3233–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. 2010. The immune response during acute HIV-1 infection: clues for vaccine development. Nat. Rev. Immunol. 10:11–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Migueles SA, Connors M. 2010. Long-term nonprogressive disease among untreated HIV-infected individuals: clinical implications of understanding immune control of HIV. JAMA 304:194–201 [DOI] [PubMed] [Google Scholar]
- 55. Migueles SA, et al. 2008. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 29:1009–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Migueles SA, et al. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. U. S. A. 97:2709–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Migueles SA, et al. 2009. Defective human immunodeficiency virus-specific CD8+ T-cell polyfunctionality, proliferation, and cytotoxicity are not restored by antiretroviral therapy. J. Virol. 83:11876–11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Navis M, et al. 2007. Viral replication capacity as a correlate of HLA B57/B5801-associated nonprogressive HIV-1 infection. J. Immunol. 179:3133–3143 [DOI] [PubMed] [Google Scholar]
- 59. Novitsky V, et al. 2003. Association between virus-specific T-cell responses and plasma viral load in human immunodeficiency virus type 1 subtype C infection. J. Virol. 77:882–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O'Connell KA, Bailey JR, Blankson JN. 2009. Elucidating the elite: mechanisms of control in HIV-1 infection. Trends Pharmacol. Sci. 30:631–637 [DOI] [PubMed] [Google Scholar]
- 61. Pelak K, et al. 2010. Host determinants of HIV-1 control in African Americans. J. Infect. Dis. 201:1141–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pereyra F, et al. 2008. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J. Infect. Dis. 197:563–571 [DOI] [PubMed] [Google Scholar]
- 63. Pereyra F, et al. 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Peters B, et al. 2005. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol. 3:e91 doi:10.1371/journal.pbio.0030091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rao X, Hoof I, Costa AI, van Baarle D, Keşmir C. 2011. HLA class I allele promiscuity revisited. Immunogenetics 63:691–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rehr M, et al. 2008. Emergence of polyfunctional CD8+ T cells after prolonged suppression of human immunodeficiency virus replication by antiretroviral therapy. J. Virol. 82:3391–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sáez-Cirión A, et al. 2011. Restriction of HIV-1 replication in macrophages and CD4+ T cells from HIV controllers. Blood 118:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sáez-Cirión A, et al. 2007. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc. Natl. Acad. Sci. U. S. A. 104:6776–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sáez-Cirión A, Pancino G, Sinet M, Venet A, Lambotte O. 2007. HIV controllers: how do they tame the virus? Trends Immunol. 28:532–540 [DOI] [PubMed] [Google Scholar]
- 70. Sáez-Cirión A, et al. 2009. Heterogeneity in HIV suppression by CD8 T cells from HIV controllers: association with Gag-specific CD8 T cell responses. J. Immunol. 182:7828–7837 [DOI] [PubMed] [Google Scholar]
- 71. Scheid JF, et al. 2009. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 458:636–640 [DOI] [PubMed] [Google Scholar]
- 72. Schmitz JE, et al. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857–860 [DOI] [PubMed] [Google Scholar]
- 73. Schneidewind A, et al. 2009. Maternal transmission of human immunodeficiency virus escape mutations subverts HLA-B57 immunodominance but facilitates viral control in the haploidentical infant. J. Virol. 83:8616–8627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shacklett BL, et al. 2003. Optimization of methods to assess human mucosal T-cell responses to HIV infection. J. Immunol. Methods 279:17–31 [DOI] [PubMed] [Google Scholar]
- 75. Simpson EH. 1949. Measurement of diversity. Nature 163:688 [Google Scholar]
- 76. Streeck H, et al. 2008. Antigen load and viral sequence diversification determine the functional profile of HIV-1-specific CD8+ T cells. PLoS Med. 5:e100 doi:10.1371/journal.pmed.0050100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Streeck H, et al. 2009. Human immunodeficiency virus type 1-specific CD8+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD4+ T cells. J. Virol. 83:7641–7648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Streeck H, et al. 2007. Recognition of a defined region within p24 gag by CD8+ T cells during primary human immunodeficiency virus type 1 infection in individuals expressing protective HLA class I alleles. J. Virol. 81:7725–7731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Streeck H, Nixon DF. 2010. T cell immunity in acute HIV-1 infection. J. Infect. Dis. 202(Suppl. 2):S302–S308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Turnbull EL, et al. 2006. HIV-1 epitope-specific CD8+ T cell responses strongly associated with delayed disease progression cross-recognize epitope variants efficiently. J. Immunol. 176:6130–6146 [DOI] [PubMed] [Google Scholar]
- 81. Turnbull EL, et al. 2009. Kinetics of expansion of epitope-specific T cell responses during primary HIV-1 infection. J. Immunol. 182:7131–7145 [DOI] [PubMed] [Google Scholar]
- 82. Wang YE, et al. 2009. Protective HLA class I alleles that restrict acute-phase CD8+ T-cell responses are associated with viral escape mutations located in highly conserved regions of human immunodeficiency virus type 1. J. Virol. 83:1845–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wong JT, Colvin RB. 1987. Bi-specific monoclonal antibodies: selective binding and complement fixation to cells that express two different surface antigens. J. Immunol. 139:1369–1374 [PubMed] [Google Scholar]
- 84. Wong JT, Colvin RB. 1991. Selective reduction and proliferation of the CD4+ and CD8+ T cell subsets with bispecific monoclonal antibodies: evidence for inter-T cell-mediated cytolysis. Clin. Immunol. Immunopathol. 58:236–250 [DOI] [PubMed] [Google Scholar]
- 85. Yang OO. 2009. Candidate vaccine sequences to represent intra- and inter-clade HIV-1 variation. PLoS One 4:e7388 doi:10.1371/journal.pone.0007388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yang OO, et al. 2005. Genetic and stochastic influences on the interaction of human immunodeficiency virus type 1 and cytotoxic T lymphocytes in identical twins. J. Virol. 79:15368–15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yang OO, Daar ES, Ng HL, Shih R, Jamieson BD. 2011. Increasing CTL targeting of conserved sequences during early HIV-1 infection is correlated to decreasing viremia. AIDS Res. Hum. Retroviruses 27:391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.