

Abstract
Ac34 and its homologs are highly conserved in all sequenced alphabaculoviruses. In this paper, we show that ac34 transcripts were detected from 6 to 48 h postinfection (p.i.) in Autographa californica nucleopolyhedrovirus (AcMNPV)-infected Sf9 cells. Ac34 localized to both the cytoplasm and the nuclei of infected cells but was not a viral structural protein. To determine the function of ac34 in the viral life cycle, an ac34 knockout AcMNPV (vAc34KO) was constructed. Compared with wild-type and repair viruses, vAc34KO exhibited an approximately 100-fold reduction in infectious virus production. Further investigations showed that the ac34 deletion did not affect the replication of viral DNA, polyhedron formation, or nucleocapsid assembly but delayed the expression of late genes, such as vp39, 38k, and p6.9. Bioassays revealed that vAc34KO was unable to establish a fatal infection in Trichoplusia ni larvae via per os inoculation. Few infectious progeny viruses were detected in the hemolymph of the infected larvae, indicating that the replication of vAc34KO was attenuated. These results suggest that Ac34 is an activator protein that promotes late gene expression and is essential for the pathogenicity of AcMNPV.
INTRODUCTION
Members of the family Baculoviridae have double-stranded circular supercoiled DNA genomes that vary in size from approximately 80 to 180 kb (44). Typical alphabaculoviruses have a biphasic infection process and produce two forms of virions that are genotypically identical but phenotypically distinct. Budded viruses (BVs) are required for the spread of infection from cell to cell, whereas occlusion-derived viruses (ODVs), which are embedded within occlusion bodies (OBs), are responsible for horizontal transmission between insect hosts via oral infection (61).
Gene expression of baculoviruses is mainly regulated at the transcriptional level and is typically categorized into three classes: early, late, and very late (17). Early promoters of baculovirus genes are recognized and regulated by the host RNA polymerase II (17), while the late promoters are recognized by a virus-encoded RNA polymerase that is α-amanitin insensitive (40, 44). In general, the expression of early genes is required for viral DNA replication and/or late gene expression; in contrast, late genes encode viral structural proteins.
Based on phylogenetic evidence, baculoviruses are divided into four genera: Alphabaculovirus (lepidopteran nucleopolyhedroviruses [NPVs]), Betabaculovirus (lepidopteran granuloviruses [GVs]), Gammabaculovirus (hymenopteran NPVs), and Deltabaculovirus (dipteran NPVs) (25). Two lepidopteran baculovirus genera, the lepidopteran NPVs and GVs, show major differences in the morphologies of their occlusion bodies. GVs are normally packaged as single nucleocapsids per envelope, and NPVs are packaged either as single nucleocapsids per envelope or multiple nucleocapsids per envelope. Dozens of virions are occluded in a polyhedron, while only a single virion is occluded in a granulum (15). Polyhedra are approximately 800 to 2,000 nm in diameter, whereas granules are ovoid and are typically 500 nm long and 200 nm wide (1, 49). In addition, the cytopathology of the lepidopteran NPVs also differs from that of the GVs (15). Cells infected with NPVs appear to clump together (20, 66), and their nuclear membranes remain intact until cell lysis and OB release (15). However, in GV-infected cells, the nuclear membrane disintegrates completely, and the cytoplasm and nucleoplasm become intermixed prior to the assembly of ODVs (18). After nuclear lysis, the infected cells dissociate from each other (15). Lepidopteran NPV-specific genes could be responsible for the biological differences between these two baculovirus genera. The determination of the relationship between the distinct phenotypes and gene contents of GVs and NPVs requires further characterization of genus-specific genes.
All sequenced lepidopteran NPVs share 5 specific genes: ac9 (pp78/83), ac24 (protein kinase-interacting protein [pkip]), ac34, ac104 (vp80), and ac131 (pp34) (44). Four of these genes have been characterized. A temperature-sensitive pkip mutant exhibits a defect in very late transcription and a delay in the shutoff of host protein synthesis (37). PKIP interacts with and is required for the essential function of a virus-encoded protein kinase in viral very late transcription (12). The phosphoprotein PP34 is involved in the morphogenesis of the polyhedron envelope and the part of the OB carbohydrate envelope called the calyx (29, 60). PP78/PP83 and VP80 localize to one end of nucleocapsids and are associated with both BVs and ODVs (19, 36, 45). These proteins interact with F-actin and are required for the transportation of nucleocapsids (19, 36, 39, 41). The only lepidopteran NPV-specific gene of unknown function is ac34. In a microarray assay, ac34 was grouped in the ac31-to-ac35 cluster of genes with synchronized expression patterns. Its transcription began in the early phase, and maximum transcription levels were observed in the late phase of infection (26).
In this study, an ac34 knockout Autographa californica nucleopolyhedrovirus (AcMNPV) mutant (vAc34KO) was generated to investigate the role of ac34 in AcMNPV infection in vitro and in vivo. We found that ac34 was not essential for viral replication, and its deletion had no negative effect on viral DNA replication. However, the replication of vAc34KO was impaired in Spodoptera frugiperda (Sf9) cells; the virus formed only small plaques on a cell monolayer, and BV production was reduced approximately 100-fold. The expression of late genes was delayed in vAc34KO-infected cells. In vivo assays revealed that the deletion mutant was unable to establish fatal infection in Trichoplusia ni larvae.
MATERIALS AND METHODS
Viruses and cells.
Sf9 cells were maintained at 27°C in TNM-FH medium (Invitrogen) supplemented with 10% fetal bovine serum, penicillin (100 μg/ml), and streptomycin (30 μg/ml). bMON14272 (Invitrogen), which contained an AcMNPV genome (34), was used to generate recombinant viruses. The BV titer was determined by 50% tissue culture infective dose (TCID50) assays in Sf9 cells.
Reverse transcription (RT)-PCR and rapid amplification of cDNA 5′ end (5′ RACE) analyses.
Sf9 cells (1.0 × 106) were infected with AcMNPV at a multiplicity of infection (MOI) of 5 TCID50/cell and harvested at the indicated time points. Total RNA was isolated using the RNeasy Protect Mini Kit (Qiagen) according to the manufacturer's protocol. cDNA synthesis was performed using the iScript cDNA Synthesis Kit (Bio-Rad). The primers 3451 (5′-GACTTTGTTACACGCACGCCGACTG-3′) and 3431 (5′-TTACTCAAAGTCCATCAATTCGTAC-3′) were used to detect the ac34 transcripts.
5′ RACE was carried out to map the transcription start site of ac34. Total RNA was isolated from AcMNPV-infected cells at 6 and 24 h postinfection (p.i.). The 5′ RACE reactions were conducted according to the manufacturer's protocol using a FirstChoice RLM-RACE Kit (Ambion). Two ac34-specific primer pairs, 34sp1 (5′-TTGCAGCTTAGATTCCGCGTTTGAT-3′)/5′ outer primer (5′-GCTGATGGCGATGAATGAACACTG-3′) and 34sp2 (5′-CGTCGTCAGCCTCTATAATCTCGGG-3′)/5′ inner primer (5′-CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG-3′), were used to amplify the 5′ end of ac34-specific transcripts. The PCR products were cloned into a pMD18-T vector (TaKaRa) and sequenced.
Construction of recombinant viruses.
A 501-bp fragment flanking the 5′ end of ac34 (AcMNPV, nucleotides [nt] 28693 to 29193) and a 508-bp fragment flanking the 3′ end of ac34 (AcMNPV, nt 27940 to 28447) were amplified from the genome of AcMNPV. The primer pairs 34US1 (5′-AAGCTTAATACCCTCCTCGTAATCG-3′ [HindIII site underlined])/34US2 (5′-CTGCAGTATAATCTCGGGCAACAGC-3′ [PstI site underlined]) and 34DS1 (5′-GGATCCTTACTGTCTGTCGTCGTGTAATT-3′ [BamHI site underlined])/34DS2 (5′-GGTACCCTTTCGATGCTCATCCTTGT-3′ [KpnI site underlined]) were used. The 5′ and 3′ PCR fragments were digested with PstI/HindIII and KpnI/BamHI, respectively, and inserted into pUC18-Cm (64) to generate the transfer vector pac34UCD, in which a chloramphenicol resistance (Cm) gene was flanked by the 5′ and 3′ ends of ac34. The replacement of the 245-bp ac34 (AcMNPV, nt 28448 to 28692) gene with Cm via RecE/RecT (ET) recombination was performed as described previously (64) to generate the ac34 knockout recombinant bacmid (bAc34KO).
To facilitate the examination of viral infection, the polyhedrin (polh) and enhanced green fluorescence protein (egfp) genes were inserted into the polh locus of bAc34KO by a site-specific transposon (64) to generate vAcac34KO-PH-GFP (vAc34KO). A wild-type control virus, vAcPH-GFP (vAcWT), was generated by the insertion of polh and egfp into bMON14272.
To generate an ac34 repair virus, a donor plasmid (pFB-ac34HA-PG) was constructed. A simian vacuolating virus 40 (SV40) polyadenylation signal was amplified from pFastBac1 (Invitrogen) and cloned into the BamHI and XbaI sites of pUC18 to generate pUC18-SV40. The primers SV40-F (5′-GGATCCGATCATAATCAGCCATACCACATTTGTAGAG-3′ [BamHI site underlined]) and SV40-R (5′-TCTAGAGATCCAGACATGATAAGATACATTGATGAG-3′ [XbaI site underlined]) were used. The ac34 promoter sequence and ac34 open reading frame (ORF) containing a hemagglutinin (HA) sequence in the N-terminal region were amplified and inserted into pUC18-SV40 to generate pUC34HA-SV40. The primer pairs 34p5 (5′-GTCGACTGGTTAGCGATAATACACAAGA-3′ [SacI site underlined])/34p3 (5′-GGTACCTTATAAGTAATAGTGTAAA-3′ [KpnI site underlined]) and 34NHA5 (5′-GGTACCATGTACCCCTACGACGTGCCCGACTACGCCACAACGGTTGCTGTGAATGCGC-3′ [KpnI site underlined])/343 (5′-GGATCCTTACTCAAAGTCCATCAATTCGTAC-3′ [BamHI site underlined]), were used. pUC34HA-SV40 was digested with SacI/XbaI and inserted into pFB1-PH-GFP (64) to generate the final plasmid pFB1-ac34HA-PG. pFB1-ac34HA-PG was then transformed into bAc34KO to generate the repair bacmid vAcHA:Ac34-PH-GPF (vHA:Ac34).
The replacement of ac34 with the Cm gene and the transposon-mediated insertion of genes into the polh locus were confirmed with PCR analysis. Bacmid DNA was extracted and purified using a Qiagen Large-Construct Kit and quantified with optical density.
Viral growth curve and plaque assays.
Sf9 cells (1 × 106) were transfected with 1.0 μg of bacmid DNA or infected with BVs at an MOI of 1 TCID50/cell. Supernatants were harvested at various times to determine titers. Plaque assays were carried out according to the method of Wu et al. (64). A total of 1.8 × 106 Sf9 cells in a monolayer were transfected with 0.2 μg of bacmid DNA. At 120 h posttransfection (p.t.), plaques were photographed and measured.
qPCR.
Quantitative PCR (qPCR) was performed to determine viral DNA replication, as previously described (54) with modifications. Sf9 cells (1 × 106) were transfected with 0.5 μg of vAc34KO or vAcWT bacmid DNA and harvested at various time points. Total DNA was isolated from each sample using a Universal Genomic DNA Extraction Kit (TaKaRa) and resuspended in 100 μl of sterile water. Equal amounts of total DNA from each time point were digested with 20 U of DpnI (New England BioLabs) in a 40-μl reaction volume at 37°C overnight. A 5-μl aliquot of the digested DNA was mixed with Super Mix (Invitrogen) and the PCR primers in a 20-μl reaction volume. The qPCR was performed under the following conditions using an iQ5 instrument (Bio-Rad): 95°C for 10 s, followed by 40 cycles of 95°C for 15 s and 60°C for 35 s.
Transmission electron microscopy (TEM).
Sf9 cells (1.0 × 106) were transfected with 1.0 μg of vAc34KO or vAcWT bacmid DNA. At 72 h p.t., the cells were rinsed in phosphate-buffered saline (PBS) and centrifuged at 5,000 rpm for 5 min. The cells were then fixed, dehydrated, embedded, sectioned, and stained as described previously (31). Images were collected using a JEOL JEM-1400 transmission electron microscope.
Immunofluorescence.
Immunofluorescence assays were performed as described previously (67). Sf9 cells were infected with vHA:Ac34 at an MOI of 5 TCID50/cell, probed with mouse monoclonal anti-HA antibodies (1:400; Abmart), and subsequently incubated with Alexa 555-conjugated goat anti-mouse secondary antibody (1:1,000; Invitrogen). The cell nuclei were stained with Hoechst 33342 (Sigma) before observation. The cells were observed and imaged using a Leica TCS SP5 laser scanning confocal microscope.
Western blot analysis.
Western blotting was performed to analyze the expression and localization of Ac34 in infected cells. Sf9 cells were infected with vHA:Ac34 at an MOI of 5 TCID50/cell. At various time points, the cells were harvested and fractionated into cytoplasmic and nuclear fractions using a Nuclear/Cytosol Fractionation Kit (BioVision). BVs and ODVs were purified from vHA:Ac34-infected Sf9 cells as previously described (7).
To evaluate the effects of ac34 on viral protein expression, Sf9 cells were infected with vAc34KO or vAcWT at an MOI of 1 TCID50/cell. The cells were collected, lysed in RIPA buffer (Thermo Scientific) for 30 min on ice, and centrifuged for 10 min at 14,000 × g at 4°C. The supernatants were collected and subjected to immunoblotting.
Protein samples from the nuclear/cytoplasmic fractions, purified virions, or cell lysates were mixed with 4× NuPAGE LDS sample buffer (Invitrogen) and incubated at 72°C for 10 min. Samples were separated with 12% NuPAGE Novex Bis-Tris Gels with MOPS (morpholinepropanesulfonic acid) SDS running buffer and transferred onto nitrocellulose membranes (Schleicher and Schuell) according to the NuPAGE electrophoresis system protocol (Invitrogen). Western blotting was performed as described previously (67). The primary antibodies used were the following: (i) mouse monoclonal anti-HA antibody (1:4,000), (ii) mouse monoclonal anti-IE1 antibody (1:4,000), (iii) mouse monoclonal AcMNPV GP64 antibody (1:4,000; eBioscience), (iv) rabbit polyclonal AcMNPV VP39 antibody (1:1,000), (v) rabbit polyclonal AcMNPV P6.9 antibody (1:2,000), (vi) rabbit polyclonal AcMNPV 38K antibody (1:1,000), and (vii) mouse monoclonal actin antibody (1:4,000; Abmart). The anti-rabbit horseradish peroxidase (HRP) antibody (1:5,000; GE) or goat anti-mouse HRP secondary antibodies (1:4,000; Amersham Biosciences) were used. The signals were detected using the ECL System (Amersham Biosciences) according to the manufacturer's instructions.
qRT-PCR.
Sf9 cells were infected with vAc34KO or vAcWT at an MOI of 1 TCID50/cell and harvested at various time points. Total RNA isolation and cDNA synthesis were performed as described above. The RNA extracted from uninfected cells was used to determine the background level of expression of each gene. qPCR was performed with the following primer pairs: qie1F (5′-TTGTGATAAACAACCCAACGA-3′) and qie1R (5′-GTTAACGAGTTGACGCTTGC-3′) for ie1, qgp64F (5′-ATCAAACGCTCGTCCACC-3′) and qgp64R (5′-GGGAGACACTGCCACCAAA-3′) for gp64, q38KF (5′-GCCATACGACCACAAGACT-3′) and q38kR (5′-CATAACCGAAGAGGAGCAA-3′) for 38k, qp6.9F (5′-GGCGACCTGTCGATGAA-3′) and qp6.9R (5′-CGCAGAAGCTCGGGTTA-3′) for p6.9, and qvp39F (5′-TTGCGCAACGACTTTATACC-3′) and qvp39R (5′-TAGACGGCTATTCCTCCACC-3′) for vp39. The ORFs of the above-mentioned genes were PCR amplified and cloned into the pMD18-T vector. The number of plasmid copies was calculated based on the DNA concentration. Standard curves were generated using serial dilutions of each plasmid.
Bioassays.
OBs were obtained from vAcWT- or vAc34KO-infected Sf9 cells. Bioassays were conducted with 3rd-instar T. ni larvae. Newly molted larvae were starved for 6 h and administered doses of 100, 1,000, or 10,000 OBs/larva delivered in the diet as described previously (22). Infected larvae were reared individually and monitored daily until all of the larvae had either pupated or died.
To further investigate viral replication in the hemolymph, 4th-instar larvae were administered 20,000 OBs/larva or hemocoelically injected with 250 TCID50/larva. At 48 and 72 h p.i., hemolymph was collected to determine the viral titer.
RESULTS
Sequence analysis of Ac34 and its homologs.
Ac34 and its homologs are highly conserved in all sequenced alphabaculoviruses. Amino acid alignments show that the C-terminal region is more highly conserved than the N-terminal region (Fig. 1A). A conserved-domain search of the CDD database (35) revealed that Ac34 homologs constitute a DUF1247 superfamily with unknown function. HHpred (50) analysis showed that Trp127-Ala195 of Ac34 is similar to Trp55-Lys131 of S-phase kinase-associated protein 1 (SKP1) (Fig. 1B). SKP1 is a component of the SKP1–Cullin–F-box (SCF) complex and serves as an adapter that links the F-box and Cullin proteins (2, 48). The SCF complex targets G1 cyclins and cyclin-dependent kinase inhibitors for ubiquitylation to regulate G1/S transition (2, 16, 48). Some putative posttranslational modification sites, including three N-linked glycosylation sites, four casein kinase II phosphorylation sites, and two protein kinase C phosphorylation sites, are present in the Ac34 homologs.
Fig 1.

Sequence analysis of Ac34 and its homologs. (A) Multiple-sequence alignment of 10 selected Ac34 homologs. (B) Alignment of Ac34 (Trp127-Ala195) and Homo sapiens SKP1 (Trp55-Lys131) (GenBank accession no. 1FS1_B). The alignment was performed with the AlignX module of Vector NTI Advance 11 and edited with GeneDoc software. Black, dark gray, and light gray indicate 100%, 80%, and 60% conservation, respectively.
Transcription of ac34 is regulated by both early and late promoters.
The temporal transcription pattern of ac34 during AcMNPV infection of Sf9 cells was investigated with RT-PCR. ac34 transcripts were detected from 6 to 48 h p.i. (Fig. 2A). This transcription pattern was similar to that of gp64 (data not shown). To map the transcription start site of ac34, 5′ RACE was performed. Specific products were amplified from RNA isolated from AcMNPV-infected Sf9 cells at 6 and 24 h p.i. Sequencing showed that ac34 transcription began at the second T of the atypical early promoter motif TTCTG at 6 h p.i. and at the second A of the typical late promoter motif ATAAG at 24 h p.i. (Fig. 2B), indicating that ac34 is both an early and a late gene.
Fig 2.
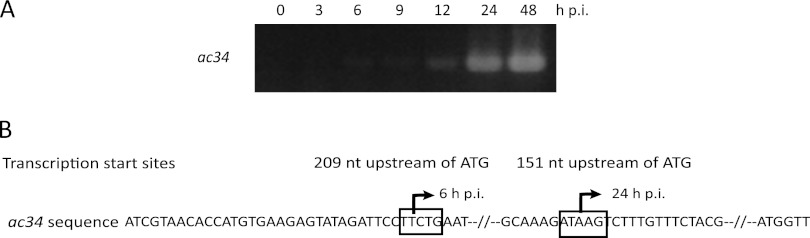
Transcription of ac34 in Sf9 cells infected with AcMNPV. (A) RT-PCR analysis of ac34 transcripts. Total RNA was isolated from AcMNPV-infected Sf9 cells at the time points indicated above the lanes. (B) 5′ RACE analysis of the ac34 transcriptional start sites. The early promoter motif TTCTG and the late promoter motif ATAAG are boxed. The arrows indicate the locations of the ac34 mRNA transcription initiation sites.
ac34 is required for efficient BV production in Sf9 cells.
To investigate the role of ac34 in the AcMNPV infection cycle, a recombinant bacmid with a partial deletion of the ac34 ORF, designated bAc34KO, was generated via ET recombination. To facilitate the examination of viral infection, polh and egfp genes were inserted into the polh locus of bAc34KO by a transposon to generate vAc34KO. The repair bacmid vHA:Ac34, in which an HA tag sequence was fused to the N terminus of ac34, was constructed to confirm that the observed phenotype was due to the deletion of ac34 (Fig. 3).
Fig 3.

Construction of recombinant viruses. A 245-bp fragment of the ac34 ORF was replaced with the Cm gene via ET homologous recombination in E. coli to generate bAc34KO. The upper portion of the figure is a schematic of vAcWT, which was generated by gene insertion into the polh locus of bMON14272. The lower portion of the figure shows the genes inserted into the polh locus of bAc34KO to generate vAc34KO and vHA:Ac34. The inserted genes are indicated in each bacmid.
Sf9 cells were transfected with vAc34KO, vHA:Ac34, or vAcWT and observed with fluorescence microscopy. At 24 h p.t., the transfected cells displayed equal proportions of fluorescent cells (approximately 10%), indicating comparable transfection efficiencies (Fig. 4A). At 48 h p.t., most of the cells transfected with vAcWT or vHA:Ac34 were infected. In contrast, the vAc34KO-transfected cells exhibited only a slight increase in the number of infected cells from 24 h p.t. to 48 h p.t., suggesting that vAc34KO was capable of generating infectious BVs from the initial transfected cells, but not as proficiently as vAcWT or the repair virus. At 72 h p.t., all of the vAcWT- or vHA:Ac34-transfected cells were infected, and the majority exhibited polyhedron formation at 96 h p.t. (Fig. 4A). In contrast, only about 50% of the cells transfected with vAc34KO were infected at 72 h p.t., and polyhedra were observed in only some of them at 96 h p.t. (Fig. 4A). Plaque assays revealed that at 120 h p.t., vAcWT and vHA:Ac34 produced plaques of equal size (Fig. 4B, i). However, the plaques produced by vAc34KO were much smaller than those of vAcWT and vHA:Ac34 (Fig. 4B, i). A relative quantitative analysis of viral plaque diameters showed that the average diameter of plaques produced by vAc34KO was reduced by 43% compared with vAcWT plaques (P < 0.05), and the defect caused by the ac34 deletion was rescued in cells transfected with vHA:Ac34 (Fig. 4B, ii).
Fig 4.

Analysis of viral replication in Sf9 cells. (A) Fluorescence microscopy images of Sf9 cells transfected with vAcWT, vAc34KO, or vHA:Ac34 at 24, 48, and 72 h p.t. Light microscopy images show the formation of OBs in the transfected cells at 96 h p.t. (B) Plaque assays of vAcWT-, vAc34KO-. and vHA:Ac34-transfected Sf9 cells. (i) Viral plaques were visualized with fluorescence microscopy at 120 h p.t. (ii) Twenty different viral plaques of each virus were randomly selected and statistically analyzed. The error bars represent standard deviations. (C) Viral growth curves from TCID50 assays. Viral titers were determined in the supernatants of cells transfected (i) or infected (ii) with vAcWT, vAc34KO, or vHA:Ac34 at various time points. The values presented for each time point represent the averages of three independent assays, and the error bars represent the standard deviations.
To further investigate the production of infectious BVs, viral growth curves were generated. Infectious BVs were detected in vAcWT- and vHA:Ac34-transfected cells at 24 h p.t.; however, BVs were not detected in vAc34KO-transfected cells until 48 h p.t., indicating that the ac34 deletion delayed BV production in virus-transfected cells. The total number of BVs produced by vAc34KO was approximately 100-fold lower than for vAcWT and vHA:Ac34 at 96 h p.t. (Fig. 4C, i). A C-terminally HA-tagged repair bacmid (vAc34:HA) was also constructed, and the titer of vAc34:HA-transfected cells showed 10-fold reduction at 96 h p.t. (data not shown). To confirm the results obtained with bacmid DNA transfection, we performed a second time course analysis of BVs produced from bacmid-transfected cells. The BV titers of vAcWT and vHA:Ac34 were substantially increased at 24 h p.i. In contrast, the BV titer of vAc34KO was only slightly increased and was approximately 10-fold lower than for vAcWT at 24 h p.i. (Fig. 4C, ii). The BV production in vAc34KO-infected cells at 48 h p.i. was equal to that in vAcWT- and vHA:Ac34-infected cells at 24 h p.i., suggesting a lag and reduction of BV production in vAc34KO-infected cells. Similarly, BV production was reduced by approximately 100-fold in vAc34KO-infected cells at 96 h p.i. (Fig. 4C, ii). The viral replication kinetics of the repair virus were similar to those of vAcWT (Fig. 4C, ii), indicating that the phenotype resulted from the ac34 deletion and was not due to the effects of the deletion site at that genomic locus and that the N terminus of Ac34 was able to display a foreign peptide without any negative effects on viral fitness, at least in cell culture.
Taken together, these results indicated that ac34 was required for the rapid and efficient production of BVs.
The deletion of ac34 does not affect viral DNA replication or morphogenesis.
To assess whether the deletion of ac34 causes a defect in viral DNA replication, qPCR was performed to compare the onset and levels of DNA synthesis in vAcWT- and vAc34KO-transfected cells during a single replication cycle, which is generally completed within the first 24 h p.t. As shown in Fig. 5, there was no significant increase in viral DNA content between 6 and 12 h p.t. for vAcWT- or vAc34KO-transfected cells. The levels of DNA synthesis increased between 12 and 18 h p.t. and were the same at 24 h p.t. for vAcWT and vAc34KO (Fig. 5). This result indicated that the deletion of ac34 did not affect the onset or levels of viral DNA synthesis in infected cells.
Fig 5.

qPCR analysis of viral DNA replication. Sf9 cells were transfected with 0.5 μg of vAcWT or vAc34KO bacmid DNA. At various time points, total cellular DNA was extracted, digested with DpnI, and analyzed with qPCR. The values presented for each time point represent the averages of two independent replication assays. The error bars represent standard deviations.
To determine whether the deletion of ac34 interferes with the assembly and egress of nucleocapsids, Sf9 cells transfected with vAc34KO or vAcWT were examined by transmission electron microscopy. The results showed that there was no discernible morphological effect on the nucleocapsid for the two bacmids (Fig. 6). The vAc34KO-transfected cells exhibited cytological changes and viral morphogenesis typical of baculovirus infection, including enlarged nuclei, electron-dense virogenic stroma containing rod-shaped nucleocapsids, and intact nuclear envelopes (Fig. 6A). Enveloped virions containing multiple nucleocapsids prior to occlusion and polyhedra containing ODVs were found within the ring zone (Fig. 6B and C). Similar characteristics were observed in cells transfected with vAcWT (Fig. 6D, E, and F), indicating that the deletion of ac34 did not affect nucleocapsid assembly or occlusion body morphogenesis.
Fig 6.

Transmission electron microscopy analysis of Sf9 cells transfected with vAc34KO (A, B, and C) or vAcWT (D, E, and F) at 72 h p.t. (B and E) Higher-magnification images of the regions boxed in panels A and D, respectively. The labels indicate the virogenic stroma (VS), nuclei (Nu), and polyhedra (p). Scale bar, 500 nm.
Expression of Ac34 during infection.
vHA:Ac34 and vAcWT had identical growth kinetics (Fig. 4C), which allowed the use of the anti-HA antibody to detect the expression of Ac34 during the infection cycle. Sf9 cells were infected with vHA:Ac34 at an MOI of 5 TCID50/cell, collected at various time points, and subjected to Western blot analysis. Ac34 expression was first detected at low levels at 12 h p.i., increased dramatically at 24 and 48 h p.i., and decreased at 72 and 96 h p.i. (Fig. 7). ac34 is predicted to encode a 215-amino-acid protein with a molecular mass of 24.9 kDa. The molecular mass of the N-terminally tagged HA:Ac34 was predicted to be approximately 26 kDa. The detected protein was approximately 30 kDa, 4 kDa larger than predicted (Fig. 7). Ac34 might be posttranslationally modified; the protein sequence contains nine potential modification sites, i.e., three N-glycosylation sites, four casein kinase II phosphorylation sites, and two protein kinase C phosphorylation sites. In addition, the HA tag was not detected in vAc34:HA-infected cells using the anti-HA antibody (data not shown).
Fig 7.

Temporal expression analysis of Ac34. Sf9 cells were infected with vHA:Ac34 at an MOI of 5 TCID50/cell. The cells were harvested at 6, 12, 24, 48, 72, and 96 h p.i., and samples were subjected to immunoblotting. The membrane was probed with anti-HA antibody to detect HA:Ac34. An anti-actin antibody was used as a loading control.
Ac34 is distributed in the cytoplasm and nucleus and is not packaged into virions.
Immunofluorescence was used to investigate the subcellular localization of Ac34. Sf9 cells infected with vHA:Ac34 were probed with mouse monoclonal anti-HA antibody and examined by confocal microscopy. At 12 h p.i., Ac34 localized to both the cytoplasm and nucleus with a low expression level (Fig. 8A). The amount of Ac34 increased at 24 h p.i., and the protein was predominantly distributed in the cytoplasm (Fig. 8A). This localization pattern was maintained at 48 h p.i. Cells infected with vAcWT (mock) did not show background fluorescence, indicating the specificity of the anti-HA antibody (Fig. 8A).
Fig 8.

Localization of Ac34 in vHA:Ac34-infected cells and purified virions. (A) Immunofluorescence analysis of the subcellular localization of Ac34. Sf9 cells were infected with vAcWT (Mock) or vHA:Ac34 at an MOI of 5 TCID50/cell. At various time points, the cells were fixed, probed with mouse monoclonal anti-HA antibody to detect HA:Ac34, and visualized using Alexa 555-conjugated goat anti-mouse antibody (violet). Nuclei were stained with Hochest33342 (blue). All of the images are shown at the same magnification. (B) Western blotting of the subcellular localization of Ac34. Sf9 cells were infected with vHA:Ac34 at an MOI of 5 TCID50/cell, and cytoplasmic and nuclear fractions were prepared at the indicated time points. The samples were subjected to immunoblotting with anti-HA, anti-IE1, or anti-GP64 antibodies. (C) Analysis of Ac34 in purified virions. Lysates of vHA:Ac34-infected cells and purified vHA:Ac34 virions (BVs, ODVs, and OBs) were subjected to immunoblotting using anti-HA and anti-VP39 antibodies.
To further demonstrate subcellular localization of Ac34, cells infected with vHA:Ac34 were fractionated into cytoplasmic and nuclear fractions, which were then subjected to Western blot analysis. As shown in Fig. 8B, Ac34 was detected in both the cytoplasmic and nuclear fractions of vHA:Ac34-infected Sf9 cells collected from 12 to 48 h p.i. The levels of Ac34 appeared to be greater in the cytoplasm than in the nucleus (Fig. 8B). The nuclear protein IE1 and the cytoplasmic protein GP64 were exclusively detected in the nuclear and cytoplasmic fractions, respectively, demonstrating the efficiency of the cellular fractionation (Fig. 8B). Taken together, these results suggested that Ac34 was distributed in both the cytoplasm and the nucleus during infection.
To determine whether Ac34 is a component of virions, BVs, OBs, and ODVs purified from vHA:Ac34-infected cells were subjected to immunoblotting. Ac34 was not detected in virions but was discernible in vHA:Ac34-infected cell lysates (Fig. 8C). To verify the loading samples, the same membrane was reprobed with anti-VP39 antibody, and VP39 was detected in all of the samples (Fig. 8C). These results indicated that Ac34 is not a structural protein.
Late gene expression is affected by the deletion of ac34.
As demonstrated above, Ac34 is not a structural protein; therefore, it is possible that Ac34 functions as a regulatory protein. To test this hypothesis, Sf9 cells infected with vAcWT or vAc34KO were subjected to immunoblotting. The results showed that the immediate-early gene product IE1 and the early-late gene product GP64 were detected beginning at 6 h p.i. in vAc34KO- and vAcWT-infected cells. However, the late proteins P6.9 and 38K were not detected until 24 h p.i. in vAc34KO-infected cells, 6 h later than when they were first detected in vAcWT-infected cells (Fig. 9A). Another late protein, VP39, exhibited a 12-h delay in expression in vAc34KO-infected cells (Fig. 9A). These results indicated that the deletion of ac34 did not affect IE1 or GP64 expression but delayed the expression of late proteins, including VP39, 38K, and P6.9.
Fig 9.

Analysis of the effect of ac34 deletion on viral gene expression. (A) Time course analysis of IE1, GP64, 38K, P6.9, and VP39 synthesis. Sf9 cells were infected with vAcWT or vAc34KO at an MOI of 1 TCID50/cell and harvested at different time points. The cell lysates were subjected to immunoblotting with anti-IE1, anti-GP64, anti-38K, anti-P6.9, or anti-VP39 antibody. An anti-actin antibody was used as a loading control. (B and C) qRT-PCR analysis of viral gene transcription. The effects of ac34 deletion on the transcription of several genes were measured with qRT-PCR at 12 (B) and 24 (C) h p.i. The transcript level of each gene was normalized to the genome copy number. The effect of ac34 deletion on gene transcription is shown for each gene as the percentage of detected transcripts relative to vAcWT. The error bars represent the standard deviations from the mean.
The delay in late gene product expression in vAc34KO-infected cells could be due to a delay or reduction in the transcription of viral genes. To determine whether the transcription of viral genes was affected by the deletion of ac34, qRT-PCR was performed to analyze the transcript levels of selected genes. The transcripts of three late genes (38k, p6.9, and vp39) were substantially reduced in vAc34KO-infected cells (Fig. 9B and C). At 12 h p.i., 38k, p6.9, and vp39 transcripts in vAc34KO-infected cells were typically reduced to 5 to 10% of the levels in vAcWT-infected cells (Fig. 9B). However, the transcript levels of gp64 and ie1 in vAc34KO-infected cells were not significantly different from those in vAcWT-infected cells at 12 and 24 h p.i. (Fig. 9B and C). These results suggested that Ac34 is an activator protein that promotes late gene expression.
vAc34KO lacks virulence in T. ni larvae.
Because the deletion of ac34 impairs viral replication in vitro, the role of ac34 in viral replication in vivo is also of interest. Therefore, bioassays were performed on 3rd-instar T. ni larvae. These results showed that the mortality of larvae infected with vAc34KO was similar to that of control larvae (Table 1). Larvae infected with vAc34KO failed to develop NPV disease, indicating that the OB of vAc34KO essentially lost its infectivity in T. ni larvae. In contrast, 100 OBs/larva of vAcWT were sufficient to generate >70% mortality in virus-infected larvae (Table 1).
Table 1.
Infectivity of vAc34KO and vAcWT in the 3rd instar of T. ni larvae
| Virus | Dose (no. of OBs/larva) | Replication 1 |
Rep. 2 |
||||
|---|---|---|---|---|---|---|---|
| No. treated | No. dead | Mortality (%) | No. treated | No. dead | Mortality (%) | ||
| vAc34KO | 100 | 24 | 1 | 4.2 | 24 | 2 | 8.3 |
| 1,000 | 24 | 1 | 4.2 | 24 | 1 | 4.2 | |
| 10,000 | 24 | 0 | 0.0 | 24 | 2 | 8.3 | |
| vAcWT | 100 | 24 | 17 | 70.8 | 24 | 18 | 75.0 |
| 1,000 | 24 | 24 | 100.0 | 24 | 23 | 95.8 | |
| 10,000 | 24 | 24 | 100.0 | 24 | 24 | 100.0 | |
| Control | 24 | 2 | 8.3 | 24 | 1 | 4.2 | |
We also determined the titer of BVs in the hemolymph of insects infected with vAc34KO or vAcWT. Fourth-instar larvae were orally infected with a high dose (20,000 OBs/larva) of vAc34KO or vAcWT. Hemolymph was collected from these larvae at 48 and 72 h p.i. Few infectious BVs were detected in the hemolymph of larvae inoculated with vAc34KO (Fig. 10), whereas the BV titers of the hemolymph of larvae treated with vAcWT were almost 108 TCID50/ml. Fluorescence microscopy analysis showed that the majority of hemocytes were infected in vAcWT-infected larvae, but not in vAc34KO-infected larvae (data not shown). To further investigate the biological activity of vAc34KO BVs in vivo, fourth-instar T. ni larvae were injected with BVs from vAc34KO or vAcWT. At 48 and 72 h p.i., hemolymph was collected and titrated. We found that the titers in the hemolymph of vAc34KO-injected larvae approached zero, but the titers in the hemolymph of vAcWT-injected larvae were >107 TCID50/ml (Fig. 10). These results suggested that vAc34KO is avirulent in T. ni larvae.
Fig 10.

BV titers in the hemolymph of larvae infected with vAcWT or vAc34KO. Fourth-instar T. ni larvae were infected with vAcWT and vAc34KO per os at 20,000 OBs/larva or hemocoelically injected with 250 TCID50/larva. At 48 and 72 h p.i., the hemolymph was collected to determine the virus titer. The error bars represent standard deviations.
DISCUSSION
The conservation of Ac34 in alphabaculoviruses may imply that the protein serves an important function in the viral life cycle. To elucidate its function, we generated an ac34 knockout AcMNPV and found that the deletion mutant showed severely impaired viral replication and delayed late gene expression in Sf9 cells. In vivo assays revealed that the deletion mutant could not establish a fatal infection in T. ni larvae.
Transcriptional analysis of ac34 showed that ac34 transcripts were detected from 6 to 48 h p.i. and that ac34 transcription was regulated by both early and late promoters (Fig. 1). This transcription profile is consistent with the findings of Jiang et al. (26), who examined the temporal transcription patterns of AcMNPV through a microarray. We used a repair virus containing an HA tag at the Ac34 N terminus to investigate the expression pattern and localization of Ac34. Ac34 localized to both the cytoplasm and nucleus during infection (Fig. 8), suggesting that it might function in both compartments. We also constructed a C-terminally HA-tagged repair bacmid (vAc34:HA). Interestingly, this tagged protein could not be detected in vAc34:HA-infected cells with anti-HA immunoblotting. In addition, vAc34:HA caused a 10-fold reduction of BV production in Sf9 cells (data not shown), suggesting that the C terminus plays a critical role in Ac34 function. Moreover, the C terminus was highly conserved in Ac34 homologs (Fig. 1).
The deletion of ac34 resulted in a significant decrease in BV production but did not negatively affect DNA replication. A similar negative effect on BV production was observed in early studies of ac66 and exon0 (9, 27). The deletion of either exon0 or ac66 resulted in a greater than 100-fold reduction in BV production (9, 14, 27). Neither the exon0 nor the ac66 deletion mutant was essential for nucleocapsid assembly, but both were required for efficient nucleocapsid egress (9, 14, 27). Electron microscopy analysis demonstrated that the ac34 deletion did not affect nucleocapsid assembly (Fig. 6). However, we did not observe any nucleocapsid accumulation within the nuclei, which differed from the typical phenotype of ac66 and exon0 deletion mutant-transfected cells (14, 27). In addition, unlike Ac66 and EXON0, which were reported to be associated with BVs and ODVs (6, 14, 56), Ac34 was not detected in virions. There are similarities between the phenotypes of ac34 and pp31 deletion mutants. Deletion of pp31 also resulted in a reduction in BV production and slightly downregulated some early and late gene expression levels (65). Ac34 may not be involved in nucleocapsid egress but could be associated with the regulation of viral late gene expression or the virus-host interaction.
To examine the possibility that viral gene expression is affected by the deletion of ac34, we compared the expression and transcription of several genes in vAc34KO- and vAcWT-infected cells. The temporal expression and transcription of the early gene ie1 and the early-late gene gp64 were similar in vAc34KO- and vAcWT-infected cells. The major effect of ac34 deletion appeared to be the delay of late gene expression (Fig. 9). VP39, P6.9, and 38K are essential for nucleocapsid assembly (55, 64), and the delayed expression of these proteins resulted in a lag in nucleocapsid assembly. This result was consistent with the delay of BV production observed in Sf9 cells (Fig. 4). In addition, a general reduction in BV production may have been caused by a decrease in the production of viral proteins. These findings suggest that Ac34 plays a role in viral replication, especially during the late phase of the infection cycle, which could result in a delay of the infection process.
Interestingly, the transcription level of gp64 was not affected, even at the late phase of infection (Fig. 9). gp64 has both early and late promoter elements. It was reported that at 24 h p.i., gp64 was mainly transcribed from the late promoter (4). Baculoviruses induce a dramatic shutoff of host protein synthesis that parallels late gene expression (52). It has been demonstrated that the early promoter of gp64 was active at 36 h p.i. (4). vp39, 38k, and p6.9 were expressed solely from late promoter motifs (3, 62, 64), and it is possible that they were more severely affected because late transcription is influenced by Ac34, while early transcription is independent of Ac34.
The amino acid sequence of Ac34 shares similarity with that of SKP1. Studies of SKP1 have shown that it is a component of the SCF complex that catalyzes the ubiquitylation of various proteins involved in cell cycle control and signal transduction (38, 68). SKP1 serves as an adapter that links the F-box protein to Cullin (48) and is essential for both G1/S and G2/M transitions (2). Of particular interest is the fact that cell cycle arrest has been shown to be important for the replication of many RNA and DNA viruses (10). Many viruses could take over the host ubiquitin system to dysregulate the cell cycle for viral proliferation (24, 46, 59). Baculovirus was able to produce a “viral S phase,” which is a benefit for viral replication (44). It has been reported that cells infected with baculovirus were arrested in the S or G2/M phase (5, 23, 47). It is therefore possible that Ac34 is required to optimize the cellular environment and functions as a regulator to help virus induce the “viral S phase” for increased viral proliferation. For example, Ac34 may interact with the SCF complex to regulate cyclin to promote the G1/S transition or to inhibit the G2/M transition.
Ac34 is not required for late transcription but is required for the amplification of late gene expression. In this respect, Ac34 is similar to some late expression factors, such as LEF6, LEF12, and PP31 (21, 32, 65). These genes do not appear to play essential roles in AcMNPV infection in Sf9 cells, but their presence accelerates the infection cycle and increases viral yields (21, 32, 65). In transient-expression experiments, Ac34 did not appear to be required for late gene expression. However, the transient-expression system does not perfectly mimic the course of viral infection, and it is possible that the functional importance of factors that are regulated by other viral components during virus infection could be overlooked. Studies of gene deletion mutants have provided new insights into the roles of some genes, particularly regarding their requirements for DNA replication and/or late transcription in the context of the infection cycle (13, 21, 30, 33, 37, 42, 51, 63). Our data demonstrate that Ac34 is required for efficient viral late transcription during the infection cycle. Ac34 could promote viral late transcription by inactivating virus-encoded factors that reduce transcription factor activity. Further understanding of the mechanism of Ac34 function should help to define its role in the expression of viral late genes.
Productive infection in insects may require specific viral proteins that are dispensable for viral replication in cell culture; therefore, we considered it important to study the contribution of Ac34 to AcMNPV infection in vivo. Our results showed that vAc34KO cannot establish a fatal infection in T. ni larvae through per os infection. Further analyses revealed that few infectious BVs were detected in the hemolymph of vAc34KO-infected larvae (through both per os and injection infection) (Fig. 10), indicating that the deletion of ac34 significantly impaired viral replication in T. ni larvae. Empirical evidence from studies of baculovirus pathogenesis in vivo revealed that insects have some defense mechanisms that could be effective against viral pathogens (8, 28, 53, 57). Midgut cell sloughing may be one widespread response by lepidopteran larvae to insect viruses in general (11, 15, 28). Previous studies showed that midgut cells were sloughed at the early phase of baculovirus infection, and all of the infected cells were cleared before larval ecdysis (11, 58). In addition, insects were able to recognize and respond to baculovirus infection via immune-related mechanisms (53, 57). Hemocytes could remove the virus from the hemolymph and participate in the encapsulation and melanization responses to eliminate viral infection, which were effective in blocking disease progression in baculovirus-infected larvae (43, 57). The host defense against baculovirus infection was required for rapid infection in vivo. Our results demonstrated that the absence of ac34 had a significant effect on viral replication and infectivity in T. ni larvae. It is possible that the onset of BV production is delayed and the rate of BV production is diminished in vAc34KO-infected larvae, which could account for the loss of virulence of vAc34KO in vivo.
In this study, we demonstrated that Ac34 was not essential for viral replication in cell culture but was associated with late gene expression. In addition, the ac34 deletion mutant failed to establish an effective infection in vivo. The role of Ac34 in late gene expression and virulence in vivo will be the topics of future studies. Five lepidopteran NPV-specific genes have been characterized, but none of them seem to be directly responsible for the differences between lepidopteran NPVs and GVs. There are 14 GV-specific genes in all of the sequenced GVs (44), and these genes may be the source of the biological differences between lepidopteran NPVs and GVs.
ACKNOWLEDGMENTS
We thank Linda A. Guarino (Texas A&M University) for the generous gift of the mouse monoclonal AcMNPV IE1 antibody.
This research was supported by the National Basic Research Program of China (973 Program; no. 2009CB118903), the Hi-Tech Research and Development Program of China (863 Program; no. 2011AA10A204), the Research Foundation of the Chinese Ministry of Education (grant no. 106128), and the Key Project of Guangdong Science and Technology (grant no. 2009A020101003).
Footnotes
Published ahead of print 11 July 2012
REFERENCES
- 1. Ackermann HW, Smirnoff WA. 1983. A morphological investigation of 23 baculoviruses. J. Invertebr. Pathol. 41:269–280 [Google Scholar]
- 2. Bai C, et al. 1996. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86:263–274 [DOI] [PubMed] [Google Scholar]
- 3. Blissard GW, Quant-Russell RL, Rohrmann GF, Beaudreau GS. 1989. Nucleotide sequence, transcriptional mapping, and temporal expression of the gene encoding p39, a major structural protein of the multicapsid nuclear polyhedrosis virus of Orgyia pseudotsugata. Virology 168:354–362 [DOI] [PubMed] [Google Scholar]
- 4. Blissard GW, Rohrmann GF. 1989. Location, sequence, transcriptional mapping, and temporal expression of the gp64 envelope glycoprotein gene of the Orgyia pseudotsugata multicapsid nuclear polyhedrosis virus. Virology 170:537–555 [DOI] [PubMed] [Google Scholar]
- 5. Braunagel SC, Parr R, Belyavskyi M, Summers MD. 1998. Autographa californica nucleopolyhedrovirus infection results in Sf9 cell cycle arrest at G2/M phase. Virology 244:195–211 [DOI] [PubMed] [Google Scholar]
- 6. Braunagel SC, Russell WK, Rosas-Acosta G, Russell DH, Summers MD. 2003. Determination of the protein composition of the occlusion-derived virus of Autographa californica nucleopolyhedrovirus. Proc. Natl. Acad. Sci. U. S. A. 100:9797–9802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Braunagel SC, Summers MD. 1994. Autographa californica nuclear polyhedrosis virus, PDV, and ECV viral envelopes and nucleocapsids: structural proteins, antigens, lipid and fatty acid profiles. Virology 202:315–328 [DOI] [PubMed] [Google Scholar]
- 8. Clarke TE, Clem RJ. 2002. Lack of involvement of haemocytes in the establishment and spread of infection in Spodoptera frugiperda larvae infected with the baculovirus Autographa californica M nucleopolyhedrovirus by intrahaemocoelic injection. J. Gen. Virol. 83:1565–1572 [DOI] [PubMed] [Google Scholar]
- 9. Dai X, Stewart TM, Pathakamuri JA, Li Q, Theilmann DA. 2004. Autographa californica multiple nucleopolyhedrovirus exon0 (orf141), which encodes a RING finger protein, is required for efficient production of budded virus. J. Virol. 78:9633–9644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davy C, Doorbar J. 2007. G2/M cell cycle arrest in the life cycle of viruses. Virology 368:219–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Engelhard EK, Volkman LE. 1995. Developmental resistance in fourth instar Trichoplusia ni orally inoculated with Autographa californica M nuclear polyhedrosis virus. Virology 209:384–389 [DOI] [PubMed] [Google Scholar]
- 12. Fan X, McLachlin JR, Weaver RF. 1998. Identification and characterization of a protein kinase-interacting protein encoded by the Autographa californica nuclear polyhedrosis virus. Virology 240:175–182 [DOI] [PubMed] [Google Scholar]
- 13. Fan X, Thirunavukkarasu K, Weaver RF. 1996. Temperature-sensitive mutations in the protein kinase-1 (pk-1) gene of the Autographa californica nuclear polyhedrosis virus that block very late gene expression. Virology 224:1–9 [DOI] [PubMed] [Google Scholar]
- 14. Fang M, Dai X, Theilmann DA. 2007. Autographa californica multiple nucleopolyhedrovirus EXON0 (ORF141) is required for efficient egress of nucleocapsids from the nucleus. J. Virol. 81:9859–9869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Federici BA. 1997. Baculovirus pathogenesis, p 33–59 In Miller LK. (ed), The baculoviruses. Plenum Press, New York, NY [Google Scholar]
- 16. Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. 1997. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 91:221–230 [DOI] [PubMed] [Google Scholar]
- 17. Friesen PD. 1997. Regulation of baculovrius early gene expression, p 141–170 In Miller LK. (ed), The baculoviruses. Plenum Press, New York, NY [Google Scholar]
- 18. Goldberg AV, Romanowski V, Federici BA, Sciocco de Cap A. 2002. Effects of the Epap granulovirus on its host, Epinotia aporema (Lepidoptera: Tortricidae). J. Invertebr. Pathol. 80:148–159 [DOI] [PubMed] [Google Scholar]
- 19. Goley ED, et al. 2006. Dynamic nuclear actin assembly by Arp2/3 complex and a baculovirus WASP-Like protein. Science 314:464–467 [DOI] [PubMed] [Google Scholar]
- 20. Griffiths CM, et al. 1999. In vitro host range of Autographa californica nucleopolyhedrovirus recombinants lacking functional p35, iap1 or iap2. J. Gen. Virol. 80:1055–1066 [DOI] [PubMed] [Google Scholar]
- 21. Guarino LA, Mistretta TA, Dong W. 2002. Baculovirus lef-12 is not required for viral replication. J. Virol. 76:12032–12043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hughes PR, Wood HA. 1981. A synchronous peroral technique for the bioassay of insect viruses. J. Invertebr. Pathol. 37:154–159 [Google Scholar]
- 23. Ikeda M, Kobayashi M. 1999. Cell-cycle perturbation in Sf9 cells infected with Autographa californica nucleopolyhedrovirus. Virology 258:176–188 [DOI] [PubMed] [Google Scholar]
- 24. Isobe T, et al. 2009. Adenovirus E1A inhibits SCF(Fbw7) ubiquitin ligase. J. Biol. Chem. 284:27766–27779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jehle JA, et al. 2006. On the classification and nomenclature of baculoviruses: a proposal for revision. Arch. Virol. 151:1257–1266 [DOI] [PubMed] [Google Scholar]
- 26. Jiang SS, et al. 2006. Temporal transcription program of recombinant Autographa californica multiple nucleopolyhedrosis virus. J. Virol. 80:8989–8999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ke J, Wang J, Deng R, Wang X. 2008. Autographa californica multiple nucleopolyhedrovirus ac66 is required for the efficient egress of nucleocapsids from the nucleus, general synthesis of preoccluded virions and occlusion body formation. Virology 374:421–431 [DOI] [PubMed] [Google Scholar]
- 28. Keddie BA, Aponte GW, Volkman LE. 1989. The pathway of infection of Autographa californica nuclear polyhedrosis virus in an insect host. Science 243:1728–1730 [DOI] [PubMed] [Google Scholar]
- 29. Lee SY, et al. 1996. A common pathway for p10 and calyx proteins in progressive stages of polyhedron envelope assembly in AcMNPV-infected Spodoptera frugiperda larvae. Arch. Virol. 141:1247–1258 [DOI] [PubMed] [Google Scholar]
- 30. Li L, Harwood SH, Rohrmann GF. 1999. Identification of additional genes that influence baculovirus late gene expression. Virology 255:9–19 [DOI] [PubMed] [Google Scholar]
- 31. Li Y, et al. 2005. vlf-1 deletion brought AcMNPV to defect in nucleocapsid formation. Virus Genes 31:275–284 [DOI] [PubMed] [Google Scholar]
- 32. Lin G, Blissard GW. 2002. Analysis of an Autographa californica multicapsid nucleopolyhedrovirus lef-6-null virus: LEF-6 is not essential for viral replication but appears to accelerate late gene transcription. J. Virol. 76:5503–5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin G, Blissard GW. 2002. Analysis of an Autographa californica nucleopolyhedrovirus lef-11 knockout: LEF-11 is essential for viral DNA replication. J. Virol. 76:2770–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luckow VA, Lee SC, Barry GF, Olins PO. 1993. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol. 67:4566–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marchler-Bauer A, et al. 2007. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 35:D237–D240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marek M, Merten OW, Galibert L, Vlak JM, van Oers MM. 2011. Baculovirus VP80 protein and the F-actin cytoskeleton interact connecting the viral replication factory with the nuclear periphery. J. Virol. 85:5350–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McLachlin JR, Yang S, Miller LK. 1998. A baculovirus mutant defective in PKIP, a protein which interacts with a virus-encoded protein kinase. Virology 246:379–391 [DOI] [PubMed] [Google Scholar]
- 38. Nakayama KI, Nakayama K. 2005. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin. Cell Dev. Biol. 16:323–333 [DOI] [PubMed] [Google Scholar]
- 39. Ohkawa T, Volkman LE, Welch MD. 2010. Actin-based motility drives baculovirus transit to the nucleus and cell surface. J. Cell Biol. 190:187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Passarelli AL, Guarino LA. 2007. Baculovirus late and very late gene regulation. Curr. Drug Targets 8:1103–1115 [DOI] [PubMed] [Google Scholar]
- 41. Possee RD, et al. 1991. Nucleotide sequence of the Autographa californica nuclear polyhedrosis 9.4 kbp EcoRI-I and -R (polyhedrin gene) region. Virology 185:229–241 [DOI] [PubMed] [Google Scholar]
- 42. Rapp JC, Wilson JA, Miller LK. 1998. Nineteen baculovirus open reading frames, including LEF-12, support late gene expression. J. Virol. 72:10197–10206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rivkin H, et al. 2006. Response of immunocompetent and immunosuppressed Spodoptera littoralis larvae to baculovirus infection. J. Gen. Virol. 87:2217–2225 [DOI] [PubMed] [Google Scholar]
- 44. Rohrmann GF. 2011. Baculovirus molecular biology. National Library of Medicine, National Center for Biotechology Information, Bethesda, MD [Google Scholar]
- 45. Russell RL, Funk CJ, Rohrmann GF. 1997. Association of a baculovirus-encoded protein with the capsid basal region. Virology 227:142–152 [DOI] [PubMed] [Google Scholar]
- 46. Saha A, et al. 2011. Epstein-Barr virus nuclear antigen 3C facilitates G1-S transition by stabilizing and enhancing the function of cyclin D1. PLoS Pathog. 7:e1001275 doi:10.1371/journal.ppat.1001275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saito T, Dojima T, Toriyama M, Park EY. 2002. The effect of cell cycle on GFPuv gene expression in the baculovirus expression system. J. Biotechnol. 93:121–129 [DOI] [PubMed] [Google Scholar]
- 48. Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. 1997. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91:209–219 [DOI] [PubMed] [Google Scholar]
- 49. Slack J, Arif B. 2007. The Baculoviruses occlusion-derived virus: virion structure and function. Adv. Virus Res. 69:99–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soding J. 2005. Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951–960 [DOI] [PubMed] [Google Scholar]
- 51. Stewart TM, Huijskens I, Willis LG, Theilmann DA. 2005. The Autographa californica multiple nucleopolyhedrovirus ie0-ie1 gene complex is essential for wild-type virus replication, but either IE0 or IE1 can support virus growth. J. Virol. 79:4619–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thiem SM. 2009. Baculovirus genes affecting host function. In Vitro Cell Dev. Biol. Anim. 45:111–126 [DOI] [PubMed] [Google Scholar]
- 53. Trudeau D, Washburn JO, Volkman LE. 2001. Central role of hemocytes in Autographa californica M nucleopolyhedrovirus pathogenesis in Heliothis virescens and Helicoverpa zea. J. Virol. 75:996–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vanarsdall AL, Okano K, Rohrmann GF. 2005. Characterization of the replication of a baculovirus mutant lacking the DNA polymerase gene. Virology 331:175–180 [DOI] [PubMed] [Google Scholar]
- 55. Wang M, et al. 2010. Specificity of baculovirus P6.9 basic DNA-binding proteins and critical role of the C terminus in virion formation. J. Virol. 84:8821–8828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang R, et al. 2010. Proteomics of the Autographa californica nucleopolyhedrovirus budded virions. J. Virol. 84:7233–7242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Washburn JO, Kirkpatrick BA, Volkman LE. 1996. Insect protection against viruses. Nature 383:767 [Google Scholar]
- 58. Washburn JO, Lyons EH, Haas-Stapleton EJ, Volkman LE. 1999. Multiple nucleocapsid packaging of Autographa californica nucleopolyhedrovirus accelerates the onset of systemic infection in Trichoplusia ni. J. Virol. 73:411–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Welcker M, Clurman BE. 2005. The SV40 large T antigen contains a decoy phosphodegron that mediates its interactions with Fbw7/hCdc4. J. Biol. Chem. 280:7654–7658 [DOI] [PubMed] [Google Scholar]
- 60. Whitt MA, Manning JS. 1988. A phosphorylated 34-kDa protein and a subpopulation of polyhedrin are thiol linked to the carbohydrate layer surrounding a baculovirus occlusion body. Virology 163:33–42 [DOI] [PubMed] [Google Scholar]
- 61. Williams GV, Faulkner P. 1997. Cytological changes and viral morphogenesis during baculovirus infection, p 61–107 In Miller LK. (ed), The baculoviruses. Plenum Press, New York, NY [Google Scholar]
- 62. Wilson ME, Mainprize TH, Friesen PD, Miller LK. 1987. Location, transcription, and sequence of a baculovirus gene encoding a small arginine-rich polypeptide. J. Virol. 61:661–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wu CP, et al. 2010. Autographa californica multiple nucleopolyhedrovirus LEF-2 is a capsid protein required for amplification but not initiation of viral DNA replication. J. Virol. 84:5015–5024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wu W, et al. 2006. Autographa californica multiple nucleopolyhedrovirus nucleocapsid assembly is interrupted upon deletion of the 38K gene. J. Virol. 80:11475–11485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yamagishi J, Burnett ED, Harwood SH, Blissard GW. 2007. The AcMNPV pp31 gene is not essential for productive AcMNPV replication or late gene transcription but appears to increase levels of most viral transcripts. Virology 365:34–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yu Q, Lin T, Feng G, Yang K, Pang Y. 2008. Functional analysis of the putative antiapoptotic genes, p49 and iap4, of Spodoptera litura nucleopolyhedrovirus with RNAi. J. Gen. Virol. 89:1873–1880 [DOI] [PubMed] [Google Scholar]
- 67. Yuan M, et al. 2011. Identification of Autographa californica nucleopolyhedrovirus ac93 as a core gene and its requirement for intranuclear microvesicle formation and nuclear egress of nucleocapsids. J. Virol. 85:11664–11674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zheng N, et al. 2002. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416:703–709 [DOI] [PubMed] [Google Scholar]