

Abstract
Retroviruses integrated into genomic DNA participate in long-range gene activation from as far away as several hundred kilobases. Hypotheses have been put forth to account for these phenomena, but data have not been provided to support a physical mechanism that explains long-range activation. In murine leukemia virus-induced myeloid leukemia in mice, integrated proviruses have been found upstream of c-myb in three regions, named Mml1, Mml2, and Mml3 (25, 50, and 70 kb upstream, respectively). The transcription factor c-Myb is an oncogene whose dysregulation and/or mutation can lead to human leukemia. We hypothesized that the murine c-myb upstream region contains regulatory elements accessed by the retrovirus. To identify regulatory sites in the murine c-myb upstream region, we looked by chromatin immunoprecipitation with microarray technology (ChIP-on-chip) for histone modifications implicating gene activation in normal cells. H3K4me3, H3K4me1, and H3K9/14ac were enriched at Mml1 and/or Mml2 in the myeloblastic cell line M1, which expresses c-myb. The enrichment of all of these histone marks decreased with differentiation-induced downregulation of the gene in M1 cells but increased and spread in tumor cells containing integrated provirus. Importantly, using chromosome conformation capture (3C)-quantitative PCR assays, interactions between the 5′ region, including the promoter and all Mml sites (Mml1, Mml2, and Mml3), were detected due to DNA looping in M1 cells and tumor cells with provirus in Mml1, Mml2, or Mml3. Therefore, our study provides a new mechanism of retrovirus insertional mutagenesis whereby spatial chromatin organization allows distally located provirus, with its own enhancer elements, to access the 5′ regulatory region of the gene.
INTRODUCTION
C-Myb is a transcription factor that regulates hematopoiesis by controlling essential cellular processes, such as proliferation, differentiation, and apoptosis (7, 21). A role for c-MYB in human T-cell leukemia (T-ALL) has been reported recently, where the gene was found to be involved in translocation and duplication (3, 18). Altered c-MYB expression also plays a role in human colon and breast carcinoma (6, 13, 37). These reports followed years of studies in avian and murine models which demonstrated that overexpression or mutations in c-myb can release its oncogenic potential, especially in myeloid cells (30, 40).
c-myb was a primary target of insertional mutagenesis when Moloney murine leukemia virus (M-MuLV) was inoculated intravenously into adult BALB/c mice following intraperitoneal injection of pristane to induce an inflammatory response (26, 33, 42). In this animal model, 100% of the tumors were shown to have undergone c-myb DNA rearrangements due to virus integration. Promoter insertion combined with the formation of gag-myb RNA fusions was the most common mechanism of activation. Therefore, an important feature was the ability of the enhancer/promoter region in the long terminal repeat (LTR) of the provirus to activate transcription from the c-myb locus, bypassing the normal promoter.
In a similar model where pristane-treated DBA/2 mice were injected with amphotropic 4070 virus (41), two-thirds had integrations directly into c-myb, and additional proviral integration sites were found far upstream of c-myb. These upstream integration sites were mapped to three regions, named Mml1, Mml2, and Mml3, located approximately 25, 50, and 70 kb upstream of the c-myb promoter, respectively (9, 16). Interestingly, many of these leukemia viruses have a single clonal provirus which was found in one of these regions.
The mechanism by which these proviral insertions in Mml1, Mml2, or Mml3 contributes to leukemia development has been unknown. After the failure to find additional gene-coding regions within this 100-kb region, we hypothesized that the upstream region of c-myb contains regulatory elements that control expression of c-myb at a distance, and perhaps these elements are utilized by the provirus to somehow activate gene expression. We have addressed this model by analyzing histone modifications within this region to identify sites that are potentially involved in positive gene regulation. Indeed, enrichments of histone methylation and acetylation marks, which identify enhancers, were found near proviruses and were associated with c-myb expression. Further analysis of the spatial organization of the same 100-kb region, using a quantitative chromosome conformation capture PCR (3C-qPCR) assay, revealed looping structures that, in tumors, allow integrated proviral LTRs access to the 5′ control region of c-myb. This provides the first evidence for a long-range mechanism of retrovirus gene activation through a 3-dimensional chromosome structure.
MATERIALS AND METHODS
Cell lines.
The murine myeloid cell line M1 (20) was maintained in RPMI 1640 medium with 10% (vol/vol) heat-inactivated horse serum (Invitrogen). All tumor cell lines (16) established in vitro from granuloma and/or ascites were cultured in Dulbecco's modified Eagle medium with 10% (vol/vol) fetal bovine serum. For interleukin-6 (IL-6) treatment, M1 and tumor cells were seeded at a density of 1 × 105 cells/ml in medium containing IL-6. IL-6 stocks were prepared as described previously (32).
Quantitative real-time PCR analysis.
Total RNA was isolated using TRIzol reagent (Invitrogen). cDNA was produced from 1 μg of total RNA using a cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR was performed in triplicate with predesigned c-myb gene expression assays (Mm 00501741-m1; Applied Biosystems). Data were normalized to a mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control (Applied Biosystems). Relative quantitation was carried out by the comparative threshold cycle (CT) method. Statistical analysis was performed using GraphPad Prism 5 software. The Student t test was used on measurements of c-myb expression from M1 and tumor samples from 3 experimental replicates.
ChIP-on-chip analysis.
Chromatin immunoprecipitation with microarray technology (ChIP-on-chip) was conducted as previously described (28). Cells were fixed in 0.8% formaldehyde for 6 min at room temperature. After lysis, samples were sonicated to a size range of 200 to 1,000 bp. Chromatin (150 to 200 μg) was immunoprecipitated with antibodies for H3K4me3 (ab8580; Abcam), H3K4me1 (ab8895; Abcam), H3K9/14Ac (06-599; Upstate), CTCF (ab70303; Abcam), H3K9me3 (ab8898; Abcam), H3K27me3 (17-622; Upstate), or rabbit IgG (15006; Sigma-Aldrich). A 10% aliquot was removed as an input fraction. ChIP DNA and input DNA were amplified using a WGA2 kit (Sigma-Aldrich). A total of 2.5 μg of amplified DNA was labeled with Cy3 (input) or Cy5 (IP) dUTP (PerkinElmer Life and Analytical Sciences) using the CGH labeling kit (Invitrogen).
Custom 8-by-15,000 tiling arrays (Agilent) contained probes spanning mouse chromosome 10 (chr.10) from bp 020600015 to 021199991 (genome browser-mm8; University of California–Santa Cruz). Probes were designed using eArray (Agilent) and a covered 600-kb region surrounding c-myb (∼40-bp spacing). A total of 3 μg of labeled ChIP and input DNA was cohybridized to the chip for 40 h at 65°C, washed, and scanned using an Agilent Scanner with Agilent Scan Control 7.0 software. Data were extracted with Feature Extraction 9.1 software and analyzed using ChIP Analytics 1.3 software (Agilent). Normalized and raw data files can be accessed at the GEO database with accession number GSE34770.
3C assay.
The 3C-qPCR protocol was performed as described previously (8), with minor modifications. Cross-linking was performed by incubating 1 × 107 cells in 10 ml of fresh medium supplemented with 1% formaldehyde for 10 min at room temperature. The reaction was quenched by addition of glycine to a final concentration of 0.125 M. Nuclei were harvested by lysis of the cells in ice-cold lysis buffer (10 mM Tris-HCl, pH 7.5; 10 mM NaCl; 0.2% NP-40; 1× complete protease inhibitor [11836145001; Roche]) at 4°C for 1 h. Nuclei were resuspended in buffer 2 (NEB) containing 0.3% SDS and incubated at 37°C for 1 h while being shaken. Triton X-100 was added to 2%, followed by incubation for 1 h at 37°C to sequester the SDS. The cross-linked DNA was digested overnight with 400 U HindIII. Digested DNA was diluted with ligation buffer (50 mM Tris-HCl, 10 mM MgCl2, 10 mM dithiothreitol [DTT], 1 mM ATP) to a low DNA concentration of 3 ng/μl. After addition of Triton X-100 to 1%, 1 h of incubation was performed at 37°C. DNA was ligated by using 100 U T4 DNA ligase in 7 ml 1× ligation buffer for 4 h at 16°C, followed by 30 min of incubation at room temperature. Proteinase K (300 μg final) was added and DNA was incubated overnight at 65°C to de-cross-link the samples. De-cross-linked DNA was incubated for 30 min at 37°C with RNase (300 μg final) and purified by phenol-chloroform extraction and ethanol precipitation. The purity assessment and loading adjustment were based on qPCR.
To prepare a qPCR control template in which all possible ligation products are present in equimolar amounts, a mouse K9 bacterial artificial chromosome (BAC) clone (9) was completely digested with HindIII and ligated at a high DNA concentration to reach a random ligation. The ligated products were diluted to the appropriate concentration to perform the standard curve for each test primer set by qPCR.
Quantitative PCR analysis of 3C DNA.
The TaqMan probe and bait primers were designed close to the HindIII restriction site of the c-myb promoter bait fragment. Test primers were designed close to restriction sites of each candidate interacting fragment. (Sequences of the test and bait primers and TaqMan probe are listed in Table 1.) Two hundred ng of 3C DNA and Universal PCR Master Mix (Applied Biosystems) were used for the TaqMan real-time PCR. Standard curves were performed for each run using serial dilutions of the control template prepared from the K9 BAC clone. Relative interactions were determined by the values corresponding to the quantification of the ligation product. Values were calculated using the parameters of the standard curve (b, intercept; a, slope) as 10(CT − b)/a. For normalization, values in different 3C samples were divided by the value of the ERCC3 internal cross-linking control (35).
Table 1.
Sequence of qPCR primers used for 3C-qPCR analysis of the mouse c-myb locusa
| HindIII fragment no. | Test primer sequence | Bait primer |
|---|---|---|
| −1 | TTGCGCAACTGTGCAAGG | B1 |
| 1 | TAATCATGAACCACAGGGCAGGCT | B2 |
| 2 | ACCCGAGAAATTCCACTCTGCCTT | B3 |
| 3 | GCACTTTGCCAGTGTCCC | B1 |
| 4 | TTGGGCTGTTTGAAGCCACCAT | B2 |
| 5 | CAATGGAATCAACCCGCAAGTGCT | B2 |
| 6 | AGCCTTTGACACAGACAAGGCA | B1 |
| 7 | TGCCCAGGAAGTTCTCACTGAA | B1 |
| 8 | CAGGCATGTCCCAATATGCCTAAG | B1 |
| 9 | GACAATTTGACATGAATTGCAAGCTTCT | B4 |
| 10 | AAGAACCAAGACGCCTCAGCAA | B1 |
| 11 | TCCGTTCTCAGCACAACCATGA | B1 |
| 12 | ACCTCTGTCTTCCCAACGCTCTG | B2 |
| 14 | ATCTTGCTGCCCTCAAGCAAAG | B2 |
| 15 | GCCCAGAAGATGATTCTGGAAAGC | B1 |
| 17 | AGTCATTGCCTTGGGCAGTTCT | B1 |
| 18 | ACTCCTCCATTGTTGGTGGGAT | B1 |
| 19 | AGACCAGAATCTTCTGCGGCAA | B1 |
| 20 | TGTCTCTCCTAGTTTGGGCCTT | B1 |
| 22 | TCACACAGTGAACTGAGGACCA | B1 |
| 23 | TCTGCCATCCTTACTTCTGCTC | B1 |
| 24 | GCTGTCTTTGTAGGTCACTTTCTCCAGC | B1 |
| 25 | TGGAAGAGCAGGCTATTGTGA | B1 |
| 26 | AGACAAATGGGACCTCATAAATTTGC | B1 |
| 28 | CACTGCCACTTATTCTTCTTTAAGCGTG | B1 |
| 31 | ACAGCAGTGCTTTCTGTGGAGA | B1 |
Four bait primers (B1, B2, B3, and B4) are all located within a 34-bp sequence located 100 to 134 bp downstream of the 5′ HindIII site of the c-myb promoter bait fragment. Their sequences are the following (5′ to 3′): B1, ATTATGGAGGCGAGAGAGGTGT; B2, TCATTATGGAGGCGAGAGAGGTGT; B3, ATTATGGAGGCGAGAGAGGTGTCA; and B4, TCATTCATTCATTATGGAGGCGAGAGAGG. For each ligation product, the bait primer giving the best amplification efficiency was used as indicated in the table. The ligation product-specific primers (so-called test primers) were designed downstream of the 5′ HindIII site of each restriction fragment (fragments 1 to 31). The sequence of the TaqMan probe used is 5′- 6-carboxyfluorescein [FAM]- AATCTTTGCAGCTGCCTGCCTGTCAGC-3′BGH. Internal interaction controls were performed using the following ERCC-3 primers as described before (35): forward primer (5′ to 3′), GCCCTCCCTGAAAATAAGGA; reverse primer (5′ to 3′), GACTTCTCACCTGGGCCTACA; ERCC-3 TaqMan probe, 5′- FAM-AAAGCTTGCACCCTGCTTTAGTGGCC-3′BGH.
Microarray data accession number.
Normalized and raw data files have been submitted to the GEO database under accession number GSE34770.
RESULTS
c-myb is expressed in tumor cell lines with provirus integrated in c-myb upstream regions.
c-Myb is an essential regulator of hematopoiesis, and its expression is largely restricted to progenitor cells and is downregulated as cells differentiate (7). As shown in Fig. 1A, expression of c-myb RNA is significantly decreased in myeloblastic M1 cells induced by IL-6 to differentiate into monocytes/macrophages during a period of 5 days. Furthermore, c-myb is expressed in tumor cell lines with integrated provirus in the Mml1, Mml2, or Mml3 region at levels similar to or higher than those of undifferentiated M1 cells (Fig. 1A). These tumor cells have either a very limited response to IL-6 compared to M1 cells or no response (Fig. 1B). This indicates that there is a positive correlation between the presence of provirus upstream of c-myb and c-myb expression.
Fig 1.

Expression of c-myb RNA in tumor cell lines with integrated provirus. Expression levels were determined by quantitative reverse transcription PCR. (A) Total RNA samples were prepared from M1 cells, M1 cells treated with IL-6 for 1 or 5 days, 6 tumor cell lines with virus integrated in the Mml1, Mml2, or Mml3 region, and NIH 3T3 cells. Data are normalized to GAPDH expression. Error bars represent standard deviations (SD) (n = 3). An asterisk represents significant difference of expression compared to M1 cells (P < 0.05). **, P < 0.01. (B) Response of tumor cell lines to IL-6. Total RNA was prepared from the indicated tumor cell lines and M1 cells after treatment with IL-6 for 0, 3, 6, and 24 h. Data are normalized to initial c-myb expression in individual cell lines. Error bars represent SD (n = 3).
Histone H3K4 trimethylation and histone acetylation at both the c-myb gene promoter and Mml1 are associated with active c-myb transcription.
Histone modifications are implicated in influencing gene expression and genome function. To provide evidence for upstream transcriptional regulatory regions that might be involved in positively influencing c-myb expression in normal cells without proviral integration, we analyzed for enrichments of H3K4 trimethylation (H3K4me3) and acetylation of H3K9 and H3K14 (H3K9/14ac) in M1 cells. These histone modifications have been found by others to be frequently present at gene promoters at either a poised or transcriptional active state (1, 15, 17). Distribution of these marks in M1 cells was determined by ChIP-on-chip analysis using a tiling microarray representing a 600-kb region surrounding the c-myb gene on mouse chr.10 (∼40-bp spacing) (Fig. 2A). Consistent with active c-myb in these cells, both H3K4me3 and H3K9/14ac were found at its transcription start site. Interestingly, there was also strong enrichment of these marks in the Mml1 region, with 3 peaks between −25 and −40 kb that indicate the presence of regulatory elements (Fig. 2A and B). It should be noted that in previous studies, no transcripts were found in these regions (9). To examine the enhancer activity of sequences representing the three peaks, E1 (3.8k), E2 (1.8k), and E3 (1.9k) fragments were cloned upstream of the c-myb promoter controlling a firefly luciferase reporter gene (Fig. 3A and B). Luciferase assays show that sequences within one of the regions increased luciferase activity (Fig. 3C), indicating the presence of an enhancer element. The histone modifications that were present at both c-myb and Mml1 sites in M1 cells significantly decreased when c-myb was downregulated in conjunction with IL-6 treatment (Fig. 2B), supporting their role in c-myb transcription.
Fig 2.

Histone modifications present at the c-myb gene and Mml regions correlate with c-myb expression. (A) ChIP-on-chip data of histone modifications known to represent active transcription, H3K9/14ac and H3K4me3, in M1 cells. The schematic presentation shows an ∼600-kb region surrounding the c-myb proto-oncogene on mouse chr.10. Four known genes on the locus are shown on top by solid rectangles with arrows that indicate their transcriptional orientation. Locations of viral integrations sites Mml1, Mml2, and Mml3 are shown. The area within the box, the c-myb gene through Mml1, is expanded in panel B. (B) Detailed analysis of the H3K4me3 distribution at the c-myb gene and the Mml1 region in cell line M1, M1 treated with IL-6 for 5 days, and cell lines with proviral integrations at Mml1 (30C-18 and 30-3-14) and Mml3 (30-2-7). The location of Mml1 is indicated by a red vertical arrow. (C) ChIP-on-chip data of H3K4me1 in M1 cells and detailed analysis of the H3K4me1 distribution in the c-myb 5′ end and Mml regions of the indicated cell lines. Tumor cell lines with integrations in Mml1 are 30C-18, 30A2-2-6, and 30-3-14. A tumor cell line with a provirus in Mml3 is 30-2-7. The locations of Mml1, Mml2, and Mml3 are indicated by red vertical arrows. Boxes in B and C depict the presence of major differences in enrichment between cells lines. Normalized enrichment data (IP/input ratio) are plotted on a log 2 scale.
Fig 3.

Luciferase assays of potential enhancers in the Mml1 region. Three fragments (E1, E2, and E3) marked by H3K4me3 in the Mml1 region (A) were cloned separately upstream of the c-myb promoter controlling a luciferase reporter gene (B) and transfected into NIH 3T3 and 293T cell lines. The green horizontal arrow shows the transcription orientation of the gene. One of the regions (E1) increases the luciferase transcription by 2- to 4-fold compared to the promoter-only control (C). Data are normalized to cotransfected Renilla gene expression. Error bars represent SD (n = 3).
Further analysis showed that, in tumor cells with integrations at Mml1 (30C-18 and 30-3-14) and expressing c-myb, there was an increase in H3K4me3 at the c-myb promoter and at Mml1 compared to the level found in M1 cells (Fig. 2B). In fact, the regions of enrichment were expanded in the direction of the c-myb gene. In a tumor cell line (30-2-7) with a provirus at Mml3, we observed a similar broad distribution of the H3K4me3 mark at c-myb and Mml1. We hypothesize that the presence of viral enhancers in integrated proviruses is responsible for the increase in histone modification.
H3K4me1 in the c-myb upstream region is increased and altered in location due to integrated provirus.
It was reported that the distribution of the H3K4me1 mark can predict regulatory elements, such as enhancers (11). ChIP-on-chip data showed that this modification was strikingly abundant at both Mml1 and Mml2 in M1 cells, and it was decreased at both sites when M1 cells were differentiated with IL-6 treatment for 5 days (Fig. 2C). We also observed that, in tumors with insertions in Mml1 and Mml3, enrichment of H3K4me1 was present at all Mml regions. Compared to M1 cells, the presence of provirus at either Mml1 or Mml3 caused an expansion of this histone modification throughout the upstream region of c-myb.
Changes in H3K27me3 and H3K9me3, which are reported to be associated with the transcriptional repression states, were not found to differ largely between c-myb-expressing and nonexpressing cells (data not shown).
All three retrovirus integration regions interact with the 5′ end of c-myb in tumor cell lines through DNA looping.
The histone modification data above suggest the existence of regulatory elements in the distal upstream region of c-myb that correlate with c-myb expression levels. Interestingly, the presence of provirus in one region causes increases of the enhancer-associated histone marks at a distance, suggesting that there are interactions between distal loci and the c-myb promoter. Therefore, we decided to look at the spatial organization of the chromosome by applying a quantitative chromosome conformation capture assay (3C-qPCR) (5, 8). The 3C assay, developed by Dekker et al., provides a method to detect long-range chromatin interactions in vivo (5). A region of approximately 100 kb, upstream of c-myb and covering the Mml1, Mml2, and Mml3 regions, was examined. Interaction frequency was detected by quantitative real-time PCR with a TaqMan probe designed in the bait. To relate the spatial conformation of the c-myb locus to its transcriptional status, the 3C-qPCR assay was performed in M1 cells and differentiated M1 cells with downregulated c-myb (Fig. 4A). Prominent peaks of interactions were detected around −20, −25, −50, and −73 kb. Surprisingly, all 4 peaks were stable, in that they did not disappear with downregulation of c-myb in IL-6-treated M1 cells.
Fig 4.

Long-range interactions detected between the Mml regions and the 5′ c-myb region, including the promoter. Interactions between the 5′ c-myb region and regions up to −100 kb upstream were determined by 3C-qPCR. A HindIII fragment, including the c-myb promoter, was used as the bait. (A) The 3C-qPCR assay was performed on differentiated (M1 plus IL-6) and undifferentiated M1 cells, and the data show the cross-linking frequency between the upstream regions and the bait fragment. The locations of HindIII fragments are indicated below the graph. Mml1, Mml2, and Mml3 regions are marked by red arrows. Data are normalized to the ERCC3 internal cross-linking control (means and standard errors of the means [SEM]; n = 3). (B) Sequences of retrovirus integration sites in tumor cell lines. Virus-chromosomal DNA junction sequences were determined by a shotgun cloning method as described previously (34). Genomic DNA was digested with MseI and ligated with an adaptor (the sequence is available upon request). PCR was carried out using a viral LTR primer and an adaptor primer to amplify the junction sequences, followed by TA cloning and sequencing. Virus integration site sequences in tumor cell lines are shown as indicated. Red vertical arrows mark the virus integration sites. Black horizontal arrows indicate the orientation of retrovirus sequences inserted into the genome. A green solid rectangle with an arrow depicts the c-myb gene and its transcriptional orientation. (C) Long-range interactions detected between the 5′ c-myb region and Mml regions in tumor cells. 3C-qPCR assays were performed at the same time in M1 cells and tumor cell lines containing a provirus in one of the Mml regions. The upstream HindIII fragments examined in these experiments are indicated. Integration sites in tumor cell lines are marked by red arrows. The locations of integrated proviruses in cell lines 30C-18 (Mml1), 30-2-9 (Mml2), and 30-2-7 (Mml3) are depicted, and the genomic sequences at the virus-cell junctions are presented in panel B. The Mml3 location was previously determined by Southern blot analysis. Data are normalized to the ERCC3 internal cross-linking frequency control (means and SEM; n = 3). (D) 3C-qPCR assay of NIH 3T3 cells. Data are normalized to the ERCC3 control (means and SEM; n = 3).
Remarkably, each of the interaction peaks was located proximal to a virus integration region, either Mml1, Mml2, or Mml3 (Fig. 4A), suggesting that all of the proviruses upstream of c-myb come in close proximity to the c-myb promoter and affect c-myb transcription. In the Mml1 region, two interaction peaks were detected, one at the HindIII fragment 6 (−20 kb) and the other at fragment 9 (−26 kb). We have determined the proviral/chromosomal DNA junction sequences for proviruses within these two Mml1 peaks. These sequences, from tumors (30A2-2-6 and 30-3-14), are presented in Fig. 4B, and virus insertion sites are depicted by the three arrows in the Mml1 region in Fig. 4A. Many other proviruses were mapped previously to the region by Southern blot analysis (16). The other two strong interaction peaks, corresponding to the Mml2 and Mml3 regions, were located at −50 kb (fragment 18) and −73 kb (fragment 26). The interaction frequencies of these sites are more than 4-fold higher than those of the Mml1 region. Similar to the peaks in the Mml1 region, these peaks in M1 cells did not change upon a differentiation-induced decrease in c-myb expression.
The 3C data in M1 cells suggest that provirus in the upstream region comes close to the c-myb promoter through DNA loops and thereby affects the oncogene transcription in retrovirus-induced leukemia. To determine whether the provirus indeed comes in close proximity to the c-myb promoter in tumors with retrovirus integrations, we analyzed the spatial organization of the c-myb locus in tumor cells with proviruses in Mml1 (30C-18), Mml2 (30-2-9), and Mml3 (30-2-7). The data show that the promoter interaction regions overlap all three virus integration regions in these cells (Fig. 4C). Our data indicate that in the nuclei of the tumor cells, virus comes in close vicinity to the c-myb promoter through integration into preformed, stable loops.
The spatial organization of the c-myb locus was also examined in NIH 3T3 cells, because this represents another tissue type, one which does not express c-myb (Fig. 4D). Interestingly, cross-linking frequency data for the Mml1 region indicate the absence of interaction between the Mml1 region and the promoter in NIH 3T3 cells. However, cross-linking frequencies in the vicinities of Mml2 and Mml3 are similar to that in M1 cells. Interestingly, the cross-linking frequency between the third HindIII fragment (−11 kb) and the promoter was increased in NIH 3T3 cells compared to M1 cells, indicating a new interaction that might be related to gene silencing.
CTCF is recruited to the interaction regions.
The CTCF protein is a multivalent factor with widespread regulatory functions, and it has been implicated in mediating intrachromosomal contacts and looping (29). Here, we investigated the location of CTCF binding in the mouse genomic c-myb locus to see if its binding fit our model of looping at this locus as determined by 3C-qPCR. ChIP-on-chip was carried out using antibody to CTCF and the microarray described above (Fig. 5A). Three CTCF enrichment peaks were detected upstream of c-myb, located at −31, −56, and −70 kb from the promoter (Fig. 5B). These three CTCF binding sites overlap the provirus integration regions (indicated by red vertical arrows). The ChIP data showed that all CTCF binding sites overlap long-range promoter interaction regions (Fig. 5B), indicating that CTCF plays a role in loop formation.
Fig 5.

CTCF is recruited to the promoter interaction regions. (A) ChIP-on-chip experiment using antibody specific for CTCF and the microarray described in the legend to Fig. 2 were performed on the indicated cell lines. Red vertical arrows indicate Mml1, Mml2, and Mml3. (B) Detailed locations of CTCF binding sites and the chromosome structure of the c-myb locus in M1 cells. 3C-qPCR data of the c-myb locus in M1 cells (details are given in the legend to Fig. 4). Potential matrix attachment regions (MARs) were predicted within a 100-kb region upstream of c-myb by the online program MARWIZ (http://genomecluster.secs.oakland.edu/marwiz). In silico data show that potential MARs are located at the boundaries of the loops identified in the 3C assay (alignments are shown by dotted lines).
DISCUSSION
This study describes a new mechanism for gene activation by retroviruses that insert in intergenic regions. We show here that proviruses within 100 kb upstream of the c-myb gene in murine myeloid leukemias are inserted specifically at DNA sites that interact with the 5′ region, including the promoter of the gene, through looping (Fig. 6). Since a well-accepted mechanism of insertional mutagenesis is enhancer insertion, the mechanism by which the far-upstream proviruses activate c-myb may be a modification of this mechanism (22, 27). Since it was reported that differential expression of c-myb is regulated by a transcriptional arrest mechanism in the first intron (2, 31) and the bait used in the conformation capture assay here included the promoter and a small part of intron 1, we cannot rule out the possibility that the integrated proviruses in the Mml regions act through prevention of attenuation at the elongation block.
Fig 6.
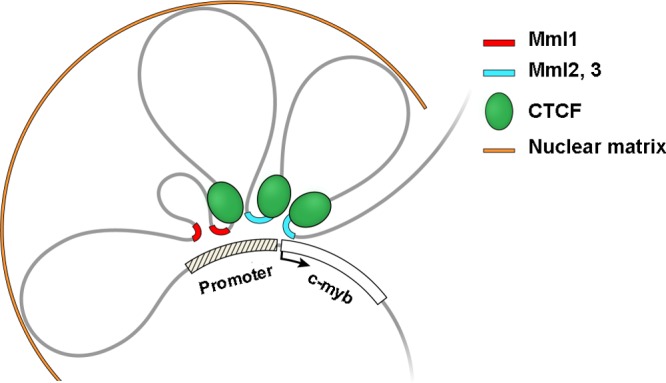
Model of the long-range interactions between retrovirus integration regions and the 5′ end of c-myb in tumor cells. Proviruses in Mml1 (red lines) or Mml2 and Mml3 (blue lines) come in close proximity to the 5′-end regulatory region of the gene by DNA looping. CTCF binding near the interaction regions is suggested to be involved in the looping formation. Potential MARs were found at the boundaries of the looping structure.
Sequences in the LTR enhancer elements presumably are responsible for activation of gene transcription in these tumors. We previously found that proviruses integrated in the Mml1 region were deleted. The total size of some of the proviruses in 4 cases was between 0.4 and 0.9 kb, and we know that the U3 LTR sequences were present (16). This indicates that in some cases, in the absence of structural genes, only an LTR remained in these proviruses. Although such a model could be predicted based upon recent studies that show that transcriptional activation in higher eukaryotes frequently involves the long-range action of regulatory elements (10, 24), this is the first time it has actually been shown that the provirus is in a position to interact with the promoter and/or control region in intron 1 through a 3-dimensional chromatin structure.
In this study, we applied a quantitative 3C assay that was recently developed by Hagege et al. (8) to include TaqMan real-time PCR technology, which provides accurate measurements of cross-linking frequencies. Our 3C data show that all three Mml integration regions are close to the promoter by loop formation in M1 cells (Fig. 4A) and in tumor cell lines with provirus in these regions (Fig. 4C). The Mml1 region contains 2 loops, giving a total of 4 loops, and provirus was found to be inserted in each of these loops. Interestingly, M1 cells that are induced to differentiate with IL-6 and do not express appreciable levels of c-myb still maintain the same chromatin looping structure observed in cells with active transcription. Therefore, this chromatin looping structure is insufficient by itself for c-myb expression and may require addition of transcription factors to result in expression. Examination of the chromatin conformation in a nonhematopoietic cell line, NIH 3T3, which does not express c-myb, revealed that differences in the looping structure at the c-myb locus exist in different cell types. Apparently, a novel interaction peak that is not found in the hematopoietic cells was present very close to the promoter at approximately −11 kb. The fact that looping structures vary between tissues is supported by a recent finding showing that murine erythroid cells have a different pattern of looping than we found here in myeloid cells (36).
The finding that CTCF binds in the vicinity of the looping structure is not surprising, in that it is known that CTCF contributes to looping associated with gene activation (12, 29). Although we discovered 4 dominant promoter-interacting loops, only 3 were associated with CTCF (Fig. 5B). The most proximal loop was not associated with this DNA binding protein. Interestingly, others have shown that enhancer and promoter sites are often bound by cohesin and mediator complexes in the absence of CTCF (14). Matrix attachment regions (MARs) are evolutionarily conserved genome sequences that anchor DNA to the nuclear matrix and are reported to act as boundary elements for chromatin functional domains (23). The MARWIZ online program was used to predict potential MARs within the region 100 kb upstream of c-myb in M1 cells. In silico data show that potential MARs are located at the boundaries of the loops identified in the 3C assay (Fig. 5B).
Integration at a site in the genome can be a consequence of both target site selection and clonal selection, and it would be reasonable to imagine that both play a role in the far-upstream region of c-myb. When murine leukemia virus (MLV) site selection is studied in vitro, in the absence of clonal selection, it has been shown that these viruses have preferences not only for the vicinity of the 5′ ends but also for insertion within a kilobase of DNase hypersensitivity sites (4, 19). This suggests that there is a preference for intergenic regions that are involved in active transcription.
Here, we mapped histone H3 modification sites in the c-myb upstream region to identify regulatory elements that might exist in normal cells. Using ChIP-on-chip, we found that Mml integration regions in myeloid cells contain active histone modifications, especially H3K4me1 and H3K4me3, which are hallmarks of enhancers (11). The enrichment of these marks was correlated with c-myb expression in the cell lines that we examined, indicating that c-myb is regulated from a distance in myeloblastic M1 cells. Interestingly, long-range regulation from upstream sequences of c-myb has been suggested by others who reported that a transgene integrated approximately 77 kb upstream of the c-myb disrupts sequences that regulate c-myb gene expression in megakaryocyte/erythrocyte lineage-restricted progenitor cells (25). In addition, studies on the human HBS1L-MYB intergenic interval associated with elevated fetal hemoglobin (HbF) levels suggest that the HBS1L-MYB intergenic region contains regulatory sequences that could be important in hematopoiesis by controlling MYB expression (38).
We did not observe a significant change in repression-associated histone modifications (H3K27me3 and H3K9me3) with downregulation of c-myb (data not shown). Perhaps these modifications are more generally involved in bivalent stem cell states and the repression of genes in early stages of development as stem cells differentiate and begin to form specialized cells (39).
This study provides a new mechanism of retroviral insertional mutagenesis whereby looping allows an integrated provirus that is distal to a gene access to the gene's promoter and 5′ control region. It is hypothesized that the enhancers of the provirus can act at the promoter in a manner similar to that when the virus is integrated directly in the 5′ end of the gene and provide enhancer activity.
ACKNOWLEDGMENTS
We thank Richard Koller in our laboratory for assistance in tumor cell culturing and Sam John and Ofir Hakim, Laboratory of Receptor Biology and Gene Expression, for their much valued and excellent advice.
The work was supported by the Intramural Program at the National Cancer Institute, Center for Cancer Research. J.M. was partially supported by grant 2/0135/09 from the Slovak Grant Agency VEGA.
Footnotes
Published ahead of print 18 July 2012
REFERENCES
- 1. Barski A, et al. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129:823–837 [DOI] [PubMed] [Google Scholar]
- 2. Bender TP, Thompson CB, Kuehl WM. 1987. Differential expression of c-myb mRNA in murine B lymphomas by a block to transcription elongation. Science 237:1473–1476 [DOI] [PubMed] [Google Scholar]
- 3. Clappier E, et al. 2007. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood 110:1251–1261 [DOI] [PubMed] [Google Scholar]
- 4. Daniel R, Smith JA. 2008. Integration site selection by retroviral vectors: molecular mechanism and clinical consequences. Hum. Gene Ther. 19:557–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dekker J, Rippe K, Dekker M, Kleckner N. 2002. Capturing chromosome conformation. Science 295:1306–1311 [DOI] [PubMed] [Google Scholar]
- 6. Drabsch Y, et al. 2007. Mechanism of and requirement for estrogen-regulated MYB expression in estrogen-receptor-positive breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 104:13762–13767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Greig KT, Carotta S, Nutt SL. 2008. Critical roles for c-Myb in hematopoietic progenitor cells. Semin. Immunol. 20:247–256 [DOI] [PubMed] [Google Scholar]
- 8. Hagege H, et al. 2007. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2:1722–1733 [DOI] [PubMed] [Google Scholar]
- 9. Haviernik P, et al. 2002. Linkage on chromosome 10 of several murine retroviral integration loci associated with leukaemia. J. Gen. Virol. 83:819–827 [DOI] [PubMed] [Google Scholar]
- 10. Heintzman ND, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459:108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heintzman ND, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39:311–318 [DOI] [PubMed] [Google Scholar]
- 12. Hou CH, Zhao H, Tanimoto K, Dean A. 2008. CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc. Natl. Acad. Sci. U. S. A. 105:20398–20403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hugo H, et al. 2006. Mutations in the MYB intron I regulatory sequence increase transcription in colon cancers. Genes Chromosomes Cancer 45:1143–1154 [DOI] [PubMed] [Google Scholar]
- 14. Kagey MH, et al. 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467:430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koch CM, et al. 2007. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 17:691–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koller R, et al. 1996. Mml1, a new common integration site in murine leukemia virus-induced promonocytic leukemias maps to mouse chromosome 10. Virology 224:224–234 [DOI] [PubMed] [Google Scholar]
- 17. Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705 [DOI] [PubMed] [Google Scholar]
- 18. Lahortiga I, et al. 2007. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat. Genet. 39:593–595 [DOI] [PubMed] [Google Scholar]
- 19. Lewinski MK, et al. 2006. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2:611–622 doi:10.1371/journal.ppat.0020060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liebermann DA, Hoffman-Liebermann B. 1989. Proto-oncogene expression and dissection of the myeloid growth to differentiation developmental cascade. Oncogene 4:583–592 [PubMed] [Google Scholar]
- 21. Lieu YK, Reddy EP. 2009. Conditional c-myb knockout in adult hematopoietic stem cells leads to loss of self-renewal due to impaired proliferation and accelerated differentiation. Proc. Natl. Acad. Sci. U. S. A. 106:21689–21694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maeda N, Fan H, Yoshikai Y. 2008. Oncogenesis by retroviruses: old and new paradigms. Rev. Med. Virol. 18:387–405 [DOI] [PubMed] [Google Scholar]
- 23. Mirkovitch J, Mirault ME, Laemmli UK. 1984. Organization of the higher-order chromatin loop: specific DNA attachment sites on nuclear scaffold. Cell 39:223–232 [DOI] [PubMed] [Google Scholar]
- 24. Morse RH. 2010. Epigenetic marks identify functional elements. Nat. Genet. 42:282–284 [DOI] [PubMed] [Google Scholar]
- 25. Mukai HY, et al. 2006. Transgene insertion in proximity to the c-myb gene disrupts erythroid-megakaryocytic lineage bifurcation. Mol. Cell. Biol. 26:7953–7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nason-Burchenal K, Wolff L. 1993. Activation of c-myb is an early bone-marrow event in a murine model for acute promonocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 90:1619–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neel BG, Hayward WS, Robinson HL, Fang J, Astrin SM. 1981. Avian leukosis virus-induced tumors have common proviral integration sites and synthesize discrete new RNAs: oncogenesis by promoter insertion. Cell 23:323–334 [DOI] [PubMed] [Google Scholar]
- 28. Paul TA, Bies J, Small D, Wolff L. 2010. Signatures of polycomb repression and reduced H3K4 trimethylation are associated with p15INK4b DNA methylation in AML. Blood 115:3098–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Phillips JE, Corces VG. 2009. CTCF: master weaver of the genome. Cell 137:1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramsay RG, Gonda TJ. 2008. MYB function in normal and cancer cells. Nat. Rev. Cancer 8:523–534 [DOI] [PubMed] [Google Scholar]
- 31. Reddy CD, Reddy EP. 1989. Differential binding of nuclear factors to the intron 1 sequences containing the transcriptional pause site correlates with c-myb expression. Proc. Natl. Acad. Sci. U. S. A. 86:7326–7330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmidt M, Nazarov V, Stevens L, Watson R, Wolff L. 2000. Regulation of the resident chromosomal copy of c-myc by c-Myb is involved in myeloid leukemogenesis. Mol. Cell. Biol. 20:1970–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen-Ong GL, Wolff L. 1987. Moloney murine leukemia virus-induced myeloid tumors in adult BALB/c mice: requirement of c-myb activation but lack of v-abl involvement. J. Virol. 61:3721–3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Slape C, et al. 2007. Retroviral insertional mutagenesis identifies genes that collaborate with NUP98-HOXD13 during leukemic transformation. Cancer Res. 67:5148–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Splinter E, et al. 2006. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 20:2349–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stadhouders R, et al. 2011. Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. EMBO J. 31:986–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thompson MA, Flegg R, Westin EH, Ramsay RG. 1997. Microsatellite deletions in the c-myb transcriptional attenuator region associated with over-expression in colon tumour cell lines. Oncogene 14:1715–1723 [DOI] [PubMed] [Google Scholar]
- 38. Wahlberg K, et al. 2009. The HBS1L-MYB intergenic interval associated with elevated HbF levels shows characteristics of a distal regulatory region in erythroid cells. Blood 114:1254–1262 [DOI] [PubMed] [Google Scholar]
- 39. Wang Z, et al. 2009. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138:1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wolff L. 1996. Myb-induced transformation. Crit. Rev. Oncogenesis 7:245–260 [DOI] [PubMed] [Google Scholar]
- 41. Wolff L, Koller R, Davidson W. 1991. Acute myeloid leukemia induction by amphotropic murine retrovirus (4070A): clonal integrations involve c-myb in some but not all leukemias. J. Virol. 65:3607–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wolff L, Mushinski JF, Shen-Ong GL, Morse HC., III 1988. A chronic inflammatory response. Its role in supporting the development of c-myb and c-myc related promonocytic and monocytic tumors in BALB/c mice. J. Immunol. 141:681–689 [PubMed] [Google Scholar]