

Abstract
Although picornavirus RNA genomes contain a 3′-terminal poly(A) tract that is critical for their replication, the impact of encephalomyocarditis virus (EMCV) infection on the host poly(A)-binding protein (PABP) remains unknown. Here, we establish that EMCV infection stimulates site-specific PABP proteolysis, resulting in accumulation of a 45-kDa N-terminal PABP fragment in virus-infected cells. Expression of a functional EMCV 3C proteinase was necessary and sufficient to stimulate PABP cleavage in uninfected cells, and bacterially expressed 3C cleaved recombinant PABP in vitro in the absence of any virus-encoded or eukaryotic cellular cofactors. N-terminal sequencing of the resulting C-terminal PABP fragment identified a 3Cpro cleavage site on PABP between amino acids Q437 and G438, severing the C-terminal protein-interacting domain from the N-terminal RNA binding fragment. Single amino acid substitution mutants with changes at Q437 were resistant to 3Cpro cleavage in vitro and in vivo, validating that this is the sole detectable PABP cleavage site. Finally, while ongoing protein synthesis was not detectably altered in EMCV-infected cells expressing a cleavage-resistant PABP variant, viral RNA synthesis and infectious virus production were both reduced. Together, these results establish that the EMCV 3C proteinase mediates site-specific PABP cleavage and demonstrate that PABP cleavage by 3C regulates EMCV replication.
INTRODUCTION
Picornaviruses contain a poly(A) tail at the 3′ end of their plus-strand RNA genomes. This genetic element plays an important role in viral biology, as its removal impairs poliovirus (PV) RNA infectivity (45). A poly(A) tract of minimal length is likewise required for encephalomyocarditis virus (EMCV) replication (13, 20, 21) and binding of the virus RNA-dependent RNA polymerase 3D (6, 7). In their eukaryotic hosts, the 3′ poly(A) tail is recognized by the cellular poly(A) binding protein (PABP), a multifunctional protein involved in mRNA metabolism, stability, and translation (33). However, the role of PABP in picornavirus biology is incompletely understood.
Upon introduction into cells, plus-strand picornavirus RNA directly serves as mRNA and is translated to produce the virus-encoded polyprotein (37). A cis-acting internal ribosome entry site (IRES) element in the 5′ untranslated region (UTR) enables cap-independent recruitment of a cellular 40S ribosome. Not only does the IRES allow ribosome recruitment without the canonical m7GTP cap ubiquitously present on cellular mRNAs, but also it provides a mechanism to sustain ongoing viral protein synthesis while translation initiation factors required for cap-dependent translation are selectively inactivated, effectively suppressing host mRNA translation in infected cells (19, 39, 49). This was first reported for poliovirus-infected cells, where the virus-encoded 2A proteinase specifically cleaved eIF4G, a key component of the multisubunit cap-binding complex, severing the eIF4E- and PABP-bound N terminus from the eIF4A-bound C-terminal fragment (9, 14, 25). The resulting C-terminal eIF4G fragment binds the IRES and retains the capacity to interact with eIF3-bound 40S subunits and eIF4A, supporting viral mRNA translation. Subsequently, enteroviruses, caliciviruses, aphthoviruses, and retroviruses have been shown to cleave eIF4G, and this has been correlated with their ability to shut off host protein synthesis (2, 5, 8, 17, 24, 29, 44).
In addition to eIF4G cleavage by their 2A proteinase, poliovirus (2A and 3C), coxsackievirus (3C), hepatitis A virus (HAV), and calicivirus (3C) proteinases also specifically cleave PABP (22, 23, 26, 27, 50). While different viral proteinases cleave PABP at discrete sites, most lie within the C-terminal linker domain separating the N-terminal RNA recognition motifs (RRMs) and eIF4G-binding site from the PABP C-terminal fragment. Along with mediating PABP oligomerization, the C-terminal fragment is thought to play a role in ribosome recycling by interacting with eRF3 and eIF4B to shuttle terminating ribosomes to the mRNA 5′ end. Bisecting PABP has therefore been suggested to interfere with the ribosome's ability to retranslate mRNAs without reengaging the initiation complex (32). Furthermore, virus-mediated site-specific PABP cleavage can also inhibit viral mRNA translation (3, 50). Suppressing translation facilitates the switch to RNA genome replication, regulating competing macromolecular processes on the same pool of mRNA template molecules (10).
Like that with other picornavirus family members, infection with the cardiovirus EMCV potently impairs host cell protein synthesis (30). This involves the virus-encoded L and 2A proteins, which differ from their enterovirus and aphthovirus counterparts in that they are not proteinases (43). While the EMCV L protein inhibits nucleocytoplasmic protein transport by associating with and impairing Ran-GTPase activity (36), EMCV 2A accumulates in nuclei and sequesters the cap-binding protein, eIF4E, away from cellular mRNAs (1, 15, 16). In addition, while eIF4G cleavage has not been detected in EMCV-infected cells, activation of the cellular cap-dependent translational repressor 4E-BP1 has been reported (12, 34). This mechanism of cellular translational shutoff is unique among picornavirus family members, as it does not rely on proteolysis of cellular translation factors. However, the impact of EMCV infection on PABP has not been evaluated. In this study, we investigated how the cellular PABP responds to EMCV infection of primary human cells. We show that full-length PABP is cleaved in EMCV-infected cells, resulting in accumulation of a 45-kDa N-terminal fragment. The virus 3C-encoded protease is necessary and sufficient to stimulate PABP cleavage in uninfected cells and site-specific PABP cleavage reconstituted using bacterially expressed 3C and PABP in vitro. 3C cleaves PABP specifically between glutamine 437 and glycine 438. While infection of cells expressing a cleavage-resistant Q473N PABP variant has no detectable effect on viral protein synthesis or host cell shutoff, it impaired EMCV replication and reduced viral RNA synthesis. Thus, unlike enteroviruses that cleave both eIF4G and PABP, the cardiovirus EMCV selectively cleaves PABP in infected cells, and this is required for wild-type levels of viral replication.
MATERIALS AND METHODS
Cells and antibodies.
All cells were propagated in 5% CO2 with Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM glutamine, 50 U of penicillin per milliliter, and 50 μg of streptomycin per milliliter, plus serum as specified below. Primary normal human dermal fibroblasts (NHDFs; Clonetics, Walkersville, MD) were cultivated in 5% fetal bovine serum (FBS) and growth arrested in 0.2% serum as described previously (48); HEK293 and HeLa cells (ATCC) were cultivated using 10% FBS. Rabbit polyclonal anti-PABP1 serum was a gift from S. Morley (University of Sussex, United Kingdom). A monoclonal antibody raised against mengovirus/EMCV 3C protease has been described previously (1). The following commercially available antibodies were purchased from the suppliers listed in parentheses: anti-eIF4G (BD Biosciences; catalog number E46520), anti-eIF4E (BD Biosciences; catalog number E27620), anti-4E-BP1 (Bethyl Labs; catalog number A300-501A), anti-Flag (Sigma; catalog number F3165), anti-6×His (Roche; catalog number 04905318001), anti-ribosomal protein S6 (anti-rpS6) (Cell Signaling; catalog number 2217), and RhoGDI (Santa Cruz Biotechnology; catalog number SC-33201).
Plasmids.
pF_3C is an EMCV pIRES-based dicistronic plasmid (Novagen) in which the first cistron encodes firefly luciferase and the second cistron encodes the EMCV 3C protease. pF_3C-H46A-C159A expresses a catalytically inactive 3C variant containing two single amino acid substitutions that inactivate the 3C protease active site. pET11-3C has been described previously (18). pET28a-(His)PABP expresses an N-terminally His-tagged PABPC1 and was a gift from R. Schneider, New York University School of Medicine. To generate a C-terminally His-tagged PABPC1, pET28a-(His)PABP was digested with NdeI and BamHI to excise the N-terminally His-tagged PABP insert. C-terminally His-tagged PABPC1 coding sequences were subsequently inserted into the pET28a vector to produce pET28a-(His)PABP. The C-terminally His-tagged PABP cassette was amplified by PCR from pET28a-(His)PABP using the following primers: 5′-CTTCTTCATATGCTCGAGTCTCCAGAGTCAC-3′ and 5′-GAGGATCCTTAGTGATGATGATGATGATGAACAGTTGGAACACCCGG-3′. To construct pCG-(Flag)PABP, PABP cDNA was PCR amplified from pET28a-(His)PABP using the following primers: 5′-GCTCTAGAAACCCCAGTGCCCCCAGC-3′ and5′-GCGGATCCTTAAACAGTTGGAACACC-3′. The resulting fragment was digested with XbaI and BamHI and cloned into the pCG expression vector.
For constructing pBABE-PABP, PABP cDNA was amplified from pET28a-(His)PABP using the following primers: 5′-CGGGATCCGCCACCATGAACCCCAGTGCCCCC-3′ and 5′-GCGGTCGACTTAAACAGTTGGAACACC-3′. The resulting amplification products were digested with BamHI and SalI and ligated into the pBABE-puromycin retrovirus expression vector that had been cleaved using the same enzymes.
3C protease cleavage site variants of PABP were generated using the Quickchange II mutagenesis kit (Stratagene) using the following primers: for Q437E, 5′-CGCTGGACTGCTGAGGGTGCCAGACCTCATCC-3′; for Q437A, 5′-CCTCGCTGGACTGCTGCAGGTGCCAGACCTCATCC-3′; and for Q437N, 5′-CCTCGCTGGACTGCTAACGGTGCCAGACCTCATCC-3′. All PCR products were validated by DNA sequencing.
Transfection and infection.
HEK293 cells were seeded at 5 × 105 cells per well in 6-well plates approximately 16 h prior to transfection. Cells were transfected with 4 μg of DNA per well using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Forty-eight hours after transfection, cell lysates were collected for analysis. For multicycle growth experiments, HeLa cells were seeded at 1.5 × 105 cells per well in a 6-well plate. Cells were transfected the following day with 8 μg of DNA per well using the calcium phosphate technique in 12.5 mM CaCl2 and 0.1× HEPES buffered saline (HBS). After 16 h, the transfection medium was removed and the cells infected with EMCV (multiplicity of infection [MOI] = 10−3). Infections were allowed to proceed for approximately 36 h. For single-cycle growth experiments, cells were infected with EMCV (MOI = 10) 48 h after transfection. High-multiplicity infections were performed in 0.4 ml per well for 1 h. After removal of the viral inoculum, the monolayer was rinsed three times with medium to remove excess unbound virus and the infection allowed to proceed for 5 h. After two freeze-thaw cycles, the amount of infectious virus was quantified by plaque assay in Vero cells. To generate NHDFs stably expressing wild-type PABP, PABP (Q437N), or enhanced green fluorescent protein (EGFP), a retrovirus containing each gene of interest was generated using the Phoenix retrovirus packaging system (35). After infection of NHDFs with retrovirus, transduced cells were selected using 2 μg/ml puromycin until all cells in the control sample (uninfected NHDFs) were killed.
Analysis of protein synthesis in EMCV-infected cells.
One hour prior to the time points indicated on the figures, EMCV-infected cells were overlaid with DMEM lacking methionine and cysteine but containing 70 μCi/ml 35S Express (Perkin-Elmer) and were metabolically labeled for 1 h at 37°C. Total protein was collected in SDS-containing sample buffer, boiled, and fractionated by SDS-PAGE. The fixed, dried gel was subsequently exposed to X-ray film.
Expression of recombinant protein.
Overnight cultures of Escherichia coli BL21(DE3) containing pET28a-(His)PABP were diluted into fresh LB medium and allowed to grow at 37°C until reaching an optical density at 600 nm (OD600) of 0.6. Cultures were subsequently induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h at 37°C. pET28a-(His)PABP was induced at room temperature for 16 h. Induced cells were spun down and flash frozen. Frozen pellets were thawed and resuspended in buffer containing 20 mM HEPES-KOH (pH 7.4), 200 mM NaCl, 1 mM EDTA, and 5% glycerol and sonicated on ice at 58% for 1 min in 5-s intervals followed by a 5-s rest using a Branson Digital Sonifier 250. The lysate was adjusted to 0.1% Triton X-100 (final concentration), rocked at 4°C for 15 min, clarified by centrifugation at 13,000 rpm in a Sorvall SS34 rotor for 5 min at 4°C, and adjusted to 1 mM dithiothreitol (DTT).
Cultures of E. coli BL21(DE3)pLysS (Invitrogen) containing pET11c-3Cpro were grown at room temperature until reaching an OD600 of 0.8 and subsequently induced with 1 mM IPTG for 4 h at room temperature. Induced cells were spun down and flash frozen. Pellets were thawed, resuspended in buffer A (18), and then sonicated at 40% for 1.5 min in 5-s intervals followed by a 5-s rest. The lysate was adjusted to 0.1% Triton X-100 and 1 mM DTT (final concentration), nutated at 4°C for 15 min, and clarified by centrifugation at 13,000 rpm in a Sorvall SS34 rotor for 15 min. All clarified bacterial lysates were snap-frozen and stored at −80°C until use.
In vitro cleavage reactions.
Two micrograms of His-PABP-containing soluble lysate (typically 1 μl of a 2-mg/ml extract) was added to various amounts of EMCV 3C proteinase-containing soluble lysate (total protein concentration of 2.0 mg/ml) or lysate lacking recombinant proteins prepared from bacteria containing the plasmid pUC19 in a manner identical to that for the 3C-containing lysate in a volume of 10 μl on ice. The total reaction volume was subsequently raised to 30 μl using buffer A, and the cleavage reaction was carried out at 30°C for 5 min. Reactions were terminated by heating to 70°C for 10 min. After addition of SDS-containing sample buffer and boiling, reaction products were fractionated by SDS-PAGE and analyzed by immunoblotting.
Purification of PABP His-tagged C-terminal cleavage fragment for N-terminal protein sequencing.
Soluble bacterial lysate containing recombinant C-terminally His-tagged PABP was bound to nickel-nitrilotriacetic acid (Ni-NTA) agarose beads (Qiagen) and incubated with a soluble bacterial lysate containing recombinant 3Cpro or a heat-inactivated, enzymatically inactive 3Cpro control for 30 min at 30°C. The beads were washed twice with excess 50 mM NaH2PO4 (pH 8.0), 1 M NaCl. 20 mM imidazole, 10% glycerol, and 1 mM DTT to remove soluble N-terminal PABP reaction products, while the His-tagged C-terminal PABP fragment remained bound to the beads. After the beads were boiled in SDS sample buffer, the bound proteins were fractionated by SDS-PAGE and visualized by staining with Coomassie blue R250. The ∼26-kDa PABP C-terminal cleavage product present in reactions that contained active 3Cpro, but not heat-inactivated 3Cpro, was excised from the gel, and the N-terminal sequence was determined by the Protein Core Facility at Columbia University Medical Center (New York, NY).
Polysome fractionation and analysis of associated proteins.
HeLa cells (106) seeded in 10-cm dishes were transfected with 40 μg of plasmid DNA using the calcium phosphate method. After 16 h, the transfection medium was removed and the cells were infected with EMCV (MOI = 10). At 5 h postinfection (hpi), cell lysates were prepared and polyribosomes isolated by sucrose gradient sedimentation as described previously (38). Following fractionation of the gradient, protein in individual fractions was precipitated using trichloroacetic acid and analyzed by immunoblotting.
Viral RNA synthesis.
HeLa cells (1.5 × 105) were seeded in 6-well dishes, transfected with plasmid DNA, and infected 48 h later with EMCV (MOI = 10). After 1 h, the inoculum in each well was removed and replaced with 1.5 ml of medium containing actinomycin D (1 μg/ml) and 6.67 μCi/ml [5,6 3H]uridine (35 to 50 Ci/mmol, catalog number NET380250UC; Perkin Elmer). Cells were collected 4 h later and lysed with 1% Triton X-100 in PBS, and tricholoroacetic acid was added to a final concentration of 20%. Acid-insoluble material was collected onto GF/C filters (Whatman) and the amount of 3H incorporated quantified by liquid scintillation counting.
RESULTS
Specific proteolytic cleavage of the cellular PABP in EMCV-infected cells.
We first sought to determine if cellular PABP levels responded to EMCV infection and to correlate any observed changes with the onset of viral protein synthesis or host cell shutoff. Since many established cell lines have deregulated or altered translational control pathways (42), these experiments were performed using primary cells. To evaluate the impact of EMCV infection on cellular protein synthesis, primary fibroblasts were infected with EMCV and metabolically labeled at various times postinfection (indicated below). Cell-free lysates were prepared at each time point, total protein was fractionated by SDS-PAGE, and newly synthesized proteins were visualized by autoradiography. In agreement with previous studies using established cell lines, infection with EMCV induced a potent suppression of new host protein synthesis that initiated between 3 and 4 hpi and became increasingly pronounced with time. In addition, this host cell shutoff is concurrent with the increased rate of viral protein synthesis (Fig. 1A). Samples from each time point were subsequently analyzed by immunoblotting using antisera specific for either eIF4G or PABP. While steady-state full-length eIF4G levels remained relatively constant through infection, the abundance of the ∼70-kDa band corresponding to the full-length PABP decreased (Fig. 1B). This was not due to a general decrease in cellular protein abundance, as steady-state tubulin levels, like eIF4G, were not observed to detectably change over time (Fig. 1B). Moreover, a faster-migrating, anti-PABP immunoreactive band was detected and began to accumulate starting around 4 hpi. Accumulation of this ∼45-kDa PABP-related protein occurred with a concomitant reduction in levels of the full-length 70-kDa protein (Fig. 1B). The accumulation of a discrete 45-kDa PABP fragment and its insensitivity to the general proteasome inhibitor MG132 (data not shown) are consistent with site-specific cleavage mediated by an MG132-insensitive protease.
Fig 1.
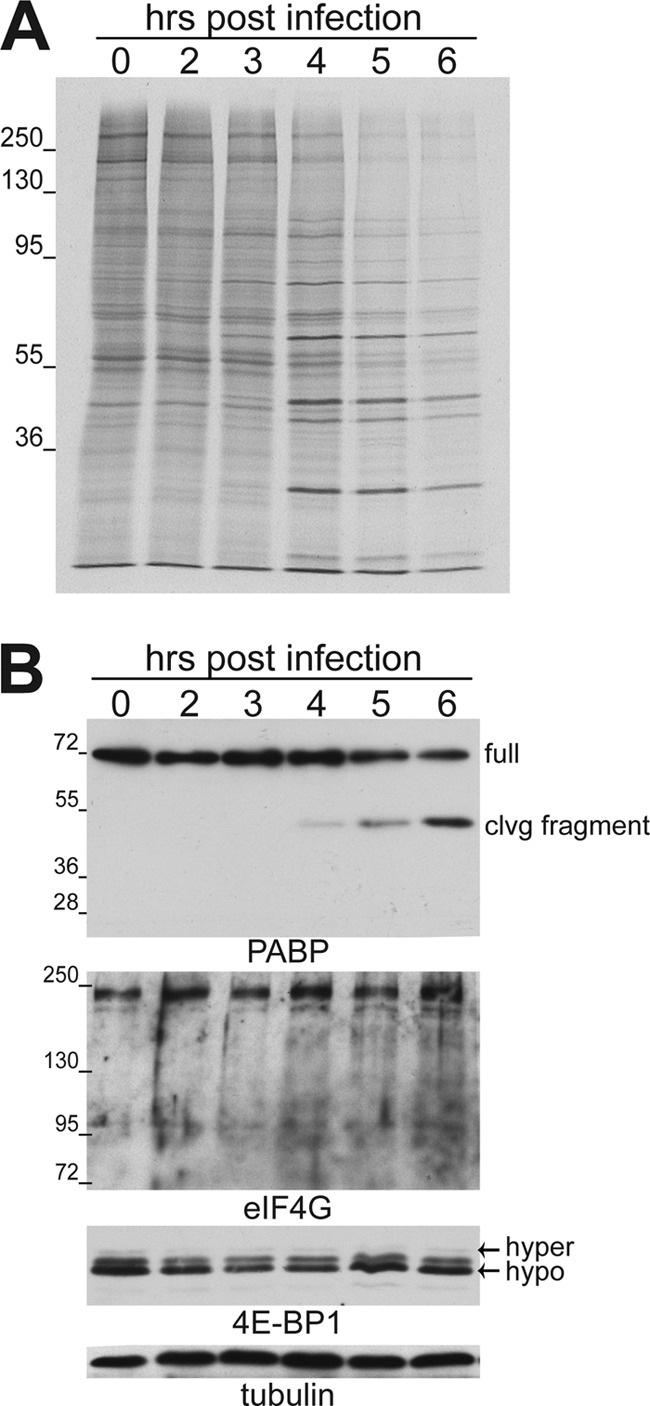
Inhibition of host protein synthesis in EMCV-infected cells correlates with accumulation of a PABP cleavage fragment. (A) Growth-arrested NHDFs were mock infected (0-h time point) or infected with EMCV (MOI = 10), metabolically labeled for 1 h with 35S-labeled cysteine and methionine, and collected at the indicated times postinfection. Total protein was isolated and fractionated by SDS-PAGE, and the fixed, dried gel was exposed to X-ray film. (B) Total protein samples (as in panel A) fractionated by SDS-PAGE were analyzed by immunoblotting using the indicated antibodies. Full-length PABP migrates with an apparent molecular mass of approximately ∼70 kDa on a 12.5% gel, whereas the cleavage fragment migrates faster, at approximately 45 kDa. Samples for the eIF4G immunoblot were fractionated on a 7.5% gel, while 4E-BP was analyzed using a 17.5% gel that resolves slower-migrating hyperphosphorylated (hyper) 4E-BP1 from the faster-migrating hypophosphorylated (hypo) form. Tubulin served as a loading control. The migration of molecular mass standards (in kilodaltons) is indicated to the left of the panels.
The phosphorylation status of the cap-dependent translation repressor, eIF4E-binding protein 1 (4E-BP1), was also analyzed throughout the course of EMCV infection. The unphosphorylated form of 4E-BP1 is able to regulate cap-dependent translation by binding to the cap-binding protein eIF4E. This prevents eIF4G binding to eIF4E and precludes the formation of a translation initiation factor complex on the mRNA 5′ end. Hyperphosphorylation of 4E-BP1 by mTORC1 inactivates the repressor's ability to bind 4E (11). Previous reports indicated that EMCV infection of a rapidly growing established cell line leads to the activation, or dephosphorylation, of 4E-BP1 (12). However, in growth-arrested primary fibroblasts, 4E-BP1 naturally accumulates in its hypophosphorylated form in uninfected or mock-infected cells (for an example, see reference 48). While host cell shutoff and EMCV protein synthesis were clearly evident (Fig. 1A), significant changes in the phosphorylation state of 4E-BP1 were not detected in growth-arrested primary cells infected with EMCV (Fig. 1B). This agrees with earlier studies that demonstrated that the mTORC1-selective inhibitor rapamycin had minimal effects on host cell shutoff and EMCV protein synthesis in cells infected with wild-type virus (47). Taken together, these data indicate that EMCV infection of primary human fibroblasts induces a change in PABP steady-state levels consistent with site-specific proteolysis, while levels of full-length eIF4G, which is cleaved in cells infected with related picornaviruses, remain unchanged.
EMCV 3C protease activity is sufficient to cleave PABP in uninfected cells.
PABP cleavage induced by poliovirus, a closely related picornavirus, is mediated by the viral protease 3C (22). Similarly, an evolutionarily related calicivirus and HAV also cleave PABP via their 3C-like proteinases (26, 50). Therefore, we hypothesized that PABP cleavage in EMCV-infected cells requires the virus-encoded proteinase 3C. To test this, HEK293 cells were transiently transfected with plasmids encoding wild-type (WT) or mutant EMCV 3C protease. Upon expression of catalytically active WT 3Cpro, a cleavage product with an apparent molecular mass of approximately 45 kDa was produced that reacted with anti-PABP antisera (Fig. 2). In contrast, the 45-kDa protein was not detected in cells transfected with a plasmid encoding a mutant 3C harboring two alanine substitutions within the enzyme's catalytic domain. Unfortunately, excessive background precluded the use of anti-3C antibody to detect 3Cpro proteins in lysates prepared from transiently transfected eukaryotic cells (data not shown). This demonstrates that expression of catalytically active EMCV 3C protease is necessary and sufficient to promote accumulation of the 45-kDa PABP-related protein without assistance from other virus-encoded or virus-induced cofactors. In addition, the 3C-induced 45-kDa PABP-related protein was similar in size to that detected in virus-infected cells (Fig. 1B). These results are consistent with site-specific cleavage of the host PABP by the EMCV 3C proteinase.
Fig 2.
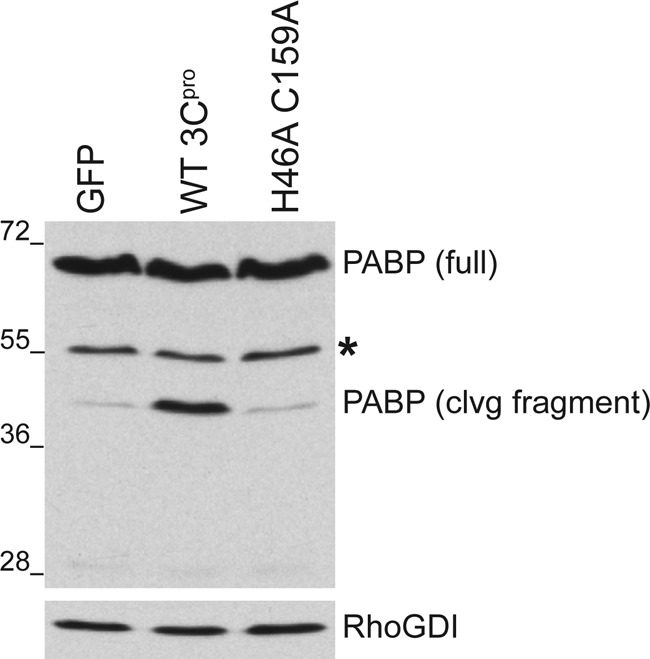
Specific cleavage of PABP by the EMCV 3C proteinase in uninfected cells. HEK293 cells were transiently transfected with mammalian expression vectors encoding EMCV WT 3C protease (WT 3Cpro), a catalytically inactive mutant form of 3C (H46A C159A), or EGFP. Forty-eight hours after transfection, total protein was isolated, fractionated by SDS-PAGE, and analyzed by immunblotting with the indicated antibodies. Full-length PABP runs around 70 kDa, whereas the cleavage (clvg) fragment migrates faster, around 45 kDa. Transfection efficiency indicated by EGFP fluorescence was above 60%. An asterisk indicates a cross-reacting band representing a cellular protein nonspecifically recognized by the polyclonal PABP antibody. The migration of molecular mass standards (in kilodaltons) is indicated to the left of the panel.
EMCV 3C cleaves PABP in vitro.
To determine if EMCV 3C is necessary and sufficient to cleave PABP without any other eukaryotic cellular factors, PABP cleavage was reconstituted in vitro using soluble lysates prepared from E. coli independently expressing 3C protease (18) or N-terminal His-tagged PABP. Recombinant PABP and 3C protease expressed in bacteria were readily detectable by immunoblotting, as anti-3C specifically detected bacterially expressed 3C in E. coli lysates (Fig. 3A, lanes 1 and 8). While the bulk of recombinant 3C partitioned into the insoluble fraction of the lysate, sufficient material remained in the soluble fraction to be detected by immunoblotting (data not shown) (18). Whereas mixing a PABP-containing bacterial lysate with a lysate prepared from bacteria transformed with a control plasmid did not detectably reduce full-length PABP levels, including increasing amounts of an EMCV 3Cpro-containing lysate progressively reduced full-length His-tagged PABP levels and resulted in the accumulation of a truncated, approximately 45-kDa N-terminal PABP cleavage product, suggesting that 3Cpro cleaves PABP within its C terminus (Fig. 3A). The minor cleavage product with a molecular mass of approximately 25 kDa, representing the C-terminal cleavage fragment, was detected upon probing of the immunoblot with anti-PABP (Fig. 3B). The corresponding sizes of the two fragments (∼45 kDa and ∼25 kDa) add up to the ∼70-kDa full-length protein. No other cleavage product was reproducibly evident within these experiments. This establishes that recombinant EMCV 3C protease was necessary and sufficient to specifically cleave bacterially expressed PABP at a single site within its C-terminal domain in vitro, giving rise to N-terminal 45-kDa and C-terminal 25-kDa fragments in the absence of any additional virus or eukaryotic cellular cofactors.
Fig 3.
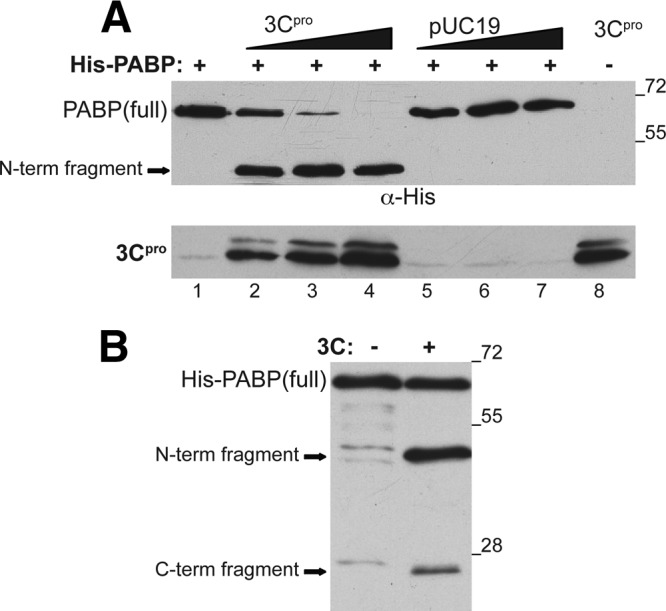
Reconstitution of EMCV 3Cpro-mediated PABP cleavage in vitro with bacterially expressed recombinant proteins. Increasing amounts of soluble lysates prepared from E. coli containing expression constructs encoding EMCV 3C protease (1) or an empty vector control (pUC19) were added to lysates containing a fixed quantity of N-terminally His-tagged human PABP. After incubation for 30 min at 30°C, reactions were terminated by adding equal parts SDS sample buffer and boiled. (A) Reaction products were fractionated by SDS-PAGE and analyzed by immunoblotting with anti-His or anti-3C. Anti-His selectively visualizes PABP derivatives with an intact N terminus. Note that slower-migrating proteins have been previously observed in preparations of 3C partially purified from bacteria (18). (B) Same as in panel A except that anti-PABP was used to visualize the C-terminal PABP cleavage product. Migration of molecular mass standards (in kilodaltons) is shown to the right of the panel.
EMCV 3Cpro cleaves PABP within the CTD at Q437/G438.
To identify the precise amino acid within the PABP polypeptide sequence proteolytically targeted by EMCV 3Cpro, we engineered a His tag epitope onto the PABP C terminus. Levels of full-length tagged PABP were reduced following incubation with a 3C-containing bacterial lysate in vitro, and the accumulation of a 25-kDa His-tagged C-terminal fragment was observed (Fig. 4A). The C-terminal His-tagged fragment was gel purified, and amino acid residues on its N terminus were identified by Edman degradation. The sequencing results revealed that the amino acid sequence GARPH was found at the amino terminus of the C-terminal cleavage fragment. The glycine corresponds to G438 within the C-terminal domain (CTD) of PABP. This suggests that EMCV 3Cpro cleaves PABP between glutamine 437 and glycine 438 with the CTD (Fig. 4B). To further confirm the precise cleavage site, single amino acid mutations were introduced at Q437 of recombinant, N-terminally His-tagged PABP. In vitro cleavage reactions were subsequently performed using bacterially expressed mutant PABP variants and analyzed by immunoblotting. PABP Q437E did not abrogate proteolysis by 3Cpro, as an ∼42-kDa cleavage product was produced similarly to the WT control. This was surprising, as a PABP Q/E mutation at residue 537, 413, or 437 is sufficient to abolish cleavage by poliovirus 3Cpro (17), and suggested that related 3C proteases within the picornavirus family exhibit variation in their recognition sequence. In contrast, only full-length PABP, and not a C-terminal cleavage fragment, was detected in reactions programmed with PABP containing an asparagine or aspartic acid substitution at residue 437 (Fig. 4C). The mutant PABP variants were also expressed in mammalian cells along with 3Cpro to further validate the cleavage site and rule out the possibility that the observed in vitro effects were not representative of in vivo findings. As expected, the cleavage product was detected in the HeLa cells overexpressing WT PABP with an N-terminal Flag tag (Fig. 4D) only in the presence of a catalytically active 3C proteinase and not with a mutant catalytically inactive 3C variant (H46A, C159A). In contrast, full-length PABP, but not PABP cleavage products, was detected when the Q437 substitution variant was introduced into HeLa cells along with 3C. This demonstrates that a single amino acid substitution at Q437 is sufficient to generate an EMCV 3Cpro-resistant form of PABP in vitro and in vivo. Moreover, no new cleavage product(s) was detected using Q437 substitution PABP derivatives under these conditions. Thus, eliminating the cleavage site at Q437 did not detectably result in utilization of a secondary cryptic site on PABP targeted by 3C. These data collectively identify the peptide bond between glutamine 437 and glycine 438 within the CTD on PABP as the single major cleavage site targeted by EMCV 3C proteinase.
Fig 4.
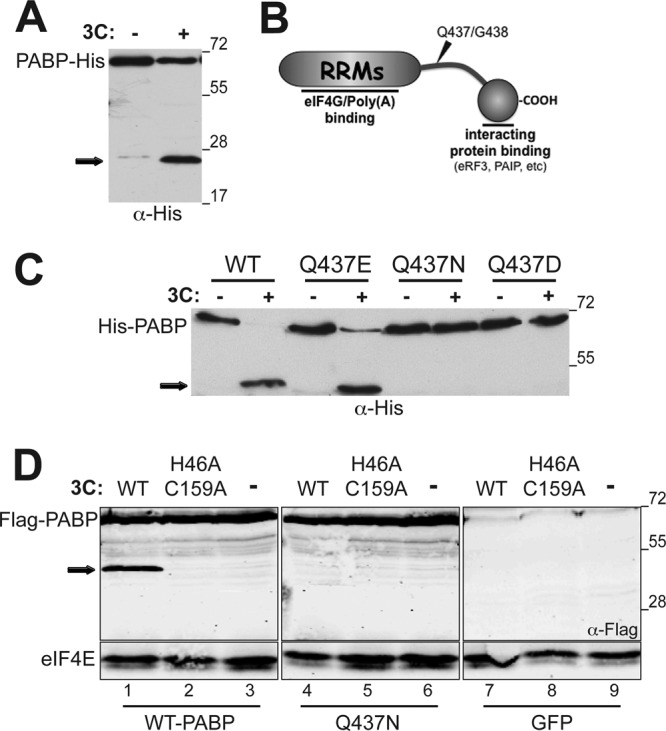
Identification and mapping of the specific PABP site cleaved by 3C. (A) In vitro cleavage reactions were performed using C-terminally His-tagged PABP (PABP-His) and analyzed as described in the Fig. 3 legend. Anti-His selectively visualizes PABP derivatives with an intact C terminus. (B) N-terminal sequencing of the 25-kDa C-terminally His-tagged PABP reaction product mapped the 3C cleavage site between Q437 and G438. Site-specific proteolytic cleavage between Q437 and G538 within the C-terminal linker region severs the RNA- and eIF4G-binding PABP N-terminal domain from the C-terminal domain that binds eIF4B, eRF4, and the Paips. (C) In vitro cleavage reactions were performed using wild-type (WT) N-terminally His-tagged PABP or variants with the indicated single amino acid substitutions at residue 437 (Q437E, Q437N, and Q437D). Reaction products were analyzed as described in the legend to Fig. 3. “+” indicates that active 3Cpro-containing lysate was present in the reaction; “−” indicates that heat-inactivated 3Cpro-containing lysate was used. (D) HeLa cells were transiently transfected with plasmids expressing Flag-tagged wild-type PABP (WT) or variants with the indicated single amino acid substitutions at residue 437 (Q437E, Q437N, and Q437D) in the absence (−) or presence of 3Cpro expression plasmids (WT or the H46A C159A 3Cpro catalytically inactive mutant). After 48 h, total protein was collected, fractionated by SDS-PAGE, and analyzed by immunoblotting with the indicated antisera. The migration of the 45-kDa N-terminal PABP cleavage product is indicated by the black arrow to the left. The migration of molecular mass standards (in kilodaltons) is indicated to the right of the panel.
Preventing site-specific PABP cleavage inhibits EMCV replication but does not detectably alter host cell shutoff.
PABP cleavage by poliovirus 3C contributes, in part, to suppression of host cell mRNA translation in virus-infected cells (3). Introduction of PV 3Cpro into cell-free HeLa extracts has been shown to attenuate translation of poly(A)-tailed RNA, and expression of PV 3Cpro in cells inhibited global protein synthesis (27). To evaluate the impact of EMCV 3Cpro expression on protein synthesis, HEK293 cells were transiently transfected with plasmids expressing WT 3Cpro, a catalytically inactive 3Cpro variant (H46A, C159A), or EGFP, and cellular protein synthesis was analyzed by metabolic labeling. Under these conditions, PABP cleavage by WT 3Cpro was readily detected (Fig. 2), although routinely incomplete, as some full-length PABP always remained. After fractionation of total protein by SDS-PAGE and autoradiography, little to no difference in the levels of newly synthesized proteins was detected in any of the transfected cultures (Fig. 5A). Furthermore, EMCV-induced host cell translational shutoff was not detectably altered when EMCV infection was carried out in cells expressing a 3Cpro cleavage-resistant mutant of PABP, Q437N, compared to WT PABP or EGFP (Fig. 5B). Importantly, similar levels of FLAG-tagged WT and Q437N PABP were detected by immunoblotting at the 0-h time point (Fig. 5C). These results suggest that catalytic activity of EMCV 3Cpro or cleavage of PABP at Q437 may not play a role in cellular translational shutoff.
Fig 5.
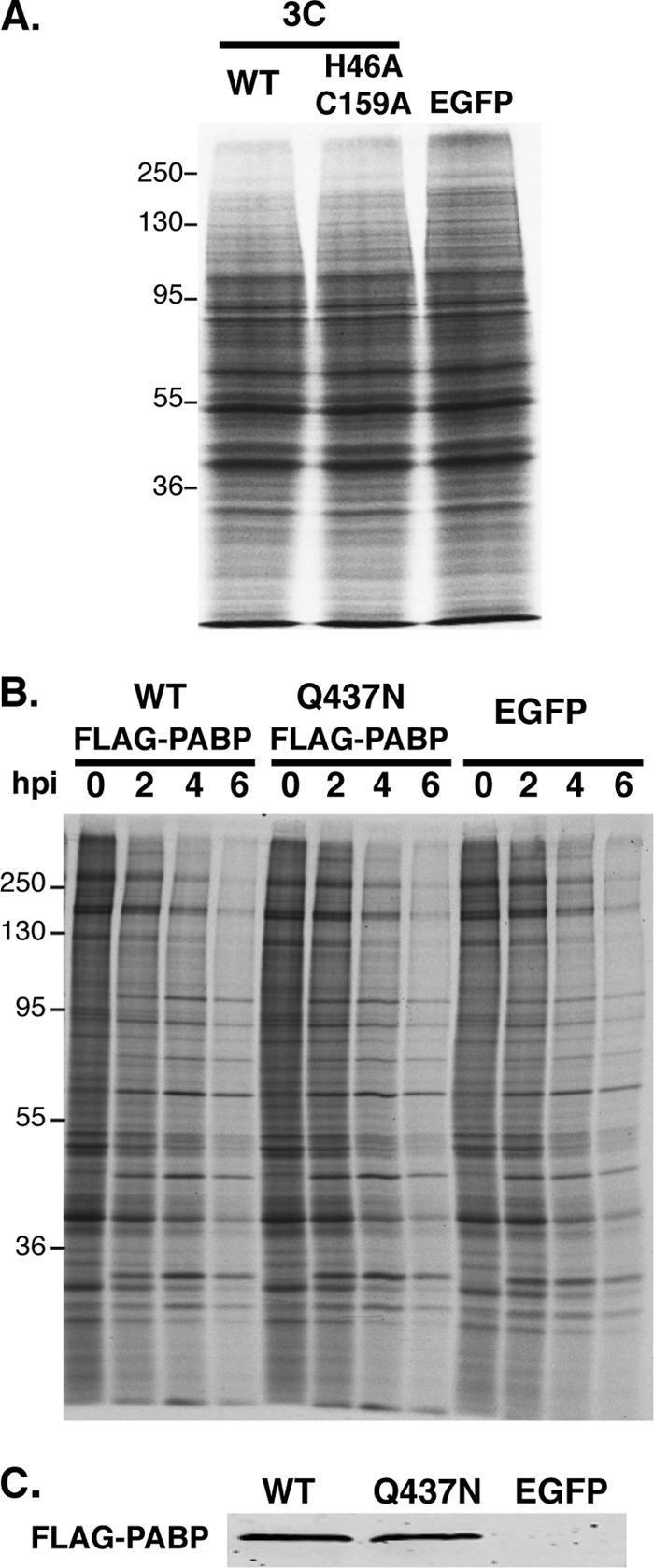
Preventing site-specific PABP cleavage does not detectably alter EMCV host cell shutoff. (A) HEK293 cells were transfected with plasmids expressing either WT 3Cpro, the H46A C159A 3Cpro catalytically inactive mutant, or EGFP. After 48 h, cells were metabolically labeled with 35S-labeled cysteine and methionine for 1 h. Total protein was isolated and analyzed as described in the legend to Fig. 1A. (B) NHDFs stably transduced with retrovirus vectors encoding WT PABP, the 3C cleavage-resistant Q473 PABP variant, or EGFP were infected with EMCV (MOI = 10). At the indicated times postinfection, total protein was harvested and analyzed as described in the legend to Fig. 1A. (C) Prior to infection with EMCV (0-hpi time point in panel B), the overall abundance of ectopically expressed PABP (FLAG-PABP) in cultures expressing Flag-tagged PABP (WT versus Q437N) or EGFP (negative control) was analyzed by immunoblotting using anti-Flag.
While we were unable to detect any changes in protein synthesis when cultures expressing noncleavable PABP alleles were infected with EMCV, PABP cleavage can influence viral replication by suppressing virus mRNA translation to allow high-level RNA synthesis required for genome replication. Indeed, PABP cleavage by poliovirus 3Cpro affects translation not only of cellular capped, poly(A)-containing messages but also of viral IRES-containing poly(A) mRNAs. This attenuation of viral protein synthesis promotes viral genome replication and infectious virion production (3). Although EMCV 3Cpro promotes site-specific PABP cleavage (Fig. 2) but does not detectably suppress ongoing host translation at the global level (Fig. 5A), it could activate virus genome replication by suppressing translation on a subset of viral RNAs destined to support RNA synthesis. As the translation process itself is known to competitively interfere with RNA synthesis on the same molecule (10), biologically, interfering with viral RNA synthesis would be expected to, in turn, reduce infectious virus production. To test this, multicycle growth experiments for EMCV replication were carried out in cells overexpressing WT versus the Q437N cleavage-resistant PABP mutant. Under these conditions, transfections performed in parallel showed that equivalent amounts of WT or Q437N Flag-PABP accumulated after 48 h (Fig. 4D; compare lanes 3 and 6). Significantly, EMCV grown on cells expressing the Q437N mutant PABP was impaired approximately 100-fold compared to virus generated in cells expressing WT PABP (Fig. 6A). This appeared not to result from mutant PABP acting in a dominant negative manner to suppress overall translation rates in uninfected cells, as global protein synthesis rates remained relatively similar between WT PABP- and Q437N PABP-overexpressing cells (Fig. 6B). Nor did it result from a failure of Q437N PABP to associate with polysomes under these conditions (Fig. 6C).
Fig 6.

Site-specific PABP cleavage stimulates EMCV RNA synthesis and infectious virus production. (A) HeLa cells transfected with plasmids expressing WT PABP, the Q437N cleavage-resistant variant (as in the Fig. 4D legend), or the plasmid vector-only control were infected with EMCV (MOI = 10−3). After 48 h, lysates were prepared by freeze-thawing, and the amount of infectious virus produced was quantified by plaque assay in Vero cells. (B) HeLa cells were transiently transfected as for panel A, transfected with an EGFP-expressing plasmid, or left untransfected. After 48 h, cultures were metabolically labeled for 1 h as for panel A. Total protein was isolated and the amount of 35S incorporated determined by trichloroacetic acid (TCA) precipitation. Acid-insoluble material was collected onto fiberglass filters and washed, and the radioactivity was quantified by counting in liquid scintillant. The relative amount of 35S incorporation in untransfected HeLa cells was normalized to 100%. Error bars indicate the standard errors of the means. (C) Cell-free lysates prepared from HeLa cells ectopically expressing cleavage-resistant Flag-tagged PABP variant (Q437N) were loaded onto sucrose gradients, which were subsequently fractionated while absorbance at 254 nm was monitored. TCA-precipitated fractions were analyzed by immunoblotting using antibodies specific for Flag or ribosomal protein s6 (rpS6). Fraction 1, free ribosome subunits; fraction 2, disomes; fractions 3 to 5, polyribosomes. (D) HeLa cells transiently transfected with empty plasmid vector or a plasmid expressing Flag-tagged PABP (WT or Q437N) were infected with EMCV (MOI = 10) for 5 h. (Top) Infections were performed in the presence of [3H]uridine and actinomycin D. Total RNA was isolated, samples were precipitated with TCA, and the amount of acid-insoluble radioactivity quantified by counting in liquid scintillant. Errors bars indicate standard errors of the means. (Bottom) The amount of infectious virus produced after a single replication cycle was quantified by plaque assay. Error bars indicate standard errors of the means.
To determine if PABP cleavage contributed to EMCV replication by promoting viral RNA synthesis, HeLa cells transiently transfected with plasmids expressing FLAG-tagged WT PABP, the Q437N cleavage-resistant PABP mutant, or vector alone were infected with EMCV and metabolically pulse-labeled with [3H]uridine in the presence of actinomycin D, which prevents RNA synthesis by host DNA-dependent RNA polymerases. After isolation of total RNA, the amount of viral RNA synthesis, as represented by acid-insoluble radioactivity, was quantified by liquid scintillation counting. While transfection of a WT PABP-expressing plasmid had little effect on RNA synthesis compared to vector alone, the Q437N PABP-expressing plasmid reduced RNA synthesis approximately 4-fold (Fig. 6D, top). This likely underrepresents the fold inhibition of viral RNA synthesis by Q437N PABP, as not all the cells in the transiently transfected population received plasmid. Under these high-MOI, single-cycle growth conditions in transiently transfected cells, a similar modest reduction in viral yield was observed in cultures expressing Flag-tagged Q437N PABP compared to WT (Fig. 6D, bottom). Taken together, these data establish a biological role for the cleavage of PABP by 3Cpro that promotes efficient EMCV replication. While interfering with PABP cleavage did not detectably impact host cell shutoff, it did suppress overall levels of viral RNA synthesis.
DISCUSSION
While a 3′ poly(A) tail of sufficient length is important for EMCV replication (6, 20, 21), the impact of EMCV infection on the host PABP remained unknown. Furthermore, although EMCV, like other picornavirus family members, induces a potent host cell shutoff that suppresses cellular protein synthesis, unlike with the Enterovirus and Aphthovirus genera, infection with the prototypical cardiovirus EMCV does not result in eIF4G cleavage. Here, we establish that EMCV infection stimulates site-specific PABP proteolysis whereby a 45-kDa N-terminal PABP fragment accumulates in virus-infected cells. Expression of a functional EMCV 3C proteinase was necessary and sufficient to stimulate PABP cleavage in uninfected cells, and bacterially expressed 3C cleaved recombinant PABP in vitro in the absence of any virus-encoded or eukaryotic cellular cofactors. The exact 3Cpro cleavage site on PABP was mapped between amino acids Q437 and G438 within the CTD, separating the RNA binding domain from the protein-interacting domain. Moreover, validation of the cleavage site using single amino acid substitution mutants suggested this site is the single, major cleavage site on PABP by EMCV 3Cpro in vitro and in vivo. Finally, while EMCV replication was impaired in cells expressing a cleavage-resistant PABP variant, ongoing protein synthesis was not detectably altered. Instead, preventing PABP cleavage reduced EMCV RNA synthesis. Together, these results demonstrate that PABP cleavage by the EMCV protease regulates EMCV replication.
Although site-specific PABP cleavage within the proline-rich bridge, severing its RNA/eIF4G binding region from its protein-interacting C-terminal domain, has been previously observed in PV (an enterovirus)- and calicivirus-infected cells (26, 27), PABP proteolysis mediated by 3C of the cardiovirus EMCV has distinctive features. Significantly, whereas only a single major cleavage site between the Q437 and G438 target is detected in EMCV-infected cells, PV 3Cpro cleaves PABP at three distinct sites within the C-terminal domain: the major site between Q537 and G538, a second site (3C alt) between Q413 and T414, and a third site (3C alt′) at Q437 and G438 (28). The third site, 3C alt′, shares target sequence homology with the site recognized by EMCV 3Cpro, yet cleavage in PV-infected cells is seldom detected, and it is considered one of two minor sites targeted by polio 3Cpro (28). In addition, a second PV proteinase, polio 2Apro, also cleaves PABP at a discrete site in the C-terminal domain (22), bringing the total number of PV-induced PABP cleavage sites to 4, versus 1 in EMCV-infected cells. Indeed, PABP cleavage at multiple sites by PV proteinases could ensure that levels of full-length PABP are effectively reduced in infected cells and potentially accounts for the observed differences in the abilities of PV and EMCV 3C to suppress ongoing translation in transiently transfected cells. Finally, while PABP Q537E substitution was resistant to PV 3Cpro (26, 27), it remained sensitive to EMCV 3Cpro, indicating differences between how the two picornavirus proteinases from the Enterovirus and Cardiovirus genera recognize this site in the PABP substrate. Differences in substrate cleavage notwithstanding, it is striking that preventing PABP cleavage in PV or EMCV-infected cells had comparable biological effects on viral RNA synthesis and replication at high MOI (reference 3 and this study).
While 3C-mediated PABP cleavage reduces full-length PABP abundance in EMCV-infected or transiently transfected cells, it does not eliminate it. This could reflect the limited stability of 3C in infected cells (31, 41), or alternatively, preferential cleavage of PABP subpopulations in vivo (40). Indeed, PABP bound to poly(A) was preferentially cleaved in PV-infected cells, whereas PABP bound to its interacting partners Paip2, eRF3, or associated with viral polyribosomes was relatively recalcitrant to site-specific proteolysis. Residual, full-length PABP may represent a fraction not bound to mRNA, associated with cellular translation machinery, or bound to interacting proteins, allowing a subpopulation of inactive (Paip2-bound) PABP, or PABP engaged in translation to be spared. This could allow templates destined for RNA synthesis to be selected from among nonpolysomal ribonucleoproteins (RNPs) with PABP bound to the 3′ end. It also suggests that in cells infected with small numbers of input genomes, some function, perhaps virus encoded or induced, is required to disengage polysomes, at least temporarily, allowing for PABP cleavage and subsequent RNA synthesis on a translationally inactive pool of template molecules.
Similar to our findings in vivo that PABP cleavage in EMCV-infected cells is substantial, yet incomplete, studies of EMCV replication in vitro utilized extracts largely depleted of PABP but not devoid of PABP (4, 46). Thus, the possibility that the remaining full-length PABP contributes to EMCV replication through mRNA translation or some other process cannot be excluded. In this regard, it is noteworthy that PABP cleavage is not detected in infected cells until after 3 hpi, whereas 35S-labeled viral proteins can be seen as early as 3 hpi. We can, however, conclude that site-specific PABP cleavage by 3C is required for wild-type levels of EMCV replication.
While the poly(A) tail enhances translation of EMCV IRES-containing reporters in vitro, it is critical for viral replication (13, 20, 21). In studies using EMCV IRES reporters, PABP depletion inhibited reporter mRNA translation in cell extracts (46), whereas PABP inhibition by Paip2 had limited effects on the EMCV IRES (4). When full-length EMCV viral mRNA was used as the template, however, PABP depletion did not detectably reduce mRNA translation in vitro (46). Our finding that the EMCV 3C protease mediates site-specific PABP cleavage in EMCV-infected cells supports the notion that full-length PABP may not be absolutely required for EMCV replication and could, in fact, potentially be deleterious, as evidenced by reduced viral replication and RNA synthesis in cells expressing a noncleavable PABP variant. Targeted site-specific cleavage of PABP by the viral 3C proteinase provides a molecular means to restrict full-length PABP binding to the poly(A) tract, perhaps creating a pool of templates competent to recruit the virus RNA-dependent RNA polymerase 3D to the template to initiate RNA synthesis (6, 7). Whether PABP cleavage is sufficient to prevent assembly of a PABP-containing RNP on the viral RNA 3′ end, or if the resulting PABP N-terminal fragment somehow functions in viral replication, remains to be determined.
ACKNOWLEDGMENTS
We thank Derek Walsh for critically commenting on the manuscript, Simon Morley for anti-PABP antisera, and Robert Schneider for the plasmid for N-terminal His-tagged PABPC1.
This work was supported by grants to I.M. (AI073898 and GM056927) and A.C.P. (AI017331 and U19 AI070503) from the NIH. M.K. acknowledges support from grants T32AI007647 and T32AI07180.
Footnotes
Published ahead of print 19 July 2012
REFERENCES
- 1. Aminev AG, Amineva SP, Palmenberg AC. 2003. Encephalomyocarditis viral protein 2A localizes to nucleoli and inhibits cap-dependent mRNA translation. Virus Res. 95:45–57 [DOI] [PubMed] [Google Scholar]
- 2. Belsham GJ, McInerney GM, Ross-Smith N. 2000. Foot-and-mouth disease virus 3C protease induces cleavage of translation initiation factors eIF4A and eIF4G within infected cells. J. Virol. 74:272–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonderoff JM, LaRey JL, Lloyd RE. 2008. Cleavage of poly(A)-binding protein by poliovirus 3C proteinase inhibits viral internal ribosome entry site-mediated translation. J. Virol. 82:9389–9399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bradrick SS, Dobrikova EY, Kaiser C, Shveygert M, Gromeir M. 2007. Poly(A)-binding protein is differentially required for translation mediated by viral internal ribosome entry sites. RNA 13:1582–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Castelló A, et al. 2009. HIV-1 protease inhibits cap- and poly(A)-dependent translation upon eIF4GI and PABP cleavage. PLoS One 4(11):e7997 doi:10.1371/journal.pone.0007997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cui T, Porter AG. 1995. Localization of binding site for encephalo-myocarditis virus RNA polymerase in the 3′-noncoding region of the viral RNA. Nucleic Acids Res. 23:377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cui T, Sankar S, Porter AG. 1993. Binding of encephalomyocarditis virus RNA polymerase to the 3′-noncoding region of the viral RNA is specific and requires the 3′-poly(A) tail. J. Biol. Chem. 268:26093–26098 [PubMed] [Google Scholar]
- 8. Etchison D, Fout S. 1985. Human rhinovirus 14 infection of HeLa cells results in the proteolytic cleavage of the p220 cap-binding complex subunit and inactivates globin mRNA translation in vitro. J. Virol. 54:634–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JWB. 1982. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000 dalton polypeptide associated with eukaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem. 257:14806–14810 [PubMed] [Google Scholar]
- 10. Gamarnik AV, Andino R. 1998. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 12:2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gingras AC, et al. 2001. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 15:2852–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gingras AC, Svitkin Y, Belsham GJ, Pause A, Sonenberg N. 1996. Activation of the translational suppressor 4E-BP1 following infection with encephalomyocarditis virus and poliovirus. Proc. Natl. Acad. Sci. U. S. A. 93:5578–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldstein AO, Paradoe IU, Burness ATH. 1976. Requirement of an adenylic acid-rich segment for the infectivity of encephalomyocarditis virus RNA. J. Gen. Virol. 31:271–276 [DOI] [PubMed] [Google Scholar]
- 14. Gradi A, Svitkin YV, Imataka H, Sonenberg N. 1998. Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc. Natl. Acad. Sci. U. S. A. 95:11089–11094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Groppo R, Brown BA, Palmenberg AC. 2011. Mutational analysis of the EMCV 2A protein identifies a nuclear localization signal and an eIF4E binding site. Virology 410:257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Groppo R, Palmenberg AC. 2007. Cardiovirus 2A protein associates with 40S but not 80S ribosome subunits during infection. J. Virol. 81:13067–13074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haghighat A, et al. 1996. The eIF4G-eIF4E complex is the target for direct cleavage by the rhinovirus 2A proteinase. J. Virol. 70:8444–8450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hall DJ, Palmenberg AC. 1996. Mengo virus 3C proteinase: recombinant expression, intergenus substrate cleavage and localization in vivo. Virus Genes 13:99–110 [DOI] [PubMed] [Google Scholar]
- 19. Hellen CU. 2009. IRES-induced conformational changes in the ribosome and the mechanism of translation initiation by internal ribosomal entry. Biochim. Biophys. Acta 1789:558–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hruby DE, Roberts WK. 1976. Encephalomyocarditis virus RNA: variations in polyadenylic acid content and biological activity. J. Virol. 19:325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hruby DE, Roberts WK. 1976. Encephalomyocarditis virus RNA. II. Polyadenylic acid requirement for efficient translation. J. Virol. 23:338–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Joachims M, Van Breugel PC, Lloyd RE. 1999. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 73:718–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kerekatte V, et al. 1999. Cleavage of poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff? J. Virol. 73:709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirchweger R, et al. 1994. Foot-and-mouth disease virus leader proteinase: purification of the Lb form and determination of its cleavage site on eIF-4 gamma. J. Virol. 68:5677–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kräusslich HG, Nicklin MJ, Toyoda H, Etchison D, Wimmer E. 1987. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J. Virol. 61:2711–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuyumcu-Martinez NM, et al. 2004. Calicivirus 3C-like proteinase inhibits cellular translation by cleavage of poly(A)-binding protein. J. Virol. 78:8172–8182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuyumcu-Martinez NM, Van Eden ME, Younan P, Lloyd RE. 2004. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol. Cell. Biol. 24:1779–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuyumcu-Martinez NM, Joachims M, Lloyd RE. 2002. Efficient cleavage of ribosome-associated poly(A)-binding protein by enterovirus 3C protease. J. Virol. 76:2062–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lamphear, et al. 1993. Mapping the cleavage site in protein synthesis initiation factor eIF4 gamma of the 2A proteases from human coxsackievirus and rhinovirus. J. Biol. Chem. 268:19200–19203 [PubMed] [Google Scholar]
- 30. Lawrence C, Thach RE. 1974. Encephalomyocarditis virus infection of mouse plasmacytoma cells. I. Inhibition of cellular protein synthesis. J. Virol. 14:598–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lawson TG, Smith LL, Palmenberg AC, Thach RE. 1989. Inducible expression of encephalomyocarditis virus 3C protease activity in transformed mouse cell lines. J. Virol. 63:5013–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lloyd RE. 2006. Translational control by viral proteinases. Virus Res. 119:76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mangus DA, Evans MC, Jacobson A. 2003. Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 4:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mosenkis J, et al. 1985. Shutoff of host translation by encephalomyocarditis virus infection does not involve cleavage of the eucaryotic initiation factor 4F polypeptide that accompanies poliovirus infection. J. Virol. 54:643–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pear WS, Nolan GP, Scott ML, Baltimore D. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U. S. A. 90:8392–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Porter FW, Bochkov YA, Albee AJ, Wiese C, Palmenberg AC. 2006. A picornavirus protein interacts with Ran GTPase and disrupts nucleo-cytoplasmic transport. Proc. Natl. Acad. Sci. U. S. A. 103:12417–12422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Racaniello VR. 2007. Picornaviridae: the viruses and their replication, p 795–838 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 38. Ramírez-Valle F, Braunstein S, Zavadil J, Formenti SC, Schneider RJ. 2008. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J. Cell Biol. 181:293–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reineke LC, Lloyd RE. 2011. Animal virus schemes for translation dominance. Curr. Opin. Virol. 1:363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rivera CI, Lloyd RE. 2008. Modulation of enteroviral proteinase cleavage of poly(A)-binding protein (PABP) by conformation and PABP-associated factors. Virology 375:59–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schlax PE, Zhang J, Lewis E, Planchart A, Lawson TG. 2007. Degradation of the encephalomyocarditis virus and hepatitis A virus 3C proteases by the ubiquitin/26S protease system in vivo. Virology 360:350–363 [DOI] [PubMed] [Google Scholar]
- 42. Silvera Formenti DSC, Schneider RJ. 2010. Translational control in cancer. Nat. Rev. Cancer 10:254–266 [DOI] [PubMed] [Google Scholar]
- 43. Skern T, et al. 2002. Structure and function of picornavirus proteases. ASM Press, Washington, DC [Google Scholar]
- 44. Sommergruber W, et al. 1994. 2A proteinases of coxsackie- and rhinovirus cleave peptides derived from eIF-4 gamma via a common recognition motif. Virology 198:741–745 [DOI] [PubMed] [Google Scholar]
- 45. Spector DH, Villa-Komaroff L, Baltimore D. 1975. Studies on the function of polyadenylic acid on poliovirus RNA. Cell 6:41–44 [DOI] [PubMed] [Google Scholar]
- 46. Svitkin YV, Costa-Mattioli M, Herdy B, Perreault S, Sonenberg N. 2007. Stimulation of picornavirus replication by the poly(A) tail in a cell-free extract is largely independent of the poly(A) binding protein (PABP). RNA 13:2330–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Svitkin Y, Hahn H, GIngras AC, Palmenberg AC, Sonenberg N. 1998. Rapamycin and wortmannin enhance replication of a defective encephalomyocarditis virus. J. Virol. 72:5811–5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walsh D, Mohr I. 2004. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 18:660–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Walsh D, Mohr I. 2011. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 9:860–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang B, Morace G, Gauss-Mueller V, Kusov Y. 2007. Poly(A) binding protein, C-terminally truncated by the hepatitis A virus proteinase 3C, inhibits viral translation. Nucleic Acids Res. 35:5975–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]