

Abstract
Tick-borne Nyamanini virus (NYMV) is the prototypic member of a recently discovered genus in the order Mononegavirales, designated Nyavirus. The NYMV genome codes for six distinct genes. Sequence similarity and structural properties suggest that genes 1, 5, and 6 encode the nucleoprotein (N), the glycoprotein (G), and the viral polymerase (L), respectively. The function of the other viral genes has been unknown to date. We found that the third NYMV gene codes for a protein which, when coexpressed with N and L, can reconstitute viral polymerase activity, suggesting that it represents a polymerase cofactor. The second viral gene codes for a small protein that inhibits viral polymerase activity and further strongly enhances the formation of virus-like particles when coexpressed with gene 4 and the viral glycoprotein G. This suggests that two distinct proteins serve a matrix protein function in NYMV as previously described for members of the family Filoviridae. We further found that NYMV replicates in the nucleus of infected cells like members of the family Bornaviridae. NYMV is a poor inducer of beta interferon, presumably because the viral genome is 5′ monophosphorylated and has a protruding 3′ terminus as observed for bornaviruses. Taken together, our results demonstrate that NYMV possesses biological properties previously regarded as typical for filoviruses and bornaviruses, respectively.
INTRODUCTION
Viruses of the order Mononegavirales possess nonsegmented negative-stranded RNA genomes ranging in size from ∼9,000 to ∼19,000 nucleotides. Based on genome organization they are assigned to four virus families, namely, Bornaviridae, Filoviridae, Paramyxoviridae, and Rhabdoviridae. Only recently were Nyamanini virus (NYMV) and Midway virus identified as members of a novel genus, designated Nyavirus, which cannot be assigned to any of the existing mononegavirus families (19). NYMV was isolated from ticks and birds in South Africa, Nigeria, Egypt, India, and Thailand (11, 13, 30). NYMV productively infects a variety of mammalian cell lines, including cells of human origin. However, to date, there have been no reported cases of NYMV infection in humans (11, 29).
The NYMV genome (GenBank accession number FJ554526) contains six genes defined by conserved transcriptional start and stop signals (19), each of which contains one major open reading frame (ORF). ORF1 and ORF6 of NYMV show sequence similarity to the N and L proteins of other negative-strand viruses, respectively (19), whereas ORF2, ORF3, ORF4, and ORF5 share no recognizable similarity to other viral or cellular proteins. A putative signal peptide and transmembrane region is present in ORF5, suggesting that it represents the viral glycoprotein G. The functions of ORF2, ORF3, and ORF4 have remained elusive to date. Based on the location of the corresponding genes in the genome, it was proposed that ORF2 might represent the viral phosphoprotein, and that either ORF3 or ORF4 might represent the matrix protein (19).
With the exception of bornaviruses, which replicate in the nucleus, all animal viruses assigned to the order Mononegavirales characterized to date replicate in the cytoplasm (reviewed in reference 33). All mononegaviruses possess a set of five conserved genes, the order of which on the viral genome is 3′-N-P-M-G-L-5′. Viruses that carry more genetic information contain additional genes upstream of the N gene, as in the case of pneumoviruses, or between the M and L genes, as seen for all members of the Paramyxovirinae. Alternatively, the coding capacity of the P gene may be extended by either RNA editing, translating overlapping ORFs, or both (reviewed in references 5 and 25). For the formation of virus particles, mononegaviruses typically employ a single matrix protein, designated M, which drives the viral budding process. If the viral glycoprotein G is coexpressed, virus-like particles (VLPs) are being formed, which can transfer viral minigenomes encoding reporter proteins to new cells by receptor-mediated membrane fusion. Viruses of the family Filoviridae require a second viral protein, designated VP24, for the efficient formation of infectious VLPs (2). The molecular details of how VP24 might serve as a minor matrix protein remain unknown. Since VP24 can associate with the viral ribonucleoprotein complex, it might facilitate the recruitment of viral nucleocapsids into budding virus particles (2, 31).
The RNA genomes of viruses assigned to the order Mononegavirales can assume panhandle-like secondary structures, and most of these viruses possess a triphosphorylated 5′ terminus. The 5′ triphosphorylated panhandle structure is efficiently recognized by retinoic acid-inducible gene I (RIG-I) and other cytoplasmic pattern recognition receptors, which in turn results in strong activation of innate immune responses and synthesis of type I and type III interferon (IFN) (reviewed in reference 12). To counteract the innate immune response, most mononegaviruses code for proteins with IFN-antagonistic activity, such as the V proteins of paramyxoviruses (7). Borna disease virus (BDV), the prototypic member of the family Bornaviridae, uses a completely different strategy for evading the innate immune response. The 5′ terminus of the BDV genome is trimmed, and the 3′ terminus is elongated on an internal template motif (16). Thus, in the panhandle conformation, the BDV genome is predicted to form a secondary structure with a monophosphorylated 5′ end and a protruding 3′ end that is not efficiently recognized by RIG-I and other cytosolic pattern recognition receptors (8, 16).
To date, the replication cycle of NYMV has not been studied thoroughly, and it is unknown which cellular compartment NYMV is using for genome multiplication, which protein of NYMV may serve as the polymerase cofactor, which viral proteins are required for particle assembly, and which strategy NYMV may use to prevent strong innate immune responses. We addressed these questions and found that (i) NYMV replicates in the nucleus and is dependent on cellular RNA polymerase II activity, (ii) ORF3 represents an essential polymerase cofactor, (iii) both ORF2 and ORF4 are required for generating infectious VLPs, and (iv) NYMV maintains a monophosphorylated genome with a protruding 3′ terminus to evade the IFN response.
MATERIALS AND METHODS
Plasmid constructions.
RNA from Vero cells infected with NYMV was reverse transcribed using the First-Strand cDNA synthesis kit (Fermentas), and PCR fragments were amplified using the Phusion DNA polymerase (New England BioLabs). The various ORFs of NYMV were inserted into pCA expression vectors. The viral leader and trailer regions were cloned and assembled in the NYMV minigenome vector pT7-NYMVmg-Gluc. All plasmid and primer sequences as well as detailed information on the cloning strategies are available on request.
Cells and viruses.
Vero cells, 293T cells, MDCKII cells, and BSR-T7 cells (a kind gift from K. K. Conzelmann) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS). The cells were kept at 37°C in a 5% CO2 humidified atmosphere. Transfections were performed using Nanofectin (PAA) according to the manufacturer's protocol. Stocks of influenza virus A/PR/8/34 (FLUAV) were generated in MDCKII cells kept in DMEM supplemented with 0.1% bovine serum albumin and 1 μg/ml trypsin. Stocks of influenza virus strain WSN were generated in MDCKII cells, and stocks of measles virus (MeV, Edmonston strain), BDV (strain BDV-LRD [1]), vesicular stomatitis virus (VSV), and NYMV (tick isolate 39) were generated in Vero cells.
Determining antiserum specificity.
293T cells were transfected with expression vectors encoding the various NYMV proteins or an empty vector as a control. At 24 h posttransfection, the cells were lysed and immunoblot analysis was performed using serum obtained from NYMV-infected mice or antiserum against β-tubulin (Sigma, T4026) as control.
Immunohistochemical analysis.
Vero cells (105) seeded onto coverslips in 12-well plates were infected with 103 50% tissue culture infective doses (TCID50s) of NYMV or were left uninfected. At 48 h postinfection, immunofluorescence analyses and 4′,6-diamidino-2-phenylindole (DAPI) staining were performed as described previously (21) using serum from NYMV-infected mice.
Cell fractionation.
Vero cells (106 per 35-mm dish) were infected with 104 TCID50s of NYMV or were left uninfected. At 48 h postinfection, the cells were washed with ice-cold Tris-buffered saline (TBS) and then lysed in either 1 ml of complete lysis buffer (50 mM Tris HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) to generate the whole-cell extract or 1 ml of sucrose buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 10 mM magnesium acetate, 3 mM CaCl2, 340 mM sucrose, and 1 mM dithiothreitol [DTT]) for subcellular fractionation. The lysates were transferred into 1.5-ml tubes and continuously incubated on ice. After 10 min of incubation, 50 μl of 10% NP-40 were added to the sucrose buffer. The lysate was vortexed for 20 s at 1,500 rpm and centrifuged for 10 min in a cooled table-top centrifuge at 3,500 × g. The supernatant was collected (cytoplasmic fraction), and the pellet was washed once in 1 ml sucrose buffer. The nuclear pellet was then resuspended in 150 μl of nucleoplasm extraction buffer (50 mM HEPES [pH 7.9], 150 mM potassium acetate, 1.5 mM MgCl2, 0.1% NP-40, and 1 mM DTT) and homogenized with 20 strokes in a douncer. The homogenate was then incubated for 20 min on a rotating wheel followed by centrifugation for 10 min at 12,000 × g. The supernatant was collected (nuclear fraction). One half of the extracts was subjected to immunoblot analyses using primary antibodies directed against histone H3 (Abcam, 1791), β-tubulin (Sigma, T4026), or serum from NYMV-infected mice. The other half was used for RNA extraction using the Peqgold RNA pure system (Peqlab) according to the manufacturer's protocol.
Inhibition of cellular RNA polymerase II activity by actinomycin D.
Vero cells (3 × 105) in 6-well plates were infected with NYMV, VSV, or WSN using an MOI of 1. The cells were washed 3 times with PBS, and culture medium containing 5 μg/ml of actinomycin D (Sigma) was added. Supernatants of VSV-infected cells were harvested at 24 h postinfection. In the case of NYMV and WSN, the cells were scraped and collected with the supernatant at 40 h postinfection before the virus was released by three freeze-thaw cycles. Viral titers were determined by the 50% tissue culture infective dose (TCID50) assay.
Minireplicon assay.
BSR-T7 cells (2 × 105) in 12-well plates were transfected with the NYMV minigenome plasmid pT7-NYMVmg-Gluc (0.4 μg), expression vectors encoding N (0.5 μg), L (0.1 μg), and the indicated other NYMV proteins (0.1 μg each). To normalize for transfection efficacy, pBST7-ffluc (0.1 μg), encoding the firefly luciferase, was cotransfected. At 72 h posttransfection, the BSR-T7 cells were lysed with 300 μl lysis buffer, and Gaussia and firefly luciferase activities were quantified using the dual-luciferase kit from Promega.
Mammalian two-hybrid assay.
293T cells were transfected with a Gal4 promoter-dependent firefly construct (0.2 μg), with pCA-VP16/ORF3 (0.4 μg) and the indicated pCA-Gal4 constructs (0.2 μg). A constitutively active Renilla luciferase reporter construct (0.05 μg) was cotransfected for normalization. At 24 h posttransfection, the 293T cells were lysed, and Renilla and firefly luciferase-mediated light emissions were quantified using the dual-luciferase kit from Promega. The firefly signal was normalized for transfection efficacy using the Renilla signal.
VLP assay.
BSR-T7 cells were transfected as described for the minireplicon assay. At 72 h posttransfection, the supernatant was collected and clarified through a 0.45-μm filter. Samples (500 μl) of the clarified supernatants were transferred onto 293T cells (5 × 104) in 24-well dishes previously transfected with pCA-N (0.25 μg), pCA-ORF3 (0.05 μg), and pCA-L (0.05 μg). At 48 h posttransfer of the supernatant, the 293T cells were lysed with 150 μl lysis buffer, and Gaussia-mediated light emission was quantified using the dual-luciferase kit from Promega.
Coimmunoprecipitation experiments.
293T cells (106) were transfected with 1 μg of pCA expression vectors encoding Flag- or hemagglutinin (HA)-tagged NYMV proteins. At 24 h posttransfection, the cells were lysed with 150 μl lysis buffer (50 mM Tris HCl [pH 7.4], with 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100), incubated for 30 min under constant agitation at room temperature, and centrifuged for 10 min at 12,000 × g. The clarified supernatant was mixed with 20 μl of washed anti-Flag M2 beads (Sigma, A2220), and the mixture was incubated overnight at 4°C under continuous rotation. The next day, the beads were washed 3 times in 500 μl cold TBS buffer (50 mM Tris HCl, with 150 mM NaCl [pH 7.4]), and bound proteins were eluted into Laemmli buffer by a 5-min incubation at 95°C. Immunoblot analyses were performed using a mouse anti-Flag M2 monoclonal antibody (MAb) (Sigma, F1804) and rabbit anti-HA serum Y-11 (Santa Cruz).
IFN-β reporter assays.
293T cells were seeded subconfluently into 24-well dishes and transfected with 0.125 μg of the firefly luciferase reporter construct p125-Luc (a kind gift of T. Fujita, Osaka, Japan) together with 0.025 μg pRL-SV40 (Roche) using 0.45 μl Fugene HD (Roche) in 50 μl Opti-MEM (Gibco-BRL). After 6 h, the cells were transfected with 0.5 μg RNA isolated from purified viral particles or infected cells using 1.5 μl of Metafectene (Biontex) in 50 μl Opti-MEM. Prior to transfection, the RNA was either mock treated or treated with shrimp alkaline phosphatase (SAP, Affymetrix). RNA isolated from uninfected Vero cells served as a negative control (mock). Viral RNA was isolated from infected cells or purified viral particles using the Direct-zol RNA miniprep kit (Zymo Research) following the manufacturer's protocol. For purification of influenza A virus particles, supernatants of infected cells were collected 2 days after infection, and particles were precipitated with polyethylene glycol as described previously (8). MeV and NYMV particles were released from the host cells by three freeze-thaw cycles at day 4 postinfection. Particles were subsequently purified by ultracentrifugation at 100,000 × g for 2 h at 4°C. The BDV particles were purified from persistently infected Vero cells as described previously (4). Reporter assays of viral RNA were performed at 18 h posttransfection by lysing the cells with 50 μl passive lysis buffer (Promega) and measuring 20-μl samples of the lysate using the dual-luciferase kit (Promega) according to the manufacturer's protocol.
Genome end analyses.
Viral RNA was purified from NYMV stocks using the Peqgold RNA pure system (Peqlab) according to the manufacturer's protocol. 5′ and 3′ rapid amplification of cDNA ends (RACE) analyses were performed as previously described for BDV (27) using NYMV-specific primers for PCR. Briefly, for 5′ RACE, a synthetic RNA oligonucleotide was ligated to the viral RNA with or without prior treatment of the RNA with pyrophosphatase. For 3′-RACE, poly(A) or poly(C) tailing was performed to terminally elongate the viral RNA. All primer sequences are available on request.
RESULTS
NYMV replicates in the nucleus.
We first determined in which cellular compartment NYMV may replicate. Since specific antibodies detecting NYMV proteins are not available yet, we infected 4-week-old C57BL/6 mice with NYMV by the intraperitoneal route. The infected animals did not develop clinical symptoms until day 14, when the serum was collected. To determine if the serum might contain virus-specific antibodies, we transfected 293T cells with expression vectors encoding the various NYMV proteins and subjected the protein extracts to immunoblotting. The serum prominently recognized the viral nucleoprotein N and ORF3 but not the other NYMV proteins (Fig. 1A). When used for immunofluorescence analysis, the serum strongly stained the nucleus of NYMV-infected, but not uninfected, Vero cells (Fig. 1B), indicating that NYMV has a nuclear replication phase. To confirm this observation, we prepared whole-cell extracts (CE) and cytoplasmic (Cy) and nuclear (Nu) protein fractions of uninfected and NYMV-infected Vero cells. Immunoblots of these samples were probed for the cytoplasmic marker protein β-tubulin and the nuclear marker histone H3 to verify successful cell fractionation (Fig. 1C). Probing with the NYMV-specific antiserum revealed a prominent ∼44-kDa double band in the nuclear fraction of NYMV-infected cells (Fig. 1C), which represents the viral nucleoprotein N and the product of viral ORF3. The cytoplasmic fraction contained much lower levels of these viral proteins.
Fig 1.

NYMV replicates in the nucleus. (A) Specificity for NYMV antigens of serum from NYMV-infected mice. 293T cells were transfected with expression vectors encoding the indicated NYMV proteins or the empty vector as a control (Ø). At 24 h posttransfection the cells were lysed, and Western blot analysis was performed. Antibodies against β-tubulin were used to control for equal loading of the gel. (B) Immunofluorescence analysis. Infected (NYMV) and uninfected (Ø) Vero cells were stained with DAPI and NYMV antiserum. (C) Subcellular distribution of NYMV proteins. Whole-cell lysates as well as cytoplasmic (Cy) and nuclear (Nu) fractions were prepared from infected and uninfected Vero cells. Samples were subjected to immunoblotting using primary antibodies against histone H3 or β-tubulin and NYMV antiserum. (D) Subcellular distribution of viral RNA. Samples of RNA extracted from whole-cell lysates or cytoplasmic and nuclear fractions were subjected to Northern blot analysis using a DNA probe corresponding to nucleotides 138 to 1971 of the NYMV antigenome, which detects RNAs containing N and ORF2. n.i., not infected.
RNA samples extracted from whole cells and from the cytoplasmic or nuclear fractions mentioned above were subjected to Northern blot analysis using a virus-specific probe. Genome-sized viral RNA was exclusively detected in the nuclear fraction (Fig. 1D). Subgenomic transcripts of NYMV were present in both cell compartments, although they were clearly more abundant in the nucleus than in the cytoplasm (Fig. 1D). Taken together, these results demonstrated that replication of the NYMV genome includes the nucleus.
We next asked whether NYMV multiplication may be dependent on the activity of cellular RNA polymerase II as in the case of other RNA viruses that replicate in the nucleus such as influenza A virus and Thogoto virus (15, 28). To check this, we infected Vero cells with NYMV in the presence of either plain medium or medium containing 5 μg/ml of actinomycin D to block the activity of RNA polymerase II. As reported by others before (6, 28), influenza A virus strain WSN, which served as a positive control in this experiment, was strongly inhibited by the actinomycin D treatment, whereas VSV, which served as a negative control, was not significantly influenced by the drug treatment (Fig. 2). In the case of NYMV, the inhibition of cellular RNA polymerase II by actinomycin D resulted in a seemingly complete block of viral multiplication. In the presence of the drug, viral titers dropped by more than 1,000-fold (Fig. 2), indicating that replication of NYMV is strongly dependent on transcriptional activity of cellular RNA polymerase II.
Fig 2.
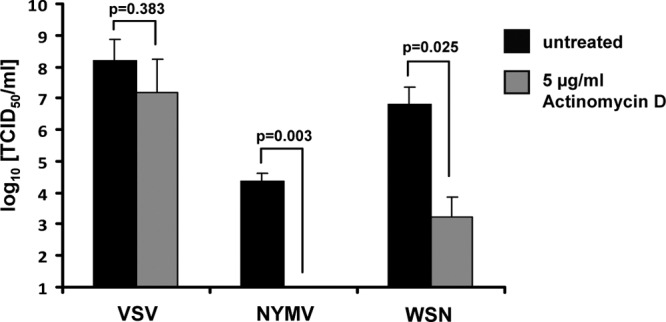
NYMV multiplication is dependent on cellular RNA polymerase II activity. Vero cells were infected with NYMV, VSV, or WSN using an MOI of 1. Immediately after infection, the cells were either treated with 5 μg/ml of actinomycin D (gray bars) or left untreated (black bars). Viral titers were determined at 24 h (for VSV) or at 40 h (for NYMV and WSN) postinfection by the 50% tissue culture infective dose (TCID50) assay. Bars represent the average values of two independent experiments. Standard deviations are shown. Significance was tested using the two-tailed t test.
ORF3 codes for a viral polymerase cofactor.
To gain more insight into the NYMV replication cycle, we determined the role of the previously uncharacterized viral proteins encoded by ORF2, ORF3, and ORF4 (Fig. 3). To analyze the contribution of these proteins to viral polymerase activity, we established a minireplicon assay based on a plasmid containing a negative-sense Gaussia luciferase reporter gene flanked by the NYMV leader and trailer sequences under the control of the T7 promoter. Only background levels of luciferase activity were detected when the NYMV minigenome was transfected into BSR-T7 cells together with expression constructs for the NYMV proteins N and L only (Fig. 3B). The same result was obtained when plasmids encoding viral ORF2 or ORF4 were coexpressed. In contrast, cotransfection of an ORF3-encoding plasmid resulted in high levels of luciferase activity (Fig. 3B). These results demonstrated that the NYMV protein encoded by ORF3, but not ORF2 and ORF4, is essential for in vitro reconstitution of functional viral polymerase complexes, suggesting that ORF3 encodes a viral polymerase cofactor.
Fig 3.

ORF3 of NYMV codes for a polymerase cofactor. (A) Schematic representation of the NYMV genome. Transcriptional start (S1–S6) and stop (T1–T6) signals are indicated. Protein-coding regions are shown as gray boxes. (B) Reconstitution of the NYMV polymerase complex. BSR-T7 cells were transfected with the NYMV minigenome encoding Gaussia luciferase, expression vectors encoding N and L, and plasmids encoding the indicated additional NYMV proteins. To normalize for transfection efficacy, a plasmid encoding firefly luciferase was cotransfected. At 72 h posttransfection, Gaussia and firefly luciferase-mediated light emissions were quantified. The normalized Gaussia luciferase RLU value obtained for the cells transfected with expression vectors for N, ORF3, and L was arbitrarily set to 100%. Bars show the average values of two independent duplicate experiments. Standard deviations are shown. (C) Interaction of the ORF3 product with other NYMV proteins in mammalian two-hybrid assays. 293T cells were transfected with a Gal4 promoter-dependent firefly luciferase construct, pCA-VP16/ORF3, and the indicated pCA-Gal4 constructs. A constitutive active Renilla luciferase reporter construct was cotransfected for normalization. At 24 h posttransfection, Renilla and firefly luciferase-mediated light emissions were quantified. BDV-P fused to the Gal4-binding domain served as the negative control; its normalized firefly RLU value was arbitrarily set to 1. Bars show the average values of three independent experiments. Standard deviations are shown. RLU = relative light units.
To determine whether the ORF3 product may interact with N and L, like P proteins of other mononegaviruses (25), mammalian two-hybrid experiments were performed (26) in which the indicated NYMV proteins were fused to the Gal4 DNA-binding domain or the VP16 transactivating domain, respectively. The P protein of BDV (BDV-P) fused to Gal4 served as a negative control in our study. We found that ORF3 specifically interacted with N, with itself, and with L (Fig. 3C). We further observed strong interaction between ORF3 and the NYMV protein encoded by ORF2 (Fig. 3C). Together with the fact that the ORF3 protein is present in the nucleus of NYMV-infected cells (Fig. 1C), we concluded from these data that ORF3 represents a polymerase cofactor of NYMV.
ORF2 codes for a negative regulator of viral polymerase activity.
The interaction of NYMV ORF3 with ORF2 prompted us to test if the latter protein can influence polymerase activity in the minireplicon system. For this, BSR-T7 cells were cotransfected with the NYMV minigenome and the indicated expression vectors, and luciferase activity was analyzed 72 h later. The luciferase signal of the positive control containing vectors encoding N, ORF3, and L only was arbitrarily set to 1 (Fig. 4A, lane 2). We found that the expression of ORF2 reduced minireplicon activity by approximately 30-fold (Fig. 4, lane 3). This inhibitory effect was dose dependent and still observed when 10-fold-higher or 10-fold-lower concentrations of the ORF2 expression vector were cotransfected (Fig. 4B). In contrast, coexpression of only ORF4 did not change minireplicon activity, whereas simultaneous expression of ORF4 together with ORF2 resulted in a substantial reduction of the luciferase signal (Fig. 4A, lanes 4 and 5). Coexpression of G moderately enhanced reporter gene expression (Fig. 4A, lane 6). Interestingly, the beneficial effect of G was most obvious when ORF2 was present in the system. Under our standard conditions, the inhibitory effect of ORF2 was attenuated if G was coexpressed alone (Fig. 4A, lane 7), and it was almost completely abolished if G was coexpressed with ORF4 (Fig. 4A, lane 9). From these data we concluded that the activity of the NYMV polymerase is negatively regulated by ORF2 and that this inhibitory effect is partially compensated by G, possibly in cooperation with ORF4.
Fig 4.

ORF2 negatively regulates NYMV polymerase activity. (A) BSR-T7 cells were transfected with the NYMV minigenome encoding Gaussia luciferase and the indicated pCA expression vectors. To normalize for transfection efficiency, a plasmid encoding firefly luciferase was cotransfected. At 72 h posttransfection, Gaussia and firefly luciferase-mediated light emissions were quantified. The normalized value obtained for the positive control (containing pCA-N, -ORF3, and -L only) was arbitrarily set to 1. Bars represent the average values of three independent experiments. Standard deviations are shown. (B) The inhibitory effect of ORF2 on viral polymerase activity is dose dependent. BSR-T7 cells were transfected as in panel A but with increasing amounts of the ORF2-encoding plasmid. The analysis was performed as described for panel A. The normalized value obtained for the positive control (containing pCA-N, -ORF3, and -L only) was arbitrarily set to 100%. Bars show the average values of three independent experiments. Standard deviations are shown. RLU = relative light units.
Formation of virus-like particles requires cooperation of G, ORF2, and ORF4.
We next asked whether ORF4 might encode the matrix protein of NYMV and stimulate viral particle formation. To address this question, we set up a VLP assay for NYMV. For this, BSR-T7 cells were transfected with the NYMV minigenome construct and expression vectors for the indicated viral proteins as described above. Three days later, supernatants were collected and transferred onto the 293T cells expressing the reconstituted NYMV polymerase complex. In this experimental system, the successful production of VLPs results in transfer of the NYMV minigenome to the recipient 293T cells, which thereby acquire the ability to synthesize luciferase.
As expected, supernatants from BSR-T7 cells transfected with expression constructs for only NYMV N, ORF3, and L failed to transfer substantial luciferase activity to 293T cells (Fig. 5, lane 1). Coexpression of G slightly stimulated VLP production (Fig. 5, lane 2). G-mediated transfer of luciferase activity was abolished if ORF2 was coexpressed (Fig. 5, lane 3). ORF4 had no negative influence but, interestingly, also failed to boost G-mediated luciferase transfer (Fig. 5, lane 4), suggesting that neither ORF2 nor ORF4 represents a functional matrix protein. Surprisingly, however, efficient transfer of the NYMV minigenome was observed if G was coexpressed together with ORF2 and ORF4 (Fig. 5, lane 5). Interestingly, this increase in luciferase activity was dependent on the presence of the reconstituted NYMV polymerase complex in the recipient cells (data not shown). These results indicated that the cooperation of ORF2 and ORF4 is required for reconstitution of a functional NYMV matrix protein and efficient transfer of the NYMV minigenome to new cells via VLPs.
Fig 5.

Proteins encoded by ORF2 and ORF4 cooperate for the production of infectious VLPs. BSR-T7 cells were transfected with the NYMV minigenome encoding Gaussia luciferase and the indicated pCA expression vectors. Supernatants were collected at 72 h posttransfection and clarified through a 0.45-μm filter. Five hundred microliters of the clarified supernatants were transferred onto 293T cells previously transfected with pCA-N, pCA-ORF3, and pCA-L. Forty-eight hours later, the 293T cells were lysed and Gaussia-mediated light emission was quantified. The value obtained for the negative control (receiving only pCA-N, -ORF3, and -L) (lane 1) was arbitrarily set to 1. Bars represent the average values of three independent experiments. Standard deviations are shown. RLU = relative light units.
To determine the interaction profile of the various NYMV proteins during VLP synthesis, we performed coimmunoprecipitation experiments (Fig. 6). Since specific antibodies are not yet available, HA- and Flag-tagged versions of the viral proteins were employed. Using the polymerase reconstitution and VLP formation assays described above, we confirmed the functionality of N-terminally tagged ORF2 protein and C-terminally tagged ORF4 and G proteins (data not shown). Our coimmunoprecipitation experiments showed that HA-tagged ORF2 specifically interacted with Flag-tagged ORF4 but not with Flag-tagged G (Fig. 6A). Similarly, HA-tagged ORF4 strongly interacted with Flag-tagged ORF2 but not with Flag-tagged G (Fig. 6B). We failed to observe specific interaction of G with ORF2 when the untagged ORF4 protein was present (Fig. 6C). Similar results were obtained when coimmunoprecipitation experiments were performed in the context of the minireplicon system with all viral proteins and the NYMV minigenome being present in the transfected cells (data not shown). These results are compatible with the view that ORF2 and ORF4 form a complex that may serve a matrix protein function. However, this complex (or its individual components) did not interact with NYMV G, presumably because membrane anchoring of G is required for association with M, as demonstrated for rabies virus (18).
Fig 6.

Products of ORF2 and ORF4 interact with each other but not with G. 293T cells were transfected with pCA expression vectors encoding HA-tagged ORF2 together with Flag-tagged ORF4 or Flag-tagged G (A) or HA-tagged ORF4 together with Flag-tagged ORF2 or Flag-tagged G (B). At 24 h posttransfection, protein extracts were subjected to immunoprecipitation using anti-Flag M2 beads. Flag- and HA-tagged proteins were detected using specific antibodies. (C) The ORF2-ORF4 protein complex does not interact with G. 293T cells were transfected with pCA expression vectors encoding HA-tagged ORF2, Flag-tagged G, and untagged ORF4. Immunoprecipitation was performed as described for panels A and B. The two bands detected for G/Flag presumably represent immature and cleaved forms of G. IP = immunoprecipitation; IB = immunoblotting.
Genomic RNA of NYMV is a poor beta interferon inducer and possesses 3′ overhangs.
Since genomic RNA of most but not all mononegaviruses is triphosphorylated at the 5′ terminus, it is readily recognized by cytoplasmic pattern recognition receptors. To address the question of whether the RNA of NYMV is sensed by these receptors, we isolated RNA from purified viral particles and determined its IFN-inducing capacity (Fig. 7). For this we employed a well-established assay (8) in which purified viral RNA is transfected into 293T cells that carry a reporter construct containing a luciferase gene under transcriptional control of the human IFN-β promoter (8). As previously reported (8), RNA from influenza A virus (FLUAV) or measles virus (MeV) particles strongly induced luciferase activity under these conditions (50- to- 70-fold induction over mock treatment), whereas RNA from BDV particles did not (Fig. 7A). Further, the IFN-β promoter-stimulating activity of FLUAV or MeV RNA was abolished if the samples were treated with shrimp alkaline phosphatase (SAP) before transfection. Only minimal activation of the luciferase reporter gene (∼5-fold) was observed if similar amounts of RNA isolated from NYMV particles were used for transfection (Fig. 7A).
Fig 7.

NYMV RNA is a poor inducer of IFN-β. Five hundred nanograms of RNA isolated from purified viral particles (A) or infected cells (B) was transfected into 293T cells that were transfected with a plasmid coding for firefly luciferase under the control of the IFN-β promoter. Where indicated, the viral RNA was treated with shrimp alkaline phosphatase (SAP) prior to transfection. Firefly luciferase activity was normalized for transfection efficacy, and the value for the mock control was arbitrarily set to 1. Bars represent the average values of three independent experiments. Standard deviations are shown. RLU = relative light units.
To verify these findings, similar experiments were performed with nonfractionated RNA isolated from virus-infected cells. As expected, we observed that RNA from FLUAV- or MeV-infected cells induced the IFN-β promoter-controlled luciferase gene very strongly (40- to 80-fold), whereas RNA isolated from BDV-infected cells did not (Fig. 7B). Transfection of RNA from NYMV-infected cells stimulated luciferase activity only weakly (∼5-fold) (Fig. 7B). Thus, unlike RNA from FLUAV and MeV, RNA derived from NYMV is an intrinsically poor IFN-β inducer.
To investigate the reason for why NYMV did not readily induce IFN, we performed 5′ and 3′ rapid amplification of cDNA ends (RACE) analyses of RNA derived from NYMV particles. For 5′ RACE, we used RNA ligase to covalently attach a short synthetic RNA of defined sequence to the 5′ end of the viral RNA before reverse transcriptase PCR. Surprisingly, this ligation reaction worked similarly well irrespective of whether or not the viral RNA was treated with tobacco acid pyrophosphatase to remove 5′-terminal triphosphates, suggesting that the bulk of NYMV genomic RNA is mono- rather than triphosphorylated. Bulk sequencing of the PCR product unambiguously revealed that the 5′-terminal sequence of NYMV genomic RNA is 5′-GCAAACC…-3′ (Fig. 8A, left panel). For 3′ RACE, we tailed the viral RNA either with poly(A) or poly(C) before RT-PCR. Bulk sequencing of the PCR products revealed that the 3′-terminal sequence is 5′-…GCUUUGCAU-3′ (Fig. 8A, middle and right panels). Thus, the extreme 5′ terminus of the NYMV genome is not complementary to the extreme 3′ terminus. In the panhandle conformation, the NYMV genome is predicted to form a secondary structure with a protruding 3′ end (Fig. 8B). Overhanging nucleotides at the 3′ termini and monophosphorylated 5′ termini are characteristic features of members of the family Bornaviridae, which are also poor IFN inducers (8, 16).
Fig 8.
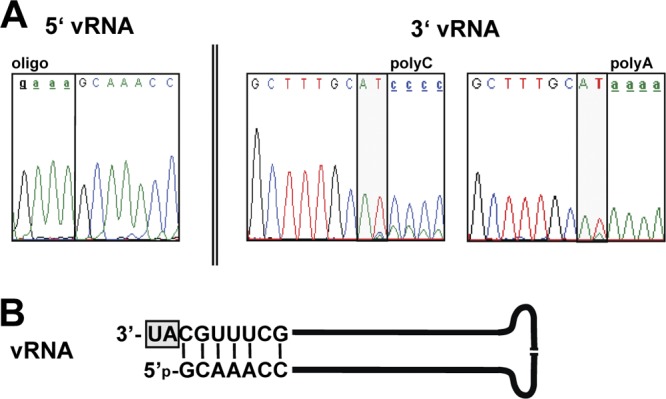
The NYMV genome is monophosphorylated and contains noncomplementary nucleotides at the 3′ end. (A) Analysis of terminal genome sequences by RACE. For 5′ RACE, a synthetic RNA (oligonucleotide) was ligated to viral RNA (vRNA) without prior pyrophosphatase treatment. For 3′ RACE, viral RNA was terminally elongated with poly(A) (right panel) or poly(C) (middle panel). Electropherograms from direct sequencing of PCR products are shown. (B) Deduced structure of the NYMV genome in the panhandle conformation. The molecule is predicted to have a monophosphate at the 5′ end and a 3′ overhang (gray box).
DISCUSSION
We demonstrate here that NYMV has several unique biological features that support the notion that this tick-borne virus indeed represents the prototype member of a new genus, designated Nyavirus, in the order Mononegavirales (19). The most peculiar features of NYMV include nuclear genome replication, the unusual location of the polymerase cofactor-encoding gene in the viral genome, the need for two rather than one matrix protein for the production of infectious particles, and evasion of the innate immune response by maintaining a monophosphorylated viral genome with a protruding 3′ end.
To date, the only other known animal viruses assigned to the order Mononegavirales that replicate in the nucleus belong to the family Bornaviridae (4, 24). Replication of bornaviruses is presumably restricted to this cell compartment for different reasons. Members of the family Bornaviridae establish a persistent infection and ensure their dissemination by close association of the viral ribonucleoprotein (RNP) complex to the host chromatin (17). Further, they use the cellular splicing machinery for generating transcripts encoding G and L (25). Splicing of NYMV transcripts is unlikely to occur, since the six known NYMV proteins encoded by nonoverlapping transcription units (Fig. 3A) are required and sufficient for efficient viral particle formation (Fig. 5). However, it remains possible that additional not-yet-recognized viral proteins are synthesized from spliced NYMV transcripts. An alternative explanation for why NYMV replicates in the nucleus of infected cells includes the possibility that viral replication requires nuclear factors. In line with this notion, we observed a strong dependence of NYMV multiplication on transcriptional activity of the cellular RNA polymerase II (Fig. 2). A similar dependence was previously reported for members of the family Orthomyxoviridae (15, 28), which use the cap structure of cellular mRNAs as primers for their own transcription (3, 14). It remains unknown whether NYMV might use a similar mechanism to cap its transcripts or whether RNA polymerase II activity is required for a different step in the NYMV replication cycle. It will also be of interest to determine whether the viral RNP complex of NYMV associates with host chromosomes to facilitate persistent intranuclear infection as recently described for BDV (17).
Our minireplicon experiments revealed that ORF3 rather than ORF2 of NYMV codes for a viral polymerase cofactor (Fig. 3). This result was unexpected, as the polymerase cofactor P is encoded by the second transcription unit in all other known viruses of the order Mononegavirales. A distinct but remotely similar gene constellation is found in bornaviruses, where both the N and X genes are located upstream of the P gene. However, unlike in NYMV, the X and P proteins of bornaviruses are translated from overlapping ORFs of a single viral mRNA (32). Some experimental evidence suggests that ORF2 of NYMV and X of bornaviruses may execute similar functions. Like the X protein of BDV (22), ORF2 of NYMV is a potent negative regulator of viral polymerase activity (Fig. 4). However, the inhibitory effect of ORF2 was strongly attenuated if ORF4 and the G protein were expressed simultaneously (Fig. 4A). A similar phenomenon was reported for X of BDV, where coexpression of plasmids coding for M and G was found to neutralize the inhibitory effect of X on viral polymerase activity (20).
Our experimental data revealed that both ORF2 and ORF4 are required for the formation of infectious VLPs and efficient transfer of the NYMV minigenome (Fig. 5), indicating that the functional NYMV matrix protein may be a two-protein complex rather than a single protein. This situation is reminiscent of the situation in filoviruses, where VP24 seems to assist VP40 in viral morphogenesis (9). VP24, however, seems to possess no classical matrix protein function. Rather, it interacts with the viral ribonucleoprotein complex, thereby inhibiting polymerase activity and presumably facilitating incorporation of the complex into budding virus particles (31). Since ORF2 of NYMV is also a negative regulator of polymerase activity and since ORF2 strongly interacts with the ORF3 polymerase cofactor, it is conceivable that VP24 of filoviruses and ORF2 of NYMV play similar roles in the viral budding process. Interestingly, this similarity can even be extended to the X protein of bornaviruses. BDV X is also a negative regulator of polymerase activity (22) that stimulates VLP formation if coexpressed with M and G (20). Antibodies specifically recognizing individual viral proteins are required for a more detailed analysis of the roles of ORF2 and ORF4 in the formation of NYMV particles.
Most negative-stranded RNA viruses are readily detected by cytoplasmic pattern recognition receptors, because the 5′ ends of their genomes are usually triphosphorylated (10). Known exceptions to this rule are Hantaan virus, Crimean-Congo hemorrhagic fever virus, and mammalian and avian bornaviruses (8, 23). We showed here that NYMV belongs to the group of viruses that do not strongly activate the innate immune system (Fig. 7), most probably because the 5′ terminus of its genome is monophosphorylated rather than triphosphorylated. Strong experimental evidence in favor of a monophosphorylated state of NYMV genomic RNA is our finding that a synthetic RNA molecule could efficiently be ligated to the 5′ terminus of untreated viral RNA. As in the case of bornaviruses (16), there was no need to unblock such RNA by treatment with pyrophosphatase prior to ligation. Moreover, sequencing of the extreme ends of the viral RNA by RACE technology revealed a 2-nucleotide overhang at the 3′ end of the NYMV genome. The predicted panhandle structure of the NYMV genome is thus imperfect (Fig. 8B), which might further contribute to poor recognition by pattern recognition receptors.
Taken together, the predicted structure of the NYMV genome termini resembles that of BDV, although the 3′ overhang is two rather than four nucleotides, as in the case of BDV (16). It is unclear at present how NYMV generates these terminal genome structures. In the case of BDV, the genomic RNA is elongated on internal template motifs after realignment of the nascent 3′ terminus. As BDV removes four nucleotides from the 5′ end of its genome to generate a monophosphorylated molecule, it appears to employ this unique mechanism of retrieving genetic information for maintaining genome integrity (8, 16). Reverse genetic technology is required to address the question of whether NYMV may use a similar mechanism to process the termini of its genomic RNA.
In conclusion, our study revealed several surprising biological features of NYMV and showed that this virus resembles bornaviruses in some aspects of its viral life cycle and filoviruses in others. It will be of interest to determine whether our findings can be extended to Midway virus, the second member of the genus Nyavirus, and whether NYMV and Midway virus exhibit similar biological properties.
ACKNOWLEDGMENTS
We thank David Wang from the Washington University School of Medicine, St. Louis, MO, for providing RNA from NYMV-infected Vero cells for initial work and Robert B. Tesh from the University of Texas Medical Branch, Galveston, TX, for providing NYMV tick isolate 39. We further thank many colleagues in our department for helpful comments on the manuscript.
This work was supported by grant SCHN 765/3-1 from the Deutsche Forschungsgemeinschaft.
Footnotes
Published ahead of print 25 July 2012
REFERENCES
- 1. Ackermann A, Kugel D, Schneider U, Staeheli P. 2007. Enhanced polymerase activity confers replication competence of Borna disease virus in mice. J. Gen. Virol. 88:3130–3132 [DOI] [PubMed] [Google Scholar]
- 2. Bamberg S, Kolesnikova L, Moller P, Klenk HD, Becker S. 2005. VP24 of Marburg virus influences formation of infectious particles. J. Virol. 79:13421–13433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bouloy M, Plotch SJ, Krug RM. 1978. Globin mRNAs are primers for the transcription of influenza viral RNA in vitro. Proc. Natl. Acad. Sci. U. S. A. 75:4886–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Briese T, de la Torre JC, Lewis A, Ludwig H, Lipkin WI. 1992. Borna disease virus, a negative-strand RNA virus, transcribes in the nucleus of infected cells. Proc. Natl. Acad. Sci. U. S. A. 89:11486–11489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Easton AJ, Domachowske JB, Rosenberg HF. 2004. Animal pneumoviruses: molecular genetics and pathogenesis. Clin. Microbiol. Rev. 17:390–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flamand A, Pese D, Bussereau F. 1977. Effect of actinomycin D and cytosine arabinoside on rabies and VSV multiplication. Virology 78:323–327 [DOI] [PubMed] [Google Scholar]
- 7. Gerlier D, Lyles DS. 2011. Interplay between innate immunity and negative-strand RNA viruses: towards a rational model. Microbiol. Mol. Biol. Rev. 75:468–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Habjan M, et al. 2008. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS One 3:e2032 doi:10.1371/journal.pone.0002032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han Z, et al. 2003. Biochemical and functional characterization of the Ebola virus VP24 protein: implications for a role in virus assembly and budding. J. Virol. 77:1793–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hornung V, et al. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997 [DOI] [PubMed] [Google Scholar]
- 11. Kaiser MN. 1966. Viruses in ticks. I. Natural infections of Argas (Persicargas) arboreus by Quaranfil and Nyamanini viruses and absence of infections in A. (p.) persicus in Egypt. Am. J. Trop. Med. Hyg. 15:964–975 [PubMed] [Google Scholar]
- 12. Kato H, Takahasi K, Fujita T. 2011. RIG-I-like receptors: cytoplasmic sensors for nonself RNA. Immunol. Rev. 243:91–98 [DOI] [PubMed] [Google Scholar]
- 13. Kemp GE, Lee VH, Moore DL. 1975. Isolation of Nyamanini and Quaranfil viruses from Argas (Persicargas) arboreus ticks in Nigeria. J. Med. Entomol. 12:535–537 [DOI] [PubMed] [Google Scholar]
- 14. Leahy MB, Dessens JT, Nuttall PA. 1997. In vitro polymerase activity of Thogoto virus: evidence for a unique cap-snatching mechanism in a tick-borne orthomyxovirus. J. Virol. 71:8347–8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mahy BW, Hastie ND, Armstrong SJ. 1972. Inhibition of influenza virus replication by-amanitin: mode of action. Proc. Natl. Acad. Sci. U. S. A. 69:1421–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martin A, Hoefs N, Tadewaldt J, Staeheli P, Schneider U. 2011. Genomic RNAs of Borna disease virus are elongated on internal template motifs after realignment of the 3′ termini. Proc. Natl. Acad. Sci. U. S. A. 108:7206–7211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matsumoto Y, et al. 2012. Bornavirus closely associates and segregates with host chromosomes to ensure persistent intranuclear infection. Cell Host Microbe 11:492–503 [DOI] [PubMed] [Google Scholar]
- 18. Mebatsion T, Weiland F, Conzelmann KK. 1999. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 73:242–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mihindukulasuriya KA, et al. 2009. Nyamanini and midway viruses define a novel taxon of RNA viruses in the order Mononegavirales. J. Virol. 83:5109–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perez M, de la Torre JC. 2005. Identification of the Borna disease virus (BDV) proteins required for the formation of BDV-like particles. J. Gen. Virol. 86:1891–1895 [DOI] [PubMed] [Google Scholar]
- 21. Poenisch M, Burger N, Staeheli P, Bauer G, Schneider U. 2009. Protein X of Borna disease virus inhibits apoptosis and promotes viral persistence in the central nervous systems of newborn-infected rats. J. Virol. 83:4297–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Poenisch M, Unterstab G, Wolff T, Staeheli P, Schneider U. 2004. The X protein of Borna disease virus regulates viral polymerase activity through interaction with the P protein. J. Gen. Virol. 85:1895–1898 [DOI] [PubMed] [Google Scholar]
- 23. Reuter A, et al. 2010. Avian bornaviruses escape recognition by the innate immune system. Viruses 2:927–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rinder M, et al. 2009. Broad tissue and cell tropism of avian bornavirus in parrots with proventricular dilatation disease. J. Virol. 83:5401–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schneider U. 2005. Novel insights into the regulation of the viral polymerase complex of neurotropic Borna disease virus. Virus Res. 111:148–160 [DOI] [PubMed] [Google Scholar]
- 26. Schneider U, Blechschmidt K, Schwemmle M, Staeheli P. 2004. Overlap of interaction domains indicates a central role of the P protein in assembly and regulation of the Borna disease virus polymerase complex. J. Biol. Chem. 279:55290–55296 [DOI] [PubMed] [Google Scholar]
- 27. Schneider U, Schwemmle M, Staeheli P. 2005. Genome trimming: a unique strategy for replication control employed by Borna disease virus. Proc. Natl. Acad. Sci. U. S. A. 102:3441–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Siebler J, Haller O, Kochs G. 1996. Thogoto and Dhori virus replication is blocked by inhibitors of cellular polymerase II activity but does not cause shutoff of host cell protein synthesis. Arch. Virol. 141:1587–1594 [DOI] [PubMed] [Google Scholar]
- 29. Takahashi M, et al. 1982. Isolation and characterization of Midway virus: a new tick-borne virus related to Nyamanini. J. Med. Virol. 10:181–193 [DOI] [PubMed] [Google Scholar]
- 30. Taylor RM, Hurlbut HS, Work TH, Kingston JR, Hoogstraal H. 1966. Arboviruses isolated from argas ticks in Egypt: Quaranfil, Chenuda, and Nyamanini. Am. J. Trop. Med. Hyg. 15:76–86 [DOI] [PubMed] [Google Scholar]
- 31. Watanabe S, Noda T, Halfmann P, Jasenosky L, Kawaoka Y. 2007. Ebola virus (EBOV) VP24 inhibits transcription and replication of the EBOV genome. J. Infect. Dis. 196(Suppl 2):S284–S290 [DOI] [PubMed] [Google Scholar]
- 32. Wehner T, et al. 1997. Detection of a novel Borna disease virus-encoded 10 kDa protein in infected cells and tissues. J. Gen. Virol. 78(Pt 10):2459–2466 [DOI] [PubMed] [Google Scholar]
- 33. Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 283:61–119 [DOI] [PubMed] [Google Scholar]