

Abstract
The receptor binding specificity of influenza A virus is one of the major determinants of viral tropism and host specificity. In general, avian viral hemagglutinin prefers to bind to α2,3-linked sialic acid, whereas the human viral hemagglutinin prefers to bind to α2,6-linked sialic acid. Here, we demonstrate that host fibronectin protein plays an important role in the life cycle of some influenza A viruses. Treating cells with anti-fibronectin antibodies or fibronectin-specific small interfering RNA can inhibit the virus replication of human H1N1 influenza A viruses. Strikingly, these inhibitory effects cannot be observed in cells infected with H5N1 viruses. By using reverse genetics techniques, we observed that the receptor binding specificity, but not the origin of the hemagglutinin subtype, is responsible for this differential inhibitory effect. Changing the binding preference of hemagglutinin from α2,6-linked sialic acid to α2,3-linked sialic acid can make the virus resistant to the anti-fibronectin antibody treatment and vice versa. Our further characterizations indicate that anti-fibronectin antibody acts on the early phase of viral replication cycle, but it has no effect on the initial binding of influenza A virus to cell surface. Our subsequent investigations further show that anti-fibronectin antibody can block the postattachment entry of influenza virus. Overall, these results indicate that the sialic acid binding preference of influenza viral hemagglutinin can modulate the preferences of viral entry pathways, suggesting that there are subtle differences between the virus entries of human and avian influenza viruses.
INTRODUCTION
Influenza A virus belongs to the Orthomyxoviridae family. It is a segmented, negative-strand RNA virus. The viral hemagglutinin (HA) protein binds to sialic acid groups of cellular surface glycoproteins to achieve viral attachment and entry. The sialic acid binding specificity of HA is one of the major determinants for controlling viral tropism and host specificity (25, 39). In general, human influenza viruses have a binding preference for α2,6-linked sialic acid, whereas avian influenza viruses have a preference for α2,3-linked sialic acid. Key amino acid positions controlling this binding specificity have been identified in the HA of seasonal human or avian viruses (10, 17, 36). After attaching to a host cell, the virus can travel to acidic endosomes for membrane fusion via clathrin- or caveolin-mediated endocytosis (29). It is also known that the virus might enter a cell by using other alternative pathways (9, 11, 12). For example, the virus is recently shown to be capable of utilizing C-type lectins to perform sialic acid-independent virus attachment and entry (34). These results demonstrate that influenza viruses can use a number of entry mechanisms to achieve viral infection. However, it is not known whether all influenza viruses can use these pathways with identical preferences.
Fibronectin (FN) exists in a soluble form in plasma and an insoluble cellular form in cells (46). The plasma form is structurally and biologically different from the cellular form. The cellular FN is an extracellular matrix glycoprotein that can be polymerized to form linear and branched meshwork on cell surface. This cellular form is an important component of the extracellular matrix, and it facilitates several cellular processes such as cell migration, surface receptor internalization, and cell signaling (46). Its pre-mRNA can undergo alternative splicing, and its mature mRNA can encode a FN monomer with a molecular mass of 230 to 250 kDa. FN is a modular protein composed of type I, II, and III repeating units. The ninth and tenth type III repeating units form the cell-binding domain of the protein for cell attachment. The protein can bind to other extracellular matrix proteins, cell surface receptors, glycosaminoglycans, and other FN molecules. Interestingly, a vast number of bacteria, protozoa, and fungi have been reported to express FN binding proteins for interacting with cellular FN (1, 22, 26). Some of these pathogens (e.g., Staphylococcus aureus) can even use cellular FN to perform intracellular invasion (37).
Previous studies have suggested that FN can facilitate the entry of gammaretrovirus, hepatitis B virus, and rhabdovirus (5, 22, 53). In addition, some of the FN-interacting partners have also been reported to have roles in the entry of other viruses (43, 51). These results have prompted us to hypothesize that FN is important in the life cycle of influenza A virus also. Here, we demonstrate that FN is involved in replication of some, but not all, influenza viruses.
MATERIALS AND METHODS
Cells and viruses.
MDCK cells (Madin-Darby canine kidney cells), 293T cells (human embryonic kidney cells), and A549 cells (human lung epithelial cells) were cultured in minimum essential medium (MEM; Gibco) supplemented with 1% penicillin-streptomycin (Invitrogen), 10% fetal bovine serum (FBS; Invitrogen), and 2.5% HEPES (Gibco). All cells were maintained in 5% CO2 at 37°C. Viral strains WSN (H1N1, A/WSN/33), PR8 (H1N1, A/Puerto Rico/8/34), A/OK/483/08 (H3N2; a generous gift from Gillian Air, Oklahoma), Indo5 (H5N1, A/Indonesia/5/05), and VN3046 (H5N1, A/Vietnam/3046/04) were grown and titrated in MDCK cells. All experiments involving high-pathogenicity H5 viruses were conducted in a biosafety level 3 facility.
Antibodies.
Rabbit polyclonal anti-human FN antibody (A0245; DakoCytomation), rabbit monoclonal IgG anti-green fluorescent protein (anti-GFP) antibody (A11122; Invitrogen), mouse monoclonal anti-human FN antibody (16E5; Santa Cruz), mouse monoclonal anti-human FN antibodies (FN1-1, FN3-8, FN9-1, and FN12-8; TaKaRa), rat monoclonal anti-human β1 integrin antibody (AIIB2; Developmental Studies Hybridoma Bank), and goat polyclonal anti-human β1 integrin antibody (N-20; Santa Cruz) were used to react with the corresponding proteins in A549 and MDCK cell cultures. These antibodies are known to cross-react with the corresponding proteins from multiple animal species. Mouse monoclonal anti-influenza virus A M2 antibody (14C2; Santa Cruz) and mouse monoclonal anti-influenza virus A NP antibody (5D8; Santa Cruz), and Alexa Fluor 488-conjugated donkey anti-mouse antibody (A21202; Invitrogen) were used to detect influenza virus-infected cells.
Viral infection.
In a typical experiment, cells were incubated with influenza viruses at multiplicity of infection (MOI) of 1 for 1 h at 37°C. Changing the incubation temperature from 37°C to room temperature (data not shown) or 4°C (see below) at this step produced similar findings. Inocula were removed by a phosphate-buffered saline (PBS) wash, and the treated cells were then cultured in MEM supplemented with 1% penicillin-streptomycin and 2 μg of TPCK (tolylsulfonyl phenylalanyl chloromethyl ketone)/ml of treated trypsin. Culture medium containing anti-FN or control antibodies was added to the cells at the indicated time points. The concentration tested in the present study was based on other studies (6, 20, 23). At a concentration of 2 μg/ml, the anti-FN antibody level is estimated to be about 7.5E+06 molecules per cell. Unless otherwise stated, cells were normally cultured in the presence of the indicated antibody throughout the postinfection period.
Immunofluorescence staining.
Infected cells grown on coverslips were washed with PBS and fixed by 4% paraformaldehyde (PFA) in PBS. Fixed cells were washed with PBS and treated with 0.2% Triton X-100 solution in PBS for 5 min. After being washed with PBS, the cells were blocked with 1% bovine serum albumin (BSA) in PBS at room temperature for 1 h. Cells were rinsed with PBS and incubated with anti-M2 (at a 1:500 dilution) or anti-NP (at a 1:500 dilution) antibody at 4°C overnight. The cells were washed with PBS for three times and incubated with Alexa Fluor 488-conjugated donkey anti-mouse antibody (at a 1:200 dilution) for 1 h at room temperature in the dark. The cells were then rinsed with PBS for three times and incubated with DAPI (4′,6′-diamidino-2-phenylindole) for 2 min. After a rinse with PBS, the stained cells were mounted on glass slides with antifade reagent. The slides were stored at 4°C until inspection. Signals from Alexa Fluor 488-conjugated and DAPI-stained cells were observed using a fluorescein isothiocyanate filter set (EX465-495, DM505, and BA515-555) and a DAPI filter set (EX340-360, DM400, and BA450-460), respectively.
Gene silencing using siRNA.
A gene knockdown experiment was performed by transfecting small interfering RNAs (siRNAs) into A549 cells in a 24-well plate format according to the manufacturer's protocol (Ambion). A549 cells were treated with trypsin immediately before transfection. Trypsinized cells were washed with PBS and resuspended in MEM supplemented with 10% FBS without antibiotics. Then, 1 μl of siPORTM NeoFX transfection agent (Ambion, catalog no. AM4511M) was diluted with 25 μl of Opti-MEM (Gibco), followed by incubation at room temperature for 10 min. Portions (25 μl) of Opti-MEM containing 1 to 20 nmol of GAPDH (Ambion, catalog no. 4390487), FN (Ambion, catalog no. 4392420), or scrambled siRNA were mixed with an equal volumes of diluted siPORT, followed by incubation at room temperature for 10 min. Each of these reactions was then mixed with 450 μl of A549 cell suspension in a culture well. Treated cells were incubated at 37°C for 24 h before viral infection or immunofluorescence staining.
Quantitative reverse transcription-PCR (RT-PCR).
The total RNA of infected cells was harvested at 4 h postincubation using TRIzol, as instructed by the manufacturer (Invitrogen). Next, 10 ng of purified RNA was reverse transcribed in a 20-μl reaction containing 4 μl of 5× first-strand buffer (Invitrogen), 2 μl of dithiothreitol (DTT; 0.1 mM), 1 μl of deoxynucleoside triphosphate (10 mM), 0.5 μl of SuperScript II reverse transcriptase (Invitrogen, 200 U/μl), 0.5 μl of RNaseOut inhibitor, and 0.5 μl of 10 nM oligonucleotide primer [oligo(dT) for β-actin mRNA or uni12 primer for M vRNA] as described previously (8). The reaction was incubated at 42°C for 1 h, followed by an enzyme inactivation step (72°C, 15 min). Three μl of the cDNA sample was mixed with 10 μl of Fast SYBR green master mix (Applied Biosystems, 4385612) and 1 μl of primers (10 nM each) and DNase/RNase-free water was used to top up the reaction volume to 20 μl. The reactions were first incubated at 50°C for 2 min and at 95°C for 20 s, then thermal-cycled for 40 cycles (at 95°C for 15 s and at 60°C for 1 min) in a quantitative PCR platform (7500 Fast real-time PCR system; Applied Biosystems). The primer sequences used to detect M vRNA are 5′-CTTCTAACCGAGGTCGAAACG-3′ and 5′-GGCATTTTGGACAAAGCGTCTA-3′, whereas the primers used to detect β-actin mRNA are 5′-CCCAAAGGCCAACCGCGAGAAGAT-3′ and 5′-GTCCCGGCCAGCCAGGTCCAG-3′. Quantitative data from the M gene assay were normalized by the corresponding data set deduced from the β-actin assay.
Reverse genetics.
Recombinant viruses were generated by using a plasmid-based reverse genetics systems (24). Wild-type WSN and Indo5 were generated as previously described (31). All mutations (WSN HA [D225G and S138A] and Indo5 HA [N182K, Q222L, and G224S]) were introduced into the corresponding HA plasmids via standard DpnI-mediated site-directed mutagenesis techniques (14). Recombinant WSN viruses with point (D225G) and double (D225G S138A) mutations and recombinant Indo5 viruses with point (N182K) and double (Q222L G224S) mutations were generated. The identities of these mutated viruses were confirmed by full-length HA gene sequencing.
TCID50 assay.
The 50% tissue culture infective dose (TCID50) virus titers were determined by Reed-Muench methods, as described previously (50).
Neutralization assay.
Neutralization was done according to standard protocols (50). In brief, 100 TCID50 of virus was mixed with an equal volume of serial dilutions of antibodies. Each dilution was performed in quadruplicate. The preincubated mixtures were added to MDCK cells in a 96-well plate format. Infected cells were incubated for 3 days, and the cytopathic effect was visually inspected under an inverted microscope.
Hemagglutination assay and hemagglutination inhibition.
Hemagglutination assay and hemagglutination inhibition were performed according to standard protocols (50). Turkey red blood cells (RBCs) were used in the hemagglutination assay. Both turkey and human RBCs were used in the hemagglutination inhibition assay.
Preparation of fluorescently labeled red blood cells.
Human and turkey RBCs were labeled with the lipid probe octadecyl rhodamine B (R18; Molecular Probes), as described elsewhere (3). A portion (15 μl) of R18 was added into 10 ml of 1% RBCs in PBS, and the mixture was incubated at room temperature in the dark for 15 min. Then, 30 ml of MEM supplemented with 10% fetal calf serum was added to the mixture to absorb unbound probes. The labeled cells were incubated at room temperature for another 20 min and then washed with 50 ml of chilled PBS six times. The cells were pelleted by a brief centrifugation after each wash. The cell pellet was finally resuspended in 100 ml of chilled PBS.
Hemadsorption assay.
MDCK cells were seeded as a monolayer on an optical fluorescent 96-well plate (Perkin-Elmer). Cells were infected with WSN at an MOI of 1 and incubated at 37°C for 24 h. The plate was washed with chilled PBS and incubated with 100 μl of 0.1% R18-labeled RBCs in the presence of anti-FN or other control antibodies. The reaction mixtures were incubated with gentle rocking (65 rpm) at 4°C for 30 min. After incubation, the supernatant (∼100 μl) containing unbound RBCs was transferred to a new 96-well optical fluoresce plate and mixed with an equal volume of 2% Triton X-100 in PBS. RBCs that bound to the MDCK cell monolayer were lysed by 200 μl of 1% Triton X-100 in PBS. Both reaction plates were incubated on ice for 2 min. The fluorescence signals (FS) of R18 from bound and unbound fractions (40) were then measured by a fluorescence spectrophotometer (Victor 3, Perkin-Elmer) (0.1 s measure time, 530 nm for excitation, 590 nm for emission) using 1% Triton X-100 in PBS as a control for background subtraction. The percentage of RBCs that bind to infected MDCK cells is calculated as follows: FSbound/(FSbound + FSunbound) × 100.
Membrane fusion assay.
R18-labeled RBCs and infected MDCK cells were prepared and mixed as described above. The amounts of RBCs bound to the infected cells under the tested conditions were confirmed to be similar (data not shown). After the 30-min incubation step, the wells were washed with cold PBS, followed by adding 100 μl of diluted citric acid into each of the well. The mixture was then incubated at 37°C for 1 min to induce fusion (38). Fluorescence signals from these acid-treated cells (FSquenching) were then measured by a plate reader (Victor 3 [Perkin-Elmer]; 0.1-s measure time, 530 nm for excitation, 590 nm for emission). After the first reading, the cells were washed with PBS and then lysed in 100 μl of 1% Triton X-100 in PBS on ice for 2 min. The FS values for the R18 dye (FStotal) released from the lysed cells were measured by a fluorescence spectrophotometer (Victor 3 [Perkin-Elmer]; 0.1-s measure time, 530 nm for excitation, 590 nm for emission) as described above. The percentage of acid-induced membrane fusion was calculated as follows: (FSquenching/FStotal) × 100.
Postattachment assay.
The viral entry assay was a modified version of a previously described protocol (41). In brief, MDCK cells cultured in a 24-well plate were incubated with WSN virus at an MOI of 2 at 4°C for 6 h. The cells were washed with ice-chilled PBS three times and then incubated with 0.2 to 2 μg of anti-FN antibody/ml at 4°C for 2 h. The antibody was removed, and fresh MEM with TPCK-trypsin was added to the monolayer. The cells were then incubated at 37°C for 8 h before harvesting them for immunohistochemistry analysis.
Removal of α2,3 sialic acids on MDCK cell surface by sialidase S.
MDCK cells were washed once with PBS and treated with sialidase S (ProZyme) at a final concentration of 100 mU/ml in cell culture medium at 37°C for 2 h. Treated cells were then washed once with PBS before carrying out normal viral infection procedures as described above. The specificity of the sialidase S treatment on MDCK was extensively described in previous investigations (13).
Desialylation and resialylation of turkey RBCs.
The desialylation and resialylation of turkey RBCs were preformed as previously described (18, 44). Briefly, RBCs were washed twice in PBS and reconstituted as 20% RBCs in 50 μl of PBS. A total of 50 mU of Vibrio cholerae neuraminidase (Roche)/ml was used to remove sialic acids in the presence of 10 mM CaCl2. After incubation of the mixture at 37°C for 1 h, the RBCs were washed twice with PBS and resuspended in 50 μl of 1% BSA in PBS. For resialylation, the 50 μl of desialylated RBC solution was incubated with 1 to 1.5 mM CMP-sialic acid (Sigma, catalog no. C8271) with either (i) 0.125 mU of α2,3-N-sialyltransferase (Calbiochem) at 37°C for 30 min or (ii) 0.192 mU of α2,6-N-sialyltransferase (Calbiochem) at 37°C for 1 h. The RBCs were then washed twice with PBS and resuspended to 0.5% in PBS.
RESULTS
Anti-FN antibody specifically inhibits H1, but not H5, viral propagation.
To determine the possible role of FN in the influenza viral life cycle, MDCK cells were treated with various concentrations of anti-FN polyclonal antibody immediately after virus incubation. Infected cells were harvested at 24 h postinfection and stained for viral NP protein. WSN (H1N1) NP viral protein expression was severely inhibited in cells treated with 1 to 2 μg of the antibody/ml (Fig. 1, upper panels). This inhibitory effect on WSN was also confirmed by using another monoclonal anti-FN antibody (FN12-8 [see below]). Strikingly, this inhibitory effect was not observed in cells infected with Indo5 (H5N1) virus. H5-infected cells treated with various concentrations of anti-FN antibody were shown to have NP expression levels comparable to those of mock-treated infected cells (Fig. 1, bottom panels). These observations were reconfirmed by detecting M2 viral protein (see below). In addition, the amounts of progeny viruses generated by these treated cells were determined by TCID50 assay. The virus titration results were in agreement with data from the above immunofluorescent staining. There was a >2 log units reduction in H1N1 virus titer from cultures treated with 2 μg of anti-FN antibody/ml (data not shown). In contrast, the virus titer of H5N1 virus was not affected by anti-FN antibody. Anti-GFP was also used as a control in the present study, and this antibody was found to have no inhibitory effect on viral replication (see below).
Fig 1.

Anti-FN antibody inhibits H1N1, but not H5N1 virus replication. MDCK cells were incubated with WSN (H1N1) or Indo5 (H5N1) virus for 1 h at an MOI of 1. Cells were treated with anti-FN antibodies at the indicated concentrations immediately after virus incubation. Cells were harvested at 24 h postincubation and fixed in 4% PFA in PBS for NP staining. Cells without viral infection (Mock) and antibody treatment were used as negative and positive controls, respectively. The samples were examined on a fluorescence microscope and photographed at a ×400 magnification.
The results presented above demonstrate that blocking cellular FN with an antibody can reduce the viral propagation of WSN virus. Cells infected with WSN virus at an MOI of 0.1 also gave similar observations (data not shown). In order to confirm that this is not a unique phenomenon for our prototype viruses, the A/PR/8/33 (H1N1), A/OK/483/08 (H3N2), and A/VN/3046/04 (H5N1) viruses were also tested in the present study. Anti-FN antibody was found to inhibit the above H1N1 and H3N2, but not H5N1, viruses (data not shown).
FN-specific siRNA molecules inhibits H1, but not H5, viral propagation.
To determine whether cellular FN is a decisive factor for WSN virus propagation, human FN-specific siRNA molecules were used to knock down the FN expression before virus infection. Human A549 cells were transfected with 1 to 20 nmol of FN-specific siRNAs for 24 h. A dose-dependent silencing effect on cellular FN protein expression was observed in cells treated with FN-specific siRNA molecules, whereas cells treated with scrambled or GAPDH siRNAs were found to have a wild-type FN expression level (data not shown). These siRNA-treated cells were infected with WSN or Indo5 at 24 h posttransfection, and the infected cells were harvested for immunofluorescence staining at 24 h postinfection. The silencing effect of FN siRNAs was found to affect WSN viral M2 expression (Fig. 2). The inhibition on WSN was correlated to the amount of FN siRNA used in the transfection. In cells treated with 20 nM FN-specific siRNAs, <50% of cells were positive for viral M2 protein (Fig. 2, rightmost upper panel; average of five random views). This inhibitory effect, however, was not observed in H5N1-infected cells. Irrespective of the FN-specific siRNA concentration used in the experiment, >95% of cells were positive for H5N1 M2 protein expression. As expected, >95% of cells treated with unrelated siRNA or mock transfection were M2 positive at 24 h postinfection. Overall, these results confirmed that FN might play a critical role in WSN, but not Indo5, virus life cycle.
Fig 2.

FN siRNA inhibits H1N1, but not H5N1 virus replication. A549 cells with mock transfection and cells treated with FN, scrambled, or GAPDH siRNA molecules were incubated with influenza virus at an MOI of 1. Cells were fixed at 24 h postinfection and stained for M2 protein (green) and nuclei (blue). The percentages of infected cells (i.e., M2 positive) were deduced from five random views. The samples were examined on a fluorescence microscope and photographed at a ×400 magnification.
The inhibitory effect of anti-FN antibody is related to the receptor binding specificity of HA.
Our subsequent analysis demonstrated that anti-FN antibody acts on the early phase of viral replication cycle. Since HA protein plays a key role in the early stage of viral infection, we hypothesized that cellular FN is involved in a viral event mediated by the HA of H1N1. To test this hypothesis, HA segments of prototype H1 (WSN) and H5 (Indo5) viruses were swapped by reverse-genetic (RG) techniques. Interestingly, RG-WSN×IndoHA was found to be resistant to the anti-FN antibody (Fig. 3). In contrast, RG-Indo5×WSNHA was found to be susceptible to the anti-FN antibody treatment. The HA titers determined from these viral cultures also confirmed these findings (data not shown).
Fig 3.
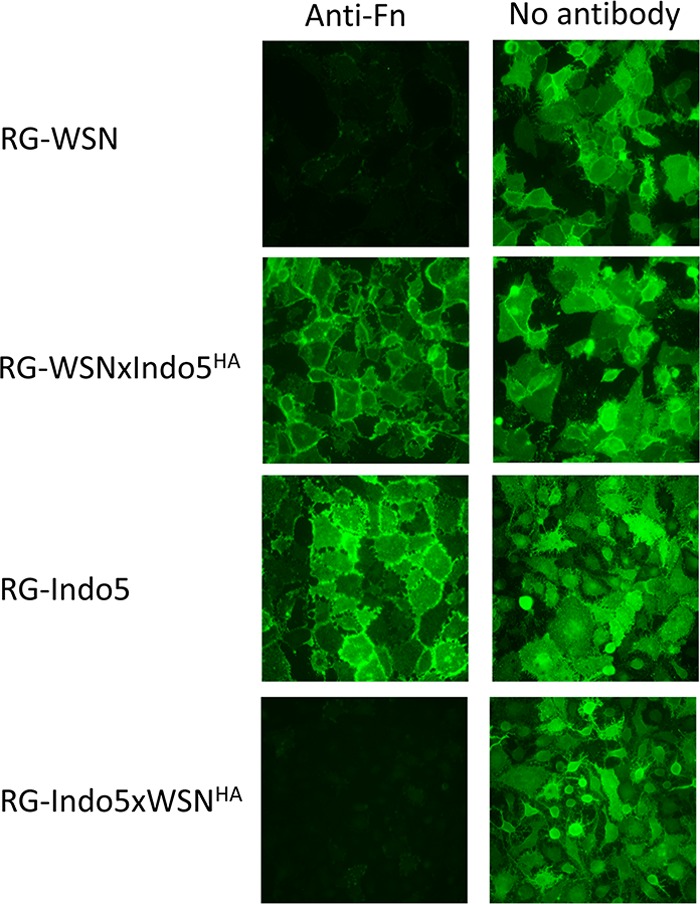
The inhibitory effect of anti-FN antibodies is determined by viral HA. MDCK cells were incubated with wild type (RG-WSN and RG-Indo5) and HA mutant (RG-WSNxIndo5HA and RG-Indo5xWSNHA) virus at an MOI of 1. Cells were immediately treated with anti-FN antibodies at a final concentration of 2 μg/ml after virus incubation (left column). Cells treated with plain virus culture medium (right column) were used as controls. The cells were harvested at 24 h postincubation and fixed in 4% PFA in PBS for M2 staining. The samples were examined on a fluorescence microscope and photographed at a ×400 magnification.
Mutations that could change (i) the binding preference of WSN from α2,6 to α2,3 and (ii) the binding preference of Indo5 from α2,3 to α2,6 were introduced into the corresponding viruses (10, 17, 36). Binding preferences of wild-type and mutant viruses were all confirmed by using resialylated RBCs with exclusively α2,3- or α2,6-linked sialic acid (Table 1). Consistent with previous findings, wild-type WSN and Indo5 were shown to have a preference for α2,6- and α2,3-linked sialic acids, respectively (10, 16). In contrast, their corresponding point and doubt mutants were all shown to have altered sialic acid binding preferences, with the WSN mutants preferring to bind to α2,3-linked sialic acid and the Indo5 mutants preferring to bind to α2,6-linked sialic acid. We also tested these viruses in MDCK cells partially depleted with α2,3-linked sialic acid by a sialidase S treatment. As expected, cells depleted with α2,3-linked sialic acid were found to be more resistant to viruses with an α2,3-linked sialic acid binding preference (data not shown).
Table 1.
Hemagglutination assay titer determined by using normal, desialylated, and resialylated 0.5% turkey RBCs
| Virus | Hemagglutination assay titera |
|||
|---|---|---|---|---|
| Untreated RBCs | Desialyated RBCs | Resialyated RBCs (2,3-N-linked SA) | Resialyated RBCs (2,6-N-linked SA) | |
| WSN | 2,048 | <2 | 64 | 512 |
| WSNHA(D225G) | 1,024 | <2 | 1,024 | 16 |
| WSNHA(S138A, D225G) | 2,048 | <2 | 256 | 32 |
| Indo5 | 1,024 | <2 | 2,048 | 32 |
| IndoHA(N182K) | 32 | <2 | 2 | 32 |
| Indo5HA(Q222L, G224S) | 16 | <2 | 2 | 16 |
Expressed as hemagglutination assay units/50 μl. SA, sialic acid.
Changing the sialic acid binding preference from a α2,6 to a α2,3 linkage made the WSNHA(D225G) and WSNHA(S138A, D225G) mutants resistant to the anti-FN antibody treatment (Fig. 4 and data not shown). Introducing a point (N182K) or a double (Q222L G224S) mutation into the HA of Indo5 resulted in an anti-FN antibody susceptible phenotype (Fig. 4 and data not shown). Our results therefore indicate that the inhibitory effect of anti-FN antibody is determined by the sialic acid binding preference of HA rather than the HA subtype.
Fig 4.

The inhibitory effect of anti-FN antibodies is determined by the receptor binding specificity of HA. Point mutants of WSN (HAD225G) and Indo5 (HAN182K) and their corresponding wild-type (WT) viruses were used to infect MDCK cells at an MOI of 1. Infected cells were treated with 2 μg of anti-FN/ml immediate after virus incubation. Cells treated with plain virus culture medium (No Ab) or medium containing anti-GFP antibodies (anti-GFP) were used as controls. Cells were harvested at 24 h postincubation and fixed in 4% PFA in PBS. Fixed cells were stained for M2 (green) and nuclei (blue). The samples were examined on a fluorescence microscope and photographed at a ×400 magnification.
Anti-FN antibody inhibits postattachment virus entry event, but not initial binding of H1N1.
To define the potential role of FN in the viral life cycle of WSN, total RNA from anti-FN antibody-treated infected MDCK cells were harvested at 4 h postinfection. Quantitative RT-PCR specific for the viral M gene was used to determine the amounts of vRNA in these infected cells. As shown in Fig. 5A, anti-FN antibody could reduce the H1N1 gene expression by >3 log units (P < 0.05). In contrast, H5-infected cells treated with or without the anti-FN antibody were found to have similar M gene expressions. These results indicated that FN might play a role in the early phase of the replication cycle of WSN virus.
Fig 5.

Anti-FN antibody inhibits early virus replication cycle. (A) M vRNA expressing in MDCK treated with anti-FN antibodies. MDCK cells were incubated with WSN or Indo5 for 1 h and were then immediately cultured in the presence or absence of anti-FN antibodies for 4 h. Total RNA samples harvested from these cells were tested by real-time RT-PCR assays for M vRNA and cellular β-actin mRNA. (B) M2 protein expression in MDCK cells treated with anti-FN antibodies at various time points. The cells were treated with antibodies before, during, and after virus incubation, as indicated. Cells pretreated with anti-FN antibodies before virus incubation were only treated with 2 μg of anti-FN/ml for 1 h immediately before virus incubation. Cells treated with anti-FN antibodies during virus incubation were incubated with anti-FN in the presence of WSN for 1 h. Except as mentioned above, no anti-FN antibody was used in other procedures in these two experimental groups. In contrast, for cells treated at 0, 1, 2, or 3 h after virus incubation, the anti-FN antibody was present in the culture medium till the end of the experiment. Infected cells were harvested at 24 h postincubation and fixed in 4% PFA in PBS. Fixed cells were stained for M2 (green) and nuclei (blue). Cells without viral infection (Mock) and infected cells with the antibody treatment (No Ab) were used as controls. The samples were examined on a fluorescence microscope and photographed at a ×200 magnification.
To further determine the effective time window of anti-FN antibody against the WSN viral infection, MDCK cells were treated with anti-FN antibody only immediately before, during, or after the viral incubation step (Fig. 5B). Cells treated with anti-FN antibody only before or during the virus incubation step were found to have M2 expressions similar to the one observed from the untreated infected cells. It should be noted that anti-FN antibody was not added in the subsequent procedures in these two experimental groups. For cells treated at different postinfection time points, the addition of anti-FN antibody was able to reduce M2 protein expression. The inhibitory effect was found to be most prominent when the antibody was added immediately after the viral incubation step. This inhibitory effect would be lost if the antibody was added at or after 3 h postincubation. In addition, coincubation H1N1 inocula and anti-FN antibody (2 μg/ml) for 1 h prior to the infection was found to have no effect on virus infectivity, indicating that the antibody itself does not directly neutralize WSN virus (data not shown).
Pretreating cellular FN by anti-FN antibody before or during the viral incubation step failed to inhibit the virus replication of WSN, suggesting that FN is not required for the initial binding between virus and cell. Two independent tests were used to test this hypothesis. First, a quantitative hemadsorption assay was used to test whether the anti-FN antibody would inhibit RBCs to bind to infected cells. Fluorescence-labeled turkey or human RBCs were incubated with WSN-infected cells in the presence of anti-FN or anti-GFP antibody. The hemadsorption activities of both control and experimental groups were found to be similar (data not shown). Second, this anti-FN polyclonal antibody and another monoclonal anti-FN antibody (FN12-8 [see below]) were tested for their abilities to inhibit HA-induced hemagglutination. None of these antibodies were found to have hemagglutination inhibition activities against H1N1 (WSN and PR8) and H5N1 (Indo5 and VN3046) viruses (data not shown). These data suggested that anti-FN does not affect the initial binding of HA to cell membrane.
A fluorescence-labeled RBC fusion assay was then used to test whether FN is involved in the postattachment event of viral entry. WSN-infected MDCK cells were first coincubated with fluorescently labeled human RBCs and anti-FN antibody. Membrane fusion was induced by a brief acidic treatment. Extensive membrane fusions were observed in the positive control (i.e., without the anti-FN antibody treatment). The number of labeled RBCs fused to the infected cells was indirectly measured by a fluorescence spectrophotometer. As shown in Fig. 6A, anti-FN antibody was found to inhibit membrane fusion in a concentration-dependent manner. In the presence of 2 μg of anti-FN antibody/ml, the fluorescence signal was reduced to 20% of the positive control. The signal from reaction treated with anti-GFP control antibody was similar to the positive control, as expected. In addition, the ability of anti-FN antibody to inhibit membrane fusion was further confirmed by a cell fusion inhibitory assay (48). In this experiment, acid-induced fusion of WSN HA-expressing HeLa cells can be inhibited by anti-FN antibody (data not shown).
Fig 6.

Anti-FN antibody inhibits postattachment events of H1N1. (A) Anti-FN antibodies inhibit membrane fusion. WSN-infected cells were incubated with various concentrations of anti-FN antibodies. Membrane fusion was induced by a brief acid wash. The amount of fused RBCs was measured, as described in Materials and Methods. Mock-infected cells (Mock), infected cells treated with 2 μg of anti-GFP/ml (anti-GFP) and infected cells with no antibody (No Ab) were used as controls in the experiment. The signal from infected cells, but without antibody treatment, was set as 100%. (B) Anti-FN can inhibit the viral entry of WSN. A viral postattachment assay was performed as described in Materials and Methods. The cells were incubated at 37°C for 8 h after antibody treatment. Fixed cells were strained for M2 (green) and DAPI (blue).
A previously described postattachment assay was used to confirm whether the anti-FN antibody is able to block viral entry even after virus attachment (41). The number of M2-expressing cells was examined at the 8-h postoperative time point. The entry of WSN virus was severely inhibited in cells treated with 2 μg of anti-FN antibody/ml (Fig. 6B, top left panel). When the antibody was used at a concentration of 1 μg/ml, the viral entry was found to be partially inhibited. Similar inhibition was not observed in cells treated with anti-GFP antibody (2 μg/ml) or anti-FN antibody at a concentration of 0.2 μg/ml. In summary, these results indicate that anti-FN antibody can inhibit the viral postattachment event but not the initial virus binding.
The entry of H1N1 virus is related to the cell-binding domain of FN but is not mediated by the β subunit of the FN receptor.
Antibodies specific for the N-terminal end (FN9-1), mid-region (FN12-8), and C-terminal end (FN12-8) of the FN protein were used to map the region important for influenza virus replication. MDCK cells were incubated with the wild-type WSN or Indo5 virus and treated with these antibodies immediately after virus incubation. FN12-8, which is specific for the cell-binding domain of FN (22), inhibited the replication of WSN virus (Fig. 7A). This inhibitory effect, however, could not be observed in infected cells treated with FN9-1 or FN12-8. The Indo5 virus, which is resistant to the polyclonal anti-FN antibody was also found to be resistant to all of these monoclonal anti-FN antibodies (Fig. 7A, left bottom half panels).
Fig 7.

The entry of H1N1 virus is related to the cell-binding domain of FN but is not mediated by the β1 subunit of FN receptor. (A) The cell-binding domain of FN is important for replication of WSN. MDCK (left panels) and A549 (right panels) cells were treated similarly to the treatments described in Fig. 3, except the anti-FN polyclonal antibody was replaced by FN-specific monoclonal antibodies (FN 1-1, FN 9-1, FN12-8, and FNH 3-8), as shown in the figure. All antibodies were used at a final concentration of 2 μg/ml. (B) The β subunit of FN receptor is not involved in inhibition caused by anti-FN antibody. MDCK (left panels) and A549 (right panels) cells were treated similar to work described in Fig. 3, except the anti-FN polyclonal antibody was replaced by different anti-β1-integrin antibodies (N20 and AIIB2) as shown. All antibodies were used at a final concentration of 2 μg/ml. Infected cells were harvested at 24 h postincubation and fixed in 4% PFA in PBS. Fixed cells were stained for M2 (green) and nuclei (blue). The samples were examined on a fluorescence microscope and photographed at a ×200 magnification.
Similar results were also observed from the A549 cell model. FN12-8 could only inhibit the expression of M2 in cells incubated with the wild-type WSN virus (Fig. 7A, right half panels). In addition, a monoclonal antibody (FNH3-8) specific for a region adjacent to the C-terminal of cell-binding domain of FN was included in the experiment. This antibody was found to have no effect on viral M2 expression. These results suggested that the cell-binding domain of FN might play a role in the postattachment event of H1N1 virus.
α5β1 integrin is a well-known FN receptor. FN uses its synergistic adhesion site and Arg-Gly-Asp motifs within the cell-binding domain to bind to this protein for cell membrane attachment (2). To investigate whether α5β1 integrin is directly involved in the postattachment event of WSN virus, two β1-integrin-specific antibodies (AIIB2 and N20) were tested individually in the MDCK and A549 cell models (21, 27). Both of these antibodies were tested at a concentration known to inhibit biological functions of α5β1 integrin in cell cultures. These two antibodies, however, failed to inhibit the replication of H1N1 or H5N1 virus (Fig. 7B). These results indicate that the inhibitory effect of anti-FN antibody might not be related to the α5β1 integrin.
DISCUSSION
In this study, we have reported that the FN might play a role in influenza virus replication. The virus replication could be inhibited by treating cells with anti-FN antibody. This effect was also confirmed by using a monoclonal antibody specific for the cell-binding domain of FN. Knocking down the FN expression by siRNA could also inhibit influenza virus replication, indicating that the inhibition is primarily due to the reduced endogenous FN expression. Our results demonstrated that FN is not involved in the early attachment event of the virus to host cells, suggesting the virus attaches to other sites initially. Our subsequent work showed that anti-FN antibody might block a postattachment event. These findings agree with the observation that a potent inhibitory effect of anti-FN antibody could only be observed when the antibody was added immediately after the virus incubation step. Such an effect, however, could not be observed in cells treated with only the antibody before or during the virus incubation step.
One of our interesting findings is that anti-FN antibodies or FN siRNA can only inhibit the replication of wild-type WSN or PR8 virus. This inhibition was not observed in cells infected with wild-type Indo5 or VN3046 virus. Changing the binding preference of these HA proteins from α2,6-linked sialic acid to α2,3-linked sialic acid could convert the virus from a susceptible to a resistant phenotype and vice versa. It is well known that the receptor binding specificity of influenza virus is modulated by multiple amino acid residues in the HA molecule. Mutating some of these positions may result in major or subtle differences in receptor binding specificity (47). The mutations tested in our studies were previously shown to be critical in modulating sialic acid binding preferences (10, 17, 36). Although our studied H1 and H5 viruses or their mutants do not have an exclusive α2,3 or α2,6 binding specificity (Table 1), changing the receptor binding preferences of the studies viruses was found to affect their phenotypes in our study. Depleting cell surface α2,3-linked sialic acids by using sialidase S had no obvious effect on the infection of wild-type WSN and PR8 (data not shown), suggesting that the α2,6-linked sialic acid binding preference is critical for their infections. Furthermore, a human H3 virus that has an exclusive 2,6 binding (19) was shown to be susceptible to anti-FN antibodies. Overall, these results indicated that the receptor binding preference of HA is a major determinant of the susceptibility of influenza viruses to anti-FN antibody but that this does not apply to the HA subtype itself. Further systematic characterization of viruses with well-defined or highly restricted binding specificities to different sialylated glycans in our model may help to reveal the inhibitory spectrum of anti-FN antibody against influenza A virus.
FN contains 4 to 9% carbohydrate, mostly in N-linked oligosaccharide chains (49). However, as demonstrated by our experiments, FN is unlikely to be the major cellular protein for initial influenza virus attachment. In addition, it should be noted that the sialic acid groups of cellular FN exist in a α2,3 linkage form (15, 42). If influenza virus can directly bind to the sialic acid groups of cellular FN, it is surprising that anti-FN antibody can only inhibit viruses with a α2,6-linked sialic acid binding preference. Hence, we speculate that influenza virus does not directly bind to FN for viral entry. However, we cannot exclude the possibility that influenza viruses with a α2,6-linked sialic acid binding preference might bind to FN via an unknown mechanism. Apart from binding to α2,3- and α2,6-linked sialic acid, influenza viruses are reported to bind to other sialic acid-containing glycans (47, 52). Moreover, recent studies have demonstrated that the attachment and entry of influenza virus can occur independently of sialic acid (33).
How does FN affect viral postattachment events? Real-time imaging analysis of influenza virus infection revealed that the viral entry can be divided into at least three stages (28). In stage 1, the attached virus moves slowly at the cell periphery for an extended period of time. This is probably one of the bottlenecks for viral entry. In fact, a great portion of viral particles (∼70%) are still found to attach to the cell surface after the postincubation period. After the stage 1 process, the attached virion then moves rapidly toward the perinuclear region of infected cell (stage 2). This is followed by an intermittent, often bidirectional virion movement in the perinuclear region (stage 3). Recently, it has been observed that the endocytosed viral particles in the perinuclear region finally move to microtubule organization center for membrane fusion after the stage 3 movement (32). FN is one of the major components of extracellular matrix and is an important regulator for extracellular matrix remodeling. The protein has a high turnover rate, and it is endocytosed for intracellular degradation. Inhibiting the cleavage of large extracellular fibronectin fibrils in the extracellular matrix can reduce the endocytosis of FN (45). In addition, FN is also known to modulate the internalization/endocytosis of cellular receptors, such as insulin receptor and integrins. It is possible that the anti-FN antibody can inhibit the extracellular matrix remodeling, thereby inhibiting the endocytosis of influenza virus. We have demonstrated here that anti-FN antibody can inhibit HA-mediated membrane fusion. It is also possible that the internalized anti-FN antibody could prevent the membrane fusion induced by viral HA. On the other hand, it should be noted that some influenza viral particles can also be rapidly internalized after the initial attachment (28). At the time of the antibody treatment in our study, it is likely that a fraction of the attached viruses might have already been internalized. FN located at the extracellular matrix at cell surface is known to affect the normal trafficking of endocytosed vesicles. For example, FN at the extracellular matrix is known to modulate the fate of endocytosed surface receptor and lipid rafts (4, 7). Thus, the anti-FN might also have some effects on the trafficking of endocytosed influenza viruses.
Influenza virus can enter host cells through various pathways. The clathrin-mediated endocytic pathway is believed to be the major pathway for influenza virus entry. Other pathways, such as caveolin-dependent and clathrin- and caveolin-independent viral entry, are also reported to be relevant in viral entry (29, 35). However, the details of influenza virus entry are still far from fully understood. Recent studies have further demonstrated that influenza virus can enter host cells by other novel mechanisms, such as macropinocytosis and DC-SIGN-mediated endocytosis (12, 34). It should be noted that many of these previous studies used human (H1 and H3) influenza viruses as models. Moreover, there was no direct comparison between the entries of human and avian influenza viruses. It is possible that the sialic acid binding preference of HA can modulate the preferences of these viral entry pathways. Our data here at least demonstrate that viruses with a α2,6-linkage sialic acid preference can enter host cells via a pathway mediated by FN. Further real-time microscopic analysis of the virus movement on the cell surface and viral endocytosis in the presence of anti-FN antibody can help to address this issue. In addition, our model might be useful for studying HA-mediated membrane fusion.
Among all of the tested monoclonal anti-FN antibodies, only FN12-8 can inhibit virus replication. This antibody specifically binds to the cell-binding domain of FN. This domain is well known to bind to α5β1 integrin, a major FN receptor for regulating the endocytosis of FN. α5β1 integrin is reported to facilitate the entry of Ebola virus and parvovirus (43, 51). Blocking the biological function of α5β1 integrin by anti-β1 antibodies, however, was found to have no effect on influenza virus replication in our studies. These data suggest that the interaction between α5β1 integrin and FN is not essential for virus infection. FN is known to bind to other integrins (e.g., αVβ3) and extracellular matrix components to facilitate virus entry (30).
In conclusion, we have discovered that FN is an essential component for the entry of influenza A viruses with a α2,6-linkage sialic acid binding preference. Our results demonstrate that influenza viruses with different sialic acid binding preferences might affect the viral entry pathway. Further investigation on this virus-host interaction might help reveal details of postattachment events of influenza virus.
ACKNOWLEDGMENTS
This project is supported by National Institutes of Health (NIAID contract HHSN266200700005C) and Area of Excellence Scheme of the University Grants Committee (AoE/M-12/06).
We thank Cynthia S.W. Leung for help during the initial phase of the study.
Footnotes
Published ahead of print 25 July 2012
REFERENCES
- 1. Alderete JF, Benchimol M, Lehker MW, Crouch ML. 2002. The complex fibronectin-Trichomonas vaginalis interactions and trichomonosis. Parasitol. Int. 51:285–292 [DOI] [PubMed] [Google Scholar]
- 2. Aota S, Nomizu M, Yamada KM. 1994. The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J. Biol. Chem. 269:24756–24761 [PubMed] [Google Scholar]
- 3. Bagai S, Lamb RA. 1995. Quantitative measurement of paramyxovirus fusion: differences in requirements of glycoproteins between simian virus 5 and human parainfluenza virus 3 or Newcastle disease virus. J. Virol. 69:6712–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Balasubramanian N, Scott DW, Castle JD, Casanova JE, Schwartz MA. 2007. Arf6 and microtubules in adhesion-dependent trafficking of lipid rafts. Nat. Cell Biol. 9:1381–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bearzotti M, et al. 1999. Fish rhabdovirus cell entry is mediated by fibronectin. J. Virol. 73:7703–7709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beer C, Pedersen L. 2007. Matrix fibronectin binds gammaretrovirus and assists in entry: new light on viral infections. J. Virol. 81:8247–8257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boura-Halfon S, Voliovitch H, Feinstein R, Paz K, Zick Y. 2003. Extracellular matrix proteins modulate endocytosis of the insulin receptor. J. Biol. Chem. 278:16397–16404 [DOI] [PubMed] [Google Scholar]
- 8. Cheung CY, et al. 2002. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360:1831–1837 [DOI] [PubMed] [Google Scholar]
- 9. Chu VC, Whittaker GR. 2004. Influenza virus entry and infection require host cell N-linked glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 101:18153–18158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chutinimitkul S, et al. 2010. In vitro assessment of attachment pattern and replication efficiency of H5N1 influenza A viruses with altered receptor specificity. J. Virol. 84:6825–6833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Vries E, et al. 2012. Influenza A virus entry into cells lacking sialylated N-glycans. Proc. Natl. Acad. Sci. U. S. A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Vries E, et al. 2011. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 7:e1001329 doi:10.1371/journal.ppat.1001329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dugan AS, Eash S, Atwood WJ. 2005. An N-linked glycoprotein with α(2,3)-linked sialic acid is a receptor for BK virus. J. Virol. 79:14442–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fisher CL, Pei GK. 1997. Modification of a PCR-based site-directed mutagenesis method. Biotechniques 23:570–574 [DOI] [PubMed] [Google Scholar]
- 15. Fukuda M, Levery SB, Hakomori S. 1982. Carbohydrate structure of hamster plasma fibronectin: evidence for chemical diversity between cellular and plasma fibronectins. J. Biol. Chem. 257:6856–6860 [PubMed] [Google Scholar]
- 16. Glaser L, Conenello G, Paulson J, Palese P. 2007. Effective replication of human influenza viruses in mice lacking a major α2,6-sialyltransferase. Virus Res. 126:9–18 [DOI] [PubMed] [Google Scholar]
- 17. Glaser L, et al. 2005. A single amino acid substitution in 1918 influenza virus hemagglutinin changes receptor binding specificity. J. Virol. 79:11533–11536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glaser L, et al. 2006. Sequence analysis and receptor specificity of the hemagglutinin of a recent influenza H2N2 virus isolated from chicken in North America. Glycoconj. J. 23:93–99 [DOI] [PubMed] [Google Scholar]
- 19. Gulati S, Smith DF, Air GM. 2009. Deletions of neuraminidase and resistance to oseltamivir may be a consequence of restricted receptor specificity in recent H3N2 influenza viruses. Virol. J. 6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo Y, Martinez-Williams C, Yellowley CE, Donahue HJ, Rannels DE. 2001. Connexin expression by alveolar epithelial cells is regulated by extracellular matrix. Am. J. Physiol. Lung Cell. Mol. Physiol. 280:L191–L202 [DOI] [PubMed] [Google Scholar]
- 21. Hall DE, et al. 1990. The α1/β1 and α6/β1 integrin heterodimers mediate cell attachment to distinct sites on laminin. J. Cell Biol. 110:2175–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Henderson B, Nair S, Pallas J, Williams MA. 2011. Fibronectin: a multidomain host adhesin targeted by bacterial fibronectin-binding proteins. FEMS Microbiol. Rev. 35:147–200 [DOI] [PubMed] [Google Scholar]
- 23. Herard AL, et al. 1996. Fibronectin and its α5β1-integrin receptor are involved in the wound-repair process of airway epithelium. Am. J. Physiol. 271:L726–L733 [DOI] [PubMed] [Google Scholar]
- 24. Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. U. S. A. 97:6108–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Imai M, Kawaoka Y. 2012. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr. Opin. Virol. 2:160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klotz SA. 1992. Fungal adherence to the vascular compartment: a critical step in the pathogenesis of disseminated candidiasis. Clin. Infect. Dis. 14:340–347 [DOI] [PubMed] [Google Scholar]
- 27. Kojima T, et al. 2003. Effect of gelatins on human cancer cells in vitro. Cancer Biother. Radiopharm. 18:147–155 [DOI] [PubMed] [Google Scholar]
- 28. Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. 2003. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. U. S. A. 100:9280–9285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lakadamyali M, Rust MJ, Zhuang X. 2004. Endocytosis of influenza viruses. Microbes Infect. 6:929–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leiss M, Beckmann K, Giros A, Costell M, Fassler R. 2008. The role of integrin binding sites in fibronectin matrix assembly in vivo. Curr. Opin. Cell Biol. 20:502–507 [DOI] [PubMed] [Google Scholar]
- 31. Li OT, et al. 2009. Full factorial analysis of mammalian and avian influenza polymerase subunits suggests a role of an efficient polymerase for virus adaptation. PLoS One 4:e5658 doi:10.1371/journal.pone.0005658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu SL, et al. 2012. Effectively and efficiently dissecting the infection of influenza virus by quantum-dot-based single-particle tracking. ACS Nano. 6:141–150 [DOI] [PubMed] [Google Scholar]
- 33. Londrigan SL, Tate MD, Brooks AG, Reading PC. 2011. Cell surface receptors on macrophages and dendritic cells for attachment and entry of influenza virus. J. Leukoc. Biol. 92:97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Londrigan SL, et al. 2011. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J. Virol. 85:2990–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luo M. 2012. Influenza virus entry. Adv. Exp. Med. Biol. 726:201–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matrosovich M, et al. 2000. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J. Virol. 74:8502–8512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Menzies BE. 2003. The role of fibronectin binding proteins in the pathogenesis of Staphylococcus aureus infections. Curr. Opin. Infect. Dis. 16:225–229 [DOI] [PubMed] [Google Scholar]
- 38. Morris SJ, Sarkar DP, White JM, Blumenthal R. 1989. Kinetics of pH-dependent fusion between 3T3 fibroblasts expressing influenza hemagglutinin and red blood cells: measurement by dequenching of fluorescence. J. Biol. Chem. 264:3972–3978 [PubMed] [Google Scholar]
- 39. Nicholls JM, Chan RW, Russell RJ, Air GM, Peiris JS. 2008. Evolving complexities of influenza virus and its receptors. Trends Microbiol. 16:149–157 [DOI] [PubMed] [Google Scholar]
- 40. Nunes-Correia I, Nir S, Pedroso de Lima MC. 2003. Kinetics of influenza virus fusion with the endosomal and plasma membranes of cultured cells: effect of temperature. J. Membr. Biol. 195:21–26 [DOI] [PubMed] [Google Scholar]
- 41. Oh HL, et al. 2010. An antibody against a novel and conserved epitope in the hemagglutinin 1 subunit neutralizes numerous H5N1 influenza viruses. J. Virol. 84:8275–8286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saren A, Virkola R, Hacker J, Korhonen TK. 1999. The cellular form of human fibronectin as an adhesion target for the S fimbriae of meningitis-associated Escherichia coli. Infect. Immun. 67:2671–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schornberg KL, et al. 2009. α5β1-integrin controls Ebola virus entry by regulating endosomal cathepsins. Proc. Natl. Acad. Sci. U. S. A. 106:8003–8008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shen S, Bryant KD, Brown SM, Randell SH, Asokan A. 2011. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J. Biol. Chem. 286:13532–13540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi F, Sottile J. 2011. MT1-MMP regulates the turnover and endocytosis of extracellular matrix fibronectin. J. Cell Sci. 124:4039–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Singh P, Carraher C, Schwarzbauer JE. 2010. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 26:397–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stevens J, et al. 2006. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J. Mol. Biol. 355:1143–1155 [DOI] [PubMed] [Google Scholar]
- 48. Sui J, et al. 2009. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16:265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tajiri M, Yoshida S, Wada Y. 2005. Differential analysis of site-specific glycans on plasma and cellular fibronectins: application of a hydrophilic affinity method for glycopeptide enrichment. Glycobiology 15:1332–1340 [DOI] [PubMed] [Google Scholar]
- 50. Webster RG, Cox N, Stöhr K. 2002. WHO manual on animal influenza diagnosis and surveillance. World Health Organization, Geneva, Switzerland [Google Scholar]
- 51. Weigel-Kelley KA, Yoder MC, Srivastava A. 2003. α5β1 integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of β1 integrin for viral entry. Blood 102:3927–3933 [DOI] [PubMed] [Google Scholar]
- 52. Wu W, Air GM. 2004. Binding of influenza viruses to sialic acids: reassortant viruses with A/NWS/33 hemagglutinin bind to α2,8-linked sialic acid. Virology 325:340–350 [DOI] [PubMed] [Google Scholar]
- 53. Yang J, et al. 2006. Fibronectin is essential for hepatitis B virus propagation in vitro: may be a potential cellular target? Biochem. Biophys. Res. Commun. 344:757–764 [DOI] [PubMed] [Google Scholar]