

Abstract
During Theiler's murine encephalomyelitis virus (TMEV) infection of macrophages, it is thought that high interleukin-6 (IL-6) levels contribute to the demyelinating disease found in chronically infected SJL/J mice but absent in B10.S mice capable of clearing the infection. Therefore, IL-6 expression was measured in TMEV-susceptible SJL/J and TMEV-resistant B10.S macrophages during their infection with TMEV DA strain or responses to lipopolysaccharide (LPS) or poly(I · C). Unexpectedly, IL-6 production was greater in B10.S macrophages than SJL/J macrophages during the first 24 h after stimulation with TMEV, LPS, or poly(I · C). Further experiments showed that in B10.S, SJL/J, and RAW264.7 macrophage cells, IL-6 expression was dependent on extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) and enhanced by exogenous IL-12. In SJL/J and RAW264.7 macrophages, exogenous IL-6 resulted in decreased TMEV replication, earlier activation of STAT1 and STAT3, production of nitric oxide, and earlier upregulation of several antiviral genes downstream of STAT1. However, neither inhibition of IL-6-induced nitric oxide nor knockdown of STAT1 diminished the early antiviral effect of exogenous IL-6. In addition, neutralization of endogenous IL-6 from SJL/J macrophages with Fab antibodies did not exacerbate early TMEV infection. Therefore, endogenous IL-6 expression after TMEV infection is dependent on ERK MAPK, enhanced by IL-12, but too slow to decrease viral replication during early infection. In contrast, exogenous IL-6 enhances macrophage control of TMEV infection through preemptive antiviral nitric oxide production and antiviral STAT1 activation. These results indicate that immediate-early production of IL-6 could protect macrophages from TMEV infection.
INTRODUCTION
Interleukin-6 (IL-6) is a pleiotropic cytokine expressed by many cell types that is induced by microbes and other cytokines. IL-6 can play a beneficial role during the immune response by contributing to neutrophil activity, nitric oxide (NO) production, and development of the Th17 CD4 T cell subset, as well as playing a beneficial role in the brain (43) and neuronal health (9, 11). In contrast, persistent IL-6 induction during infection can contribute to detrimental effects on surrounding neurons or bone and poor control of cancer or autoimmune responses (24). During microbial induction of IL-6, activation of the p38 mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) MAPK signaling pathways contributes to IL-6 expression (21, 49) and stabilization of IL-6 mRNA (2) for protein translation.
Macrophages express IL-6 when they encounter microbes, such as bacteria, fungi, or viruses (44). While the neutrophil- and Th17-promoting activities of IL-6 are critical in controlling certain bacterial or fungal infections, less is known about the role of IL-6 in viral infections. Recently, an antiviral role for IL-6 has been shown in mouse poxvirus infections, where early IL-6 production is required to control a potentially lethal infection (30), and in lymphocytic choriomeningitis virus infection, where late IL-6 production was shown to be critical for clearance of persistent viruses (12). However, failure to clear viruses from infected macrophages could result in viral spread to other tissues due to macrophage chemotaxis and could result in persistent IL-6 expression in affected tissue causing inflammatory pathologies and autoimmune diseases (16). Similarly, persistent elevation of IL-6 is associated with the development of experimental autoimmune encephalomyelitis (EAE) in mice immunized peripherally with myelin peptides, whereas mice deficient in IL-6 are resistant to development of EAE (32, 40). Such data suggest that IL-6 expression may be essential to control early viral infection, yet contributes to pathology when IL-6 is expressed persistently.
Theiler's murine encephalomyelitis virus (TMEV) causes an acute infection that is cleared through innate and adaptive immune responses in most mouse strains yet produces debilitating sequelae in susceptible mice (33). C57BL/6 and B10.S mice are prototypical TMEV-resistant strains, whereas SJL/J mice fail to clear TMEV from macrophages and dendritic cells, resulting in persistent infection (25). Subsequent infection of macrophages in the central nervous system (CNS) of SJL/J mice leads to a demyelinating disease similar to human multiple sclerosis (5). Several reports have shown that a week after infection with TMEV, CNS macrophages, microglial cells, and astrocytes from SJL/J mice express IL-6 to a greater extent than those from C57BL/6 mice (14, 17, 34). A proposed theory is that chronic production of IL-6 in the CNS of persistently infected SJL/J mice contributes to demyelinating disease in response to TMEV (17).
We propose that the dichotomy of TMEV persistence seen in resistant and susceptible macrophages depends on early differences in cytokine production. We have previously shown that B10.S macrophages produce more IL-12 p70 in response to TMEV infection than SJL/J macrophages in vitro (37). Significantly, when SJL/J macrophages are pretreated with IL-12, TMEV replication is reduced to levels comparable to those with B10.S macrophages. Paradoxically, SJL/J macrophages express more beta interferon (IFN-β) than B10.S macrophages before and during infection with TMEV (37), yet this enhanced endogenous IFN-β expression is insufficient to control TMEV replication. However, pretreatment with additional exogenous recombinant IFN-β decreases TMEV replication in SJL/J macrophages to amounts seen in B10.S macrophages (37). We found that p38 ERK MAPK pathways are activated in response to TMEV infection of B10.S or SJL/J macrophages and play a role in IL-12 and IFN-β expression (37). Therefore, differences in MAPKs, IL-12, and IFN-β may be related to disparities in susceptibility of macrophages to persistent infection with TMEV.
In order to further understand the differences between SJL/J and B10.S mice that could account for differences in TMEV persistence, we examined IL-6 expression in macrophages during the early response to TMEV. Unexpectedly, we found that during the early response to infection with TMEV, B10.S macrophages expressed significantly more IL-6 than SJL/J macrophages. Moreover, pretreatment of macrophages with exogenous IL-6 reduced TMEV replication, but neutralization of endogenous TMEV-induced IL-6 with Fab antibodies failed to affect TMEV replication in SJL/J macrophages. Additionally, we show that TMEV-induced IL-6 secretion in macrophages depends on ERK MAPK and is enhanced by IL-12. However, neither IL-6-induced nitric oxide production nor STAT1 activation is essential to IL-6 reduction of TMEV infection.
MATERIALS AND METHODS
Mice, virus, cell lines, and reagents.
The 8- to 12-week-old female B10.S and SJL/J mice were obtained from Jackson Laboratories (Bar Harbor, ME). RAW264.7 cells were obtained from the American Type Culture Collection (Rockville, MD) and maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) with 50 μg/ml gentamicin. The p38 MAPK inhibitor SB203580 and ERK MAPK inhibitor U0126 were obtained from Promega Corporation (Madison, WI), Escherichia coli O127:B8 lipopolysaccharide (LPS) was obtained from Sigma Chemical Co. (St. Louis, MO), and poly(I·C) was obtained from InvivoGen (San Diego, CA). The inducible nitric oxide synthase (iNOS) inhibitor, L-NIL, was obtained from TOCRIS Bioscience (Bristol, United Kingdom) and incubated with macrophages, where indicated, at 10 μM. The nitric oxide donor spermine NONOate was obtained from Invitrogen (Carlsbad, CA) and incubated with macrophages, where indicated, at 1 mM. Affinity-purified neutralizing rat antibody to mouse IL-6 (clone MP5-20F3) (48) was obtained from Invitrogen. Fab fragments of neutralizing anti-IL-6 were obtained after papain digestion using the Pierce Fab preparation kit (Thermo Fisher Scientific, Rockford, IL). The initial stock of the DA strain of TMEV was obtained from Kristen Drescher, Department of Medical Microbiology and Immunology, Creighton University, Omaha, NE. TMEV was grown in BHK-21 cells. The titer of stock cultures of TMEV was 2.5 × 106 PFU/ml, and macrophage cultures were infected with 2.5 × 105 PFU of TMEV unless otherwise stated.
Macrophage preparations.
Macrophages were elicited by intraperitoneal injection of 2 ml thioglycolate broth into mice. Four days later, the peritoneal cavities were each flushed with 2 ml DMEM, and the cells were incubated at 1 × 106 cells/2 ml of DMEM cell culture medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS) (Invitrogen) and 50 μg/ml gentamicin (Invitrogen). After 24 h, nonadherent cells were removed and 1 ml of culture medium was added. Adherent cells were greater than 90% Mac-1+ as determined by fluorescence-activated cell sorter (FACS) analysis. These macrophages were either untreated or pretreated for 30 min with 20 μM SB203580, 40 μM U0126, 1 ng/ml IL-12 p70 (p35/p40; BD-Pharmingen, San Diego, CA), 10 ng/ml IFN-β (Minneapolis, MN), or 10 ng/ml IL-12 p40 (BD-Pharmingen). Untreated or pretreated macrophages were uninfected, infected with 100 μl of the TMEV stock (2.5 × 105 PFU), stimulated with 1 μg/ml LPS, stimulated with 50 μg poly(I · C), or left unstimulated. After 3, 8, or 24 h of infection or stimulation, supernatants were collected for enzyme-linked immunosorbent assay (ELISA), and cell extracts were collected for RNA preparation and quantitative reverse transcription-PCR (qRT-PCR).
RNA interference.
Validated inhibitory small interfering RNA (siRNA) targeting mouse STAT1 and STAT3 was purchased from Cell Signaling, Inc. (Danvers, MA) and transfected into RAW264.7 cells according to the manufacturer's specifications using the Amaxa nucleofection kit 36 h prior to challenge with TMEV.
RNA preparation and qRT-PCR.
RNA was extracted from cells using the RNAeasy kit of Qiagen (Valencia, CA), the PerfectPure kit from 5Prime (Gaithersburg, MD), or the Purelink kit from Ambion/Invitrogen (Carlsbad, CA) according to the manufacturer's specifications. One hundred nanograms to 1 μg of RNA was reverse transcribed in 0.5 mM (each) dATP, dGTP, dTTP, and dCTP and 20 U of RNase inhibitor with Superscript II reverse transcriptase (Invitrogen) at 42°C for 1.5 h followed by 94°C for 5 min. One twenty-fifth of the cDNA sample was incubated with 0.4 μM the following primer pairs (Invitrogen): IFN-β sense 5′ ATGAACAACAGGTGGATCCTCC 3′ and antisense 5′ AGGAGCTCCTGACATTTCCGAA 3′, IL-6 sense 5′ ATGAAGTTCCTCTCTGCAAGAGACT 3′ and antisense 5′ CACTAGGTTTGCCGAGTAGATCTC 3′, IFN response factor 1 (IRF1) sense 5′ ATGCCAATCACTCGAATGCGGA 3′ and antisense 5′ GGCTGCCACTCAGACTGTTCAA 3′, IRF7 sense 5′ CCAGCGAGTGCTGTTTGGAGAC 3′ and antisense 5′ TTCCCTATTTTCCGTGGCTGGG 3′, IRF9 sense 5′ ATGGCCTCAGGCAAAGTACGCT 3′ and antisense 5′ TTCGCTTGCATGGTGATTTCTG 3′, TMEV sense 5′ CTTCCCATTCTACTGCAATG 3′ and antisense 5′ GTGTTCCTGGTTTACAGTAG 3′, or GAPDH (glyceraldehyde-3-phosphate dehydrogenase) sense 5′ TTGTCAGCAATGCATCCTGCAC 3′ and antisense 5′ ACAGCTTTCCAGAGGGGCCATC 3′. Quantitative PCRs were run on an ABI Prism 7000 thermal cycler at 50°C for 2 min and 95°C for 10 min, followed by 45 cycles of 95°C for 15 s and 60°C for 30 s. Cycle thresholds (CT) of sample were normalized to the CT of GAPDH for that sample (ΔCT) and then normalized to the average ΔCT of the control samples (ΔΔCT), after which data were expressed as relative levels of mRNA using 2−ΔΔCT. When macrophages were pretreated, TMEV RNA data are reported as percentages of TMEV RNA in nontreated but infected macrophages.
ELISAs.
ELISA plates were coated with antibodies to mouse IL-6 (MP5-20F3; BD-Pharmingen), the plates were blocked with phosphate-buffered saline (PBS)–10% FBS. After washes, cell culture supernatants or serial dilutions of recombinant IL-6 (BD-Pharmingen) were added to wells. After 2 h, biotinylated antibody to mouse IL-6 (MP5-32C11; BD-Pharmingen) was added to each well. After 1 h, streptavidin-horseradish peroxidase (1:1,000; BD-Pharmingen) was added for 30 min, and then 3,3′,5,5′-tetramethylbenzindine substrate–hydrogen peroxide solution (BD-Pharmingen) was added to each well. IL-6 was measured by determining optical densities at a 450-nm wavelength (OD450s) with a reference OD570 using an ELISA spectrophotometric plate reader.
PAGE and Western blot analysis.
Cell lysates were obtained from RAW cells challenged with TMEV with or without treatment with 10 ng/ml recombinant murine IL-6 (BD-Pharmingen, San Diego, CA) or murine recombinant IFN-β (Interferon Source, Piscataway, NJ). Twenty microliters of each sample containing 20 μg of protein in sample buffer with bromophenol blue was run on a 10% SDS–Tris-glycine-polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was treated with blocking buffer for 1 h at room temperature, followed by incubation in a 1:500 dilution of rabbit IgG anti-phospho-STAT1 (Invitrogen, Camarillo, CA), a 1:1,000 dilution of anti-phospho-STAT3 (Cell Signaling, Beverly, MA), a 1:500 dilution of anti-mouse STAT1 (Invitrogen), a 1:1,000 dilution of anti-mouse STAT3 (Invitrogen), or a 1:500 dilution of mouse anti-tubulin E7 (Developmental Studies Hybridoma Bank, University of Iowa) and then a 1:5,000 dilution of IRDye 800CW goat anti-rabbit IgG (Rockland Immunochemicals, Inc., Gilbertsville, PA) or Alexa Fluor 680-labeled anti-mouse IgG (Rockland Immunochemicals). The washed membrane was scanned with a LI-COR Odyssey infrared imaging system, and densitometric analysis was done with LI-COR imaging software.
NO assay.
Induction of nitric oxide (NO) was assayed in culture supernatants by measuring nitrite using the Greiss reagent kit from Invitrogen. Briefly, 20 μl of Griess reagent was mixed with 150 μl of supernatant plus 130 μl of deionized water and incubated for 30 min at room temperature. Color development at 570 nm, which is proportional to nitric oxide in supernatants, was measured with a spectrophotometer.
Statistical analysis.
Student's t test of the GraphPad Prism software was used to determine the significance of differences between means; a P value of <0.05 was considered significant.
RESULTS
TMEV-induced IL-6 production is greater in TMEV-resistant B10.S macrophages.
IL-6 contributes to both antiviral immunity and virus-induced pathology (3, 41). TMEV is cleared from macrophages of TMEV-resistant mice but persists in (CNS) macrophages in susceptible SJL/J mice (5). Therefore, IL-6 mRNA and protein levels from TMEV-susceptible SJL/J and TMEV-resistant B10.S macrophages were determined following challenge with TMEV. IL-6 mRNA was detected at 3 and 8 h after TMEV challenge (Fig. 1A) in both B10.S and SJL/J macrophages; however, B10.S macrophages produced more IL-6 mRNA than SJL/J macrophages at 8 h after infection. Similarly, B10.S macrophages produced significantly more IL-6 protein 8 h after TMEV challenge than SJL/J macrophages in response to TMEV (Fig. 1B). Because TMEV RNA replication is significantly higher in SJL/J macrophages than in B10.S macrophages (37), the data here suggest that enhanced early IL-6 production may contribute to better control of TMEV replication.
Fig 1.

TMEV induces IL-6 expression in macrophages. IL-6 mRNA or protein secretion was induced from SJL/J or B10.S macrophages (MΦ) 3 and 8 h after infection with TMEV. SJL/J or B10.S macrophages (1 × 106) were infected with 2 × 105 to 10 × 105 PFU of TMEV in the absence (A and B) or presence of 10 ng/ml recombinant IL-6 (rIL-6) (C and E), 0.01 to 10 ng/ml rIL-6 (D), or 3 μg/ml anti-IL-6-Fab (E), or SJL/J macrophages were treated with 10 ng/ml rIL-6, which was removed after 30 min (D), or SJLJ macrophages were treated with 10 ng/ml rIL-6 or recombinant IFN-β starting at 1, 3, 6, or 7 h p.i. (F). RNA was reverse transcribed, and relative levels of IL-6 mRNA were evaluated by real-time PCR (A), relative levels of IL-6 protein were evaluated by ELISA (B), and relative levels of TMEV were evaluated by real-time PCR (C, D, E, and F). MOI, multiplicity of infection; Thio, thioglycollate. Data are means of 3 to 5 samples per time point evaluated by Student's t test. *, P ≤ 0.05; **, P ≤ 0.01.
To determine if exogenous IL-6 could impede TMEV replication in SJL/J macrophages during early infection, recombinant IL-6 was added to SJL/J macrophages 30 min prior to TMEV infection and either left in the medium during infection or washed out by changing the medium just prior to infection. Treatment with 10 ng/ml exogenous IL-6 significantly reduced TMEV replication at 8 h postinfection (p.i.) in SJL/J macrophages (Fig. 1C). In addition, treatment with as little as 0.1 ng/ml of exogenous IL-6 significantly reduced TMEV RNA at 24 h p.i. in SJL/J macrophages (Fig. 1D). Equally significant, IL-6 preemptively triggered antiviral activity in SJL macrophages even when exogenous IL-6 was added for only 30 min and washed out prior to infection (Fig. 1D). These findings suggest that early differences in the amount of IL-6 present during the course of infection play an important role in the establishment of TMEV in SJL/J macrophages.
To examine whether TMEV-induced endogenous IL-6 contributes to control of TMEV replication in SJL/J mice, neutralizing anti-IL-6 (IgG; clone MP5-20F3) antibody was added to SJL/J macrophages at the time of TMEV infection (48). Surprisingly, addition of either whole anti-IL-6 antibody or isotype control antibody resulted in a significant reduction in TMEV replication (see Fig. S1A and S1B in the supplemental material). These findings do not preclude an antiviral role for IL-6, since interactions between the IgG antibody and Fcγ receptors may also induce antiviral immunity in macrophages. Indeed, addition of isotype antibody had a similar effect on TMEV infection of macrophages. Therefore, we treated the whole antibody with papain and removed the Fc fragments to generate Fab anti-IL-6. Addition of Fab anti-IL-6 to SJL/J macrophages at the time of infection did not decrease but also did not increase TMEV replication in infected SJL/J macrophages (Fig. 1E).
To determine the time frame at addition in which exogenous IL-6 is most effective at reducing TMEV RNA, we added 10 ng/ml recombinant IL-6 at 1, 3, 6, and 7 h or exogenous IFN-β 7 h p.i. of SJL/J macrophages with TMEV. Addition of exogenous IL-6 at 1 and 3 h p.i. significantly reduced TMEV RNA compared to the level in untreated SJL/J macrophages that were infected with TMEV (Fig. 1F). Addition of IL-6 at 6 and 7 h p.i. failed to significantly reduce TMEV RNA in SJL/J macrophages compared to the level in untreated macrophages. In contrast, addition of IFN-β at 7 h p.i. significantly reduced TMEV RNA in SJL/J macrophages. Thus, while small amounts of added IL-6 were capable of controlling TMEV replication in SJL/J macrophages in vitro, it appears that a sufficient quantity of endogenous IL-6 is not produced quickly enough to provide protection because the beneficial effects of IL-6 are only evident prior to 6 h p.i. These results confirm that the antiviral effect of IL-6 is most effective very early during the course of TMEV infection of macrophages. These findings warrant further investigation into strain differences in early TMEV-induced IL-6 expression.
TMEV-induced IL-6 expression in macrophages is dependent on ERK MAPK.
The cell signaling pathway activated by TMEV infection leading to IL-6 expression in macrophages is not well understood. We previously showed that TMEV infection of macrophages from both B10.S and SJL/J mice strongly activates ERK MAPK and weakly activates p38 MAPK (37). Therefore, macrophages from B10.S and SJL/J mice were pretreated with ERK and p38 MAPK inhibitors 30 min prior to TMEV infection. Twenty-four hours after infection, the amount of TMEV-induced IL-6 protein secreted by B10.S macrophages was significantly greater than that secreted by SJL/J macrophages (Fig. 2B). In contrast, the level of 24-h TMEV-induced IL-6 mRNA was significantly higher in SJL/J macrophages than in B10.S macrophages, suggesting translational control of IL-6. Moreover, TMEV-induced IL-6 mRNA expression was increased in macrophages treated with the p38 MAPK inhibitor SB203580 (Fig. 2A), but IL-6 protein production was unchanged (Fig. 2B). Pretreatment of macrophages with the ERK MAPK inhibitor U0126 significantly decreased TMEV induction of IL-6 mRNA (Fig. 2A) and protein (Fig. 2B) in both SJL/J and B10.S macrophages responding to TMEV. These results indicate that ERK MAPK activation is required for IL-6 expression in response to TMEV infection and suggest that TMEV-induced IL-6 expression is under translational control.
Fig 2.
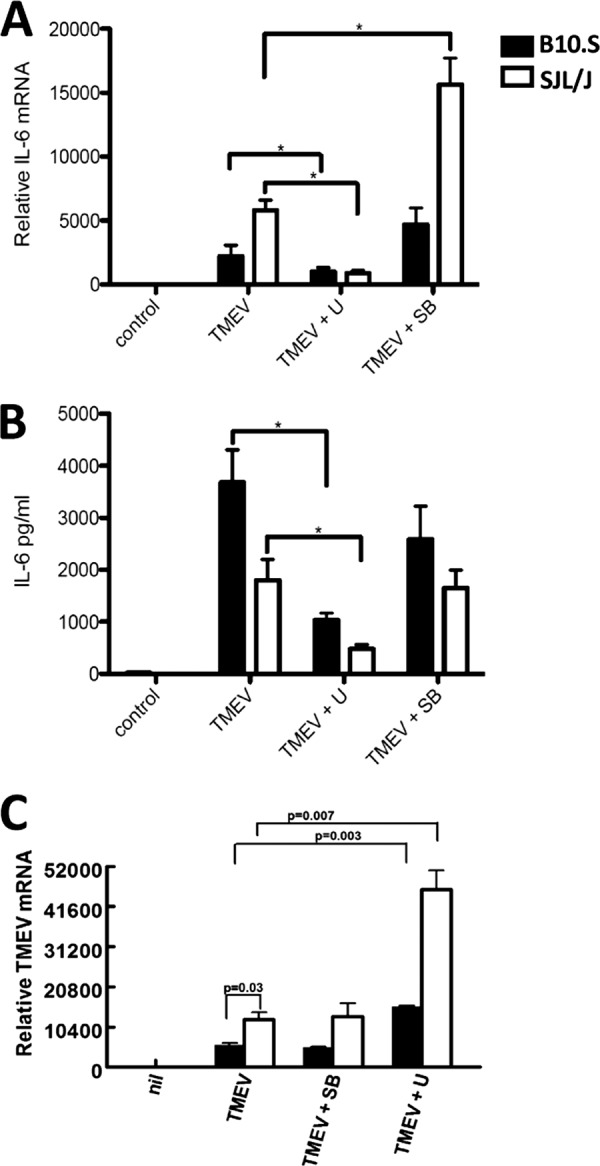
ERK MAPK activation is required for optimum TMEV-induced IL-6 expression. A total of 1 × 106 macrophages were untreated (“control” in panels A and B and “nil” in panel C) or pretreated with 20 μM SB203580 (SB) or 40 μM U0126 (U) for 30 min before infection with 2 × 105 PFU of TMEV. After 24 h, relative levels of IL-6 mRNA were evaluated by real-time PCR (A), IL-6 protein secretion was evaluated by ELISA (B), and TMEV RNA was evaluated by real-time PCR (C). Data are means of 5 samples from two experiments evaluated by Student's t test. *, P ≤ 0.05.
We have also shown that the Toll-like receptor 3 (TLR3) pathway is involved in macrophage cytokine responses to TMEV infection by inducing IFN-β, IL-12, and IL-23 (1). Therefore, we compared the responses of macrophages from each strain when treated with the TLR3 agonist poly(I · C) or the TLR4 agonist LPS. B10.S macrophages expressed significantly more IL-6 mRNA and protein than SJL/J macrophages treated with LPS (Fig. 3A and B) or poly(I · C) (Fig. 3C and D), but in macrophages from both strains, the level of IL-6 expression in response to poly(I · C) was substantially less than that in response to LPS or TMEV. Because ERK MAPK is activated by both TLR3 agonists (see Fig. S2 in the supplemental material) and TLR4 agonists (37), macrophages were also pretreated with U0126. Inhibition of ERK MAPK activation with U0126 significantly reduced IL-6 expression to the same extent in both B10.S and SJL/J macrophages responding to LPS and significantly decreased IL-6 production from SJL/J macrophages responding to poly(I · C). Therefore, ERK MAPK activation is required for IL-6 expression that occurs from activation of TLR3 and TLR4 pathways in SJL/J macrophages.
Fig 3.

Optimum IL-6 expression in B10.S compared with SJL/J macrophages following TLR4 and TLR3 stimulation depends on ERK MAPK activation. A total of 1 × 106 macrophages were untreated (control) or pretreated with U0126 (U) for 30 min before stimulation with 1 μg/ml LPS (A and B) or 50 μg/ml poly(I · C) (C and D). After 24 h, relative levels of IL-6 mRNA were evaluated by real-time PCR (A and C) and relative levels of IL-6 protein secretion (B and D) were evaluated by ELISA. Data are means of 5 samples evaluated by Student's t test. *, significantly different at P ≤ 0.05.
IL-6 expression by macrophages in response to TMEV is enhanced by IL-12.
Previously, we showed that SJL/J macrophages responding to TMEV express significantly more IFN-β and IL-12 p40 (p40/p40) but significantly less IL-12 p70 (p35/p40) than B10.S macrophages. Also, we showed that addition of IFN-β, IL-12 p40, or p70 decreases TMEV replication in SJL/J macrophages (37). To see if these cytokines could affect the level of TMEV-induced IL-6, B10.S and SJL/J macrophages were pretreated with the IL-12 p40 homodimer, bioactive IL-12 p70, or IFN-β 30 min prior to and during TMEV challenge. Treatment with IL-12 p70 or p40 significantly enhanced IL-6 expression in response to TMEV in both B10.S and SJL/J macrophages at 24 and/or 48 h post-TMEV infection (Fig. 4A and B); however, the modest increase in IL-6 expression upon pretreatment with IFN-β was not significant (Fig. 4). As we have seen before, IL-12 and IFN-β treatment reduced TMEV replication in both B10.S and SJL/J macrophages (Fig. 4C). Therefore, deficient production of IL-12 during response to TMEV could contribute to diminished IL-6 expression by SJL/J macrophages.
Fig 4.
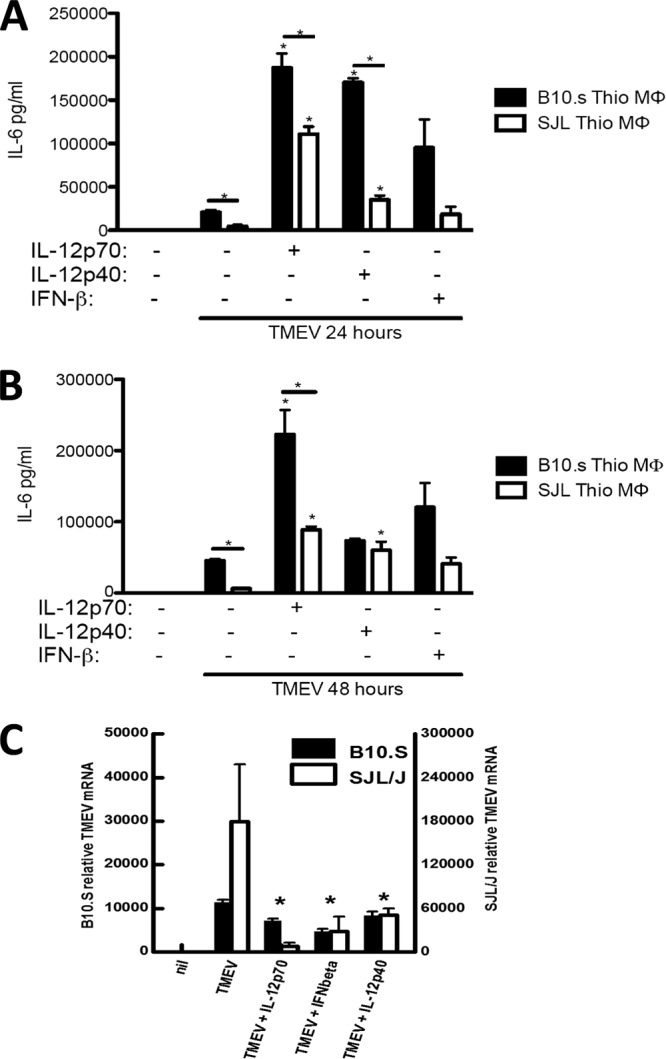
IL-12 and IFN-β enhance IL-6 expression and decrease TMEV replication during TMEV infection of B10.S and SJL/J macrophages. A total of 1 × 106 macrophages (MΦ) from SJL/J and B10.S mice were untreated (control) or treated with IL-12 p70 (1 ng/ml), IFN-β (10 ng/ml), or IL-12 p40 (10 ng/ml) 45 min before and during infection with 2 × 105 PFU of TMEV. After 24 and 48 h of infection, relative levels of IL-6 mRNA were evaluated by real-time PCR (A), IL-6 protein was evaluated by ELISA (B), and TMEV RNA was evaluated by real-time PCR (C). Thio, thioglycollate. Data are means of 3 samples, each evaluated by Student's t test. *, significantly different at P ≤ 0.05.
To gain further insight into the role of IL-12 in IL-6 expression, we used the RAW264.7 macrophage cell line, which is permissive for TMEV replication, expresses IL-6 well, expresses IL-12 poorly, and activates ERK MAPK following TMEV infection (26, 37). To confirm the effects of IL-12 and the ERK MAPK inhibitor on IL-6 expression, RAW264.7 cells were treated with IL-12 with or without U0126 during TMEV infection. Pretreatment with IL-12 enhanced IL-6 mRNA (Fig. 5A) and protein (Fig. 5B) expression following TMEV challenge of RAW264.7 cells, while pretreatment with U0126 repressed the IL-12 enhancement of IL-6 (Fig. 5A and B). Therefore, IL-12 is involved in IL-6 expression during TMEV infection of macrophages in an ERK MAPK-dependent manner.
Fig 5.
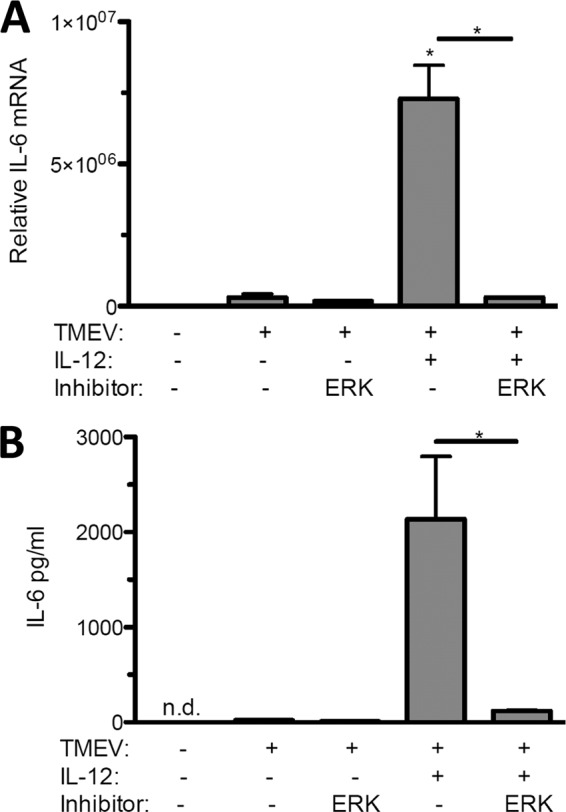
IL-12 and ERK-MAPK contribute to IL-6 expression during TMEV infection of RAW264.7 macrophages. A total of 1 × 106 RAW264.7 macrophage cells were untreated (control) or treated with IL-12 p70 (1 ng/ml) with or without 40 μM U0126 ERK MAPK inhibitor 45 min before and during infection with 1 × 106 PFU of TMEV. After 24 h of infection, relative levels of IL-6 mRNA were evaluated by real-time PCR (A) and IL-6 protein was evaluated by ELISA (B). Data are means of 3 samples each evaluated by Student's t test. This experiment was repeated three times. *, P ≤ 0.05; **, P ≤ 0.01; n.d., not detectable.
IL-6 has direct antiviral activity against TMEV replication in RAW264.7 cells.
We next set out to determine if, when, and at what quantity IL-6 represses TMEV infection in RAW264.7 cells. First, RAW264.7 cells were treated with 10 ng/ml exogenous recombinant IL-6 30 min prior to and during challenge with TMEV. Like SJL/J macrophages, treatment of RAW264.7 cells with IL-6 significantly decreased the expression of TMEV RNA at 24 h p.i. (Fig. 6A). Interestingly, as little as 0.1 ng/ml significantly reduced TMEV RNA 24 h p.i. (Fig. 6B). Furthermore, IL-6 treatment enhanced IL-6 expression (Fig. 6C) at 24 h p.i., but IL-6 treatment did not decrease TMEV RNA expression at 3 and 8 h p.i. in RAW264.7 cells (Fig. 6D), suggesting the time period in which TMEV-induced events occur in RAW264.7 cells is slightly different from that seen in SJL/J macrophages.
Fig 6.

IL-6 constrains TMEV replication during infection of RAW264.7 macrophages. A total of 1 × 106 RAW264.7 macrophage cells were untreated (control) or treated with 10 ng/ml IL-6 (A and C) or 0.01 to 10 ng/ml IL-6 (B) during infection with 1 × 106 PFU of TMEV. After 3 or 6 h (D) or 24 h (A, B, C) of infection, relative levels of TMEV (A, B, D) or IL-6 (C) mRNA were evaluated by real-time PCR. MOI, multiplicity of infection. Means are of 6 samples from two experiments, each evaluated by Student's t test. *, P ≤ 0.05; **, P ≤ 0.01.
IL-6 antiviral activity against TMEV replication is associated with activation of STAT1-related antiviral activity.
Like IFN-β, treatment of macrophages with IL-6 leads to activation of STAT3 by phosphorylation at its tyrosine 705 and activation of STAT1 at its tyrosine 701 (13). IL-6 activation of STAT1 could contribute to control of virus replication (28, 46) by inducing expression of IRF1 (22), IRF7 (23), and IRF9, which enhance IFN-β expression. To determine if IL-6 activates STAT1 and induces expression of IRFs that are downstream of STAT1 during TMEV infection of macrophages, RAW264.7 cells were treated with IL-6, IFN-β, or TMEV alone or in combination, and phospho-STAT1, as well as phospho-STAT3, activity was evaluated by Western blotting. TMEV infection alone failed to activate STAT1 by 30 min p.i. but did so by 6 h p.i. (Fig. 7A). In contrast, treatment of RAW264.7 cells with either IL-6 or IFN-β activated STAT1 and STAT3 as early as 30 min after TMEV challenge. To determine if STAT1 activation is involved in control of TMEV replication, RAW264.7 cells were transfected with small inhibitory RNA (siRNA) that has been verified to reduce STAT1 (siSTAT1) or siSTAT3. Knockdown of STAT1 resulted in significantly increased TMEV RNA, while knockdown of STAT3 failed to affect TMEV RNA replication in RAW264.7 cells (Fig. 7B). However, pretreatment with exogenous IL-6 of RAW264.7 cells with STAT1 knocked down resulted in a reduction of TMEV infection back to a level seen in RAW264.7 cells infected with TMEV. Moreover, addition of exogenous IL-6 to RAW264.7 cells with STAT3 knocked down and STAT1 and STAT3 double knocked down significantly reduced TMEV RNA replication compared with the level in untreated RAW264.7 cells infected with TMEV (Fig. 7B). Therefore, STAT1 activation contributes to control of TMEV replication and the activation of STAT3 may negatively impact the antiviral effect of STAT1. Consistent with STAT1 activation, treatment of RAW264.7 cells with IL-6 resulted in significant enhancements of expression of TMEV-induced IRF1 (Fig. 7C), IRF7 (Fig. 7D), IRF9 (Fig. 7E), and IFN-β (Fig. 7F). These results confirm that IL-6 can directly limit viral replication in macrophages early after infection with TMEV by initiating a STAT1 antiviral program.
Fig 7.

IL-6 activates STAT1 and STAT3 in macrophages. (A) Western blots of phospho-STAT1, phospho-STAT3, total STAT3, and β-tubulin at 30 min and 6 h in RAW264.7 cells either untreated or treated with IL-6 or IFN-β with or without TMEV infection. (B) TMEV RNA measured by qRT-PCR in RAW264.7 cells transfected with siSTAT1-RNA (3 pmol) or siSTAT3-RNA (3 pmol) and then infected with TMEV at a multiplicity of infection (MOI) of 1 after 36 h. (B to E) Real-time PCR of IRF1 (B), IRF7 (C), IRF9 (D), and IFN-β (E) in RAW cells treated with 10 ng/ml IL-6 prior to and during TMEV infection after 3 and 6 h of infection. Data are means of 5 samples, each evaluated by Student's t test. *, significantly different at P ≤ 0.05; n.d., not detectable.
IL-6 induces nitric oxide in RAW264.7 cells.
IL-6 (42) and activated STAT1 (29) also induce expression of nitric oxide synthase and production of nitric oxide, which is a potent antiviral factor (41). Therefore, another possibility is that IL-6 controls TMEV replication in macrophages by inducing nitric oxide. To explore this possibility, TMEV-challenged RAW264.7 cells were treated with 10 ng/ml IL-6 in the presence or absence of L-NIL, an inducible nitric oxide synthase inhibitor (27), and production of nitric oxide was measured. TMEV infection of RAW264.7 cells did not result in significant nitric oxide production at 24 h p.i. (see Fig. S3A in the supplemental material). However, addition of exogenous IL-6 to TMEV-infected RAW264.7 cells resulted in a significant enhancement of nitric oxide secretion, which was prevented by L-NIL. IL-6 or NONOate, a nitric oxide donor, repressed TMEV replication in RAW264.7 cells (see Fig. S3B). However, L-NIL did not reverse the IL-6 repression of TMEV replication in RAW264.7 cells. Thus, while IL-6 induces nitric oxide and nitric oxide is antiviral, it does not play the decisive role in the IL-6-induced antiviral effect observed in macrophages in vitro.
DISCUSSION
The data herein show that IL-6 protects macrophages early after infection with TMEV by decreasing virus replication in macrophages. These results are somewhat surprising because research on chronic TMEV infection has supported the theory that enhanced IL-6 expression may play a detrimental role in the immunopathology of TMEV in susceptible mice (14). Indeed, a recent report has shown that SJL/J mice infected with TMEV exhibit greater levels of IL-6 in the CNS than TMEV-resistant C57BL/6 mice starting at day 8 after infection (14). However, in that report, IL-6 was not measured until day 8 after infection. We show herein that the antiviral effect of IL-6 takes place within the first 6 h after infection. Therefore, the beneficial effect of IL-6 is early after TMEV infection, but not later. It is also possible that TMEV-resistant C57BL/6 mice may have a different mechanism to control TMEV infection in macrophages compared with TMEV-resistant B10.S mice. The results herein indicate that during the first day after infection in vitro, IL-6 protein production by macrophages from TMEV-resistant B10.S mice is greater than that from macrophages of TMEV-susceptible SJL/J, and this difference may play an important role in determining the severity of the infection. Moreover, significant levels of IL-6 are not produced until 8 h after TMEV infection and IL-6 is only effective at controlling TMEV replication during the first 8 h after infection. Therefore, IL-6 produced by the TMEV-infected macrophages helps to protect uninfected macrophages from subsequent infection. Altogether, the present data suggest the CNS demyelination that SJL/J mice develop several weeks to months after infection with TMEV may be due in part to an inability of SJL/J macrophages to control viral replication very early after the initial infection. Similarly, B10.S mice that do not develop demyelination following infection with TMEV may stem from better early control of TMEV replication in macrophages. Our results indicate that the ability to control TMEV replication in macrophages begins during the first 24 h after infection is to a large extent dependent on macrophage production of IL-6, which then protects uninfected cells from infection. Thus, IL-6 production by virally infected cells may have dual roles in viral infection by contributing to both antiviral immunity and subsequent pathology.
Several reports have proposed beneficial effects of exogenous IL-6 treatment in TMEV infection of SJL/J mice (35, 39). In fact, recombinant exogenous IL-6 was shown to suppress chronic demyelination and reduce virus replication in SJL/J mice infected with TMEV. Other reports have shown that IL-6 is involved in neuronal health, facilitating neuronal differentiation, neurite outgrowth, survival, regeneration (47), and oligodendrocyte differentiation (52). In stark contrast to these reports are others which have suggested that chronic expression of IL-6 in TMEV-susceptible SJL/J mice is responsible for development of demyelinating disease (17). Hou et al. (14) showed that IL-6 levels in the brain and spinal cord of SJL/J mice are elevated 8 to 90 days postinfection compared with the levels in TMEV-resistant C57BL/6 mice. It is possible that the lower early acute IL-6 response of SJL/J mice to TMEV infection is inadequate to control viral replication, thereby rendering these mice susceptible to chronic viral infection and a chronically elevated IL-6 response. Alternatively, the enhanced IL-6 level in the CNS of TMEV-infected SJL/J mice may not be a reflection of a heightened IL-6 response of infected cells to TMEV but may be due to enhanced infiltration of macrophages into the CNS, which then are infected with TMEV and produce IL-6.
The antiviral and pathological effects of IL-6 notwithstanding, the signaling pathways that lead to IL-6 expression in response to TMEV remain unclear. Previous reports showed that ERK and p38 MAPK pathways are involved in both innate antiviral immunity (7) and IL-6 expression (49). The results herein confirm that activation of ERK MAPKs contributes to IL-6 expression in response to TMEV. However, maximum ERK activation occurs within 30 min after TMEV infection of macrophages, and we have not noticed differences in the intensities of ERK activation between SJL and B10.S macrophages following TMEV infection (37, 38). Activated ERK MAPKs have been shown to phosphorylate downstream transcription factors, such as the cyclic AMP (cAMP) response element binding protein (CREB) at serine 133 (4, 51, 45), CREB response elements (CREs) are located at the IL-6 promoter (8), activated CREB binds to IL-6 promoter CRE (10), and CREB activity is required for IL-6 (36). Therefore, TMEV induction of IL-6 is likely to depend on the ERK activation of the transcription factor, CREB.
The results herein also suggest that TMEV induction of IL-6 in macrophages is in part dependent upon macrophage production of IL-12, which is also the primary inducer of Th1 development during adaptive immune responses (15). In addition, our results show that IL-12 enhancement of IL-6 expression is dependent on the ERK MAPK pathway. Other reports have shown in IL-12 p35 knockout mice that IL-12 plays a significant role in IL-6 expression during viral infection (18), IL-6 expression depends in part on activation of ERK (19), and IL-12 activates ERK MAPK (20). These results are consistent with our previous data that showed significantly more IL-12 production from B10.S macrophages than that of SJL/J macrophages following TMEV infection (37). Therefore, IL-12 expression by macrophages that is responsible for Th1 development during adaptive immune responses also contributes to IL-6 expression during innate immune responses: both roles of IL-12 are likely to be beneficial to early antiviral immunity.
Overall, the data here suggest a direct, interferon-like role for IL-6 in the suppression of TMEV replication, which is exemplified by its ability to activate STAT1 and induce expression of STAT1-dependent genes. Indeed, early reports regarding IL-6, which was originally called “interferon-beta 2,” showed it to have antiviral activity (44). While it is not clear how IL-6 decreases TMEV replication, it is known to be a potent inducer of iNOS and production of nitric oxide (31), which has antiviral properties (41). Our data confirm that IL-6 induces significant nitric oxide production from macrophages and that nitric oxide does indeed control TMEV replication. However, prevention of IL-6-induced nitric oxide production did not reverse the antiviral effect of IL-6. This is most likely because IL-6 also activated STAT1 and STAT3, which led to the induction of STAT1-downstream antiviral genes, IRF1, IRF7, and IRF9. Therefore, the data suggest that early after infection with TMEV, IL-6 controls acute viral infection by inducing multiple innate antiviral programs.
One of the discrepancies noted herein is that between IL-6 mRNA expression and IL-6 protein secretion at time points beyond 8 h postinfection. These data suggest that TMEV-induced IL-6 production is under translational or inhibitory RNA control, which is especially notable in the TMEV-susceptible SJL/J macrophages. At 24 and 28 h postinfection, SJL/J macrophages expressed more IL-6 mRNA but produced less IL-6 than B10.S macrophages. In one study, K homology (KH)-type splicing regulatory protein (KHSRP) was shown to target the AU-rich elements in the IL-6 mRNA 3′-untranslated region, which restricts its translation but does not lead to degradation of mRNA (6). In another study, microRNA 365 was shown to inhibit the translation of IL-6 mRNA without affecting IL-6 mRNA (50). Our results suggest that differences in translational or inhibitory control of IL-6 expression following infection of macrophages could contribute to susceptibility versus resistance of macrophages to TMEV.
In summary, our findings show that strain differences in onset and amount of IL-6 upregulation upon infection with TMEV correlate with previously observed differences in disease outcomes between B10.S and SJL/J mice. Whereas B10.S mice with enhanced early expression of IL-6 are capable of clearing TMEV, in SJL/J mice TMEV replication is not reduced by IL-6 unless exogenous IL-6 is added, suggesting that these mice do not produce IL-6 quickly enough or in sufficient quantity to control viral replication. The present investigation suggests that enhanced early expression of IL-6 by macrophages during infection with a macrophage-trophic virus, TMEV, is responsible for better control of viral replication in TMEV-resistant B10.S macrophages than that in TMEV-susceptible SJL/J macrophages. These results indicate that the persistent TMEV infection of macrophages in the CNS of SJL/J mice could be the result of insufficient IL-6 production that controls early TMEV infection through multiple antiviral mechanisms that include nitric oxide production and STAT1 activation. Furthermore, the present data clearly show that IL-12 and ERK MAPK play a major role in TMEV-induced IL-6. While ERK MAPK is required for IL-6 expression in response to TMEV, its activation is equivalent in both strains. In contrast, our previous studies point to differences in TLR signaling and IL-12 expression that may contribute to strain differences in TMEV-induced IL-6. These results are supported by our data showing that TMEV-induced IL-6 is also antiviral in RAW264.7 cells, IL-6 expression is enhanced by IL-12, and IL-6 leads to activation of STAT1 and expression of its downstream genes IRF1, IRF7, IRF9, and IFN-β, as well as induction of nitric oxide. These results are significant because persistent infection of SJL/J macrophages by TMEV contributes to the inflammatory autoimmune demyelination in the CNS of TMEV-infected SJL/J mice, which mimics human multiple sclerosis, is not seen in TMEV-infected B10.S mice. It remains to be determined if IL-6 is effective at controlling TMEV replication during established persistent TMEV infection of macrophage populations. Nevertheless, this study suggests therapeutic strategies that promote early IL-6 antiviral pathways could prevent chronic viral infections.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the University of Nebraska Medical Center College of Dentistry and University of Nebraska, Lincoln, School of Biological Sciences, and was supported by award no. 8P30GM10350903 and 5P20GM103489 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Published ahead of print 25 July 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Al-Salleeh F, Petro TM. 2007. TLR3 and TLR7 are involved in expression of IL-23 subunits while TLR3 but not TLR7 is involved in expression of IFN-beta by Theiler's virus-infected RAW264.7 cells. Microbes Infect. 9:1384–1392 [DOI] [PubMed] [Google Scholar]
- 2. Andoh A, et al. 2002. Extracellular signal-regulated kinases 1 and 2 participate in interleukin-17 plus tumor necrosis factor-alpha-induced stabilization of interleukin-6 mRNA in human pancreatic myofibroblasts. Biochim. Biophys. Acta 1591:69–74 [DOI] [PubMed] [Google Scholar]
- 3. Banerjee S, Narayanan K, Mizutani T, Makino S. 2002. Murine coronavirus replication-induced p38 mitogen-activated protein kinase activation promotes interleukin-6 production and virus replication in cultured cells. J. Virol. 76:5937–5948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cammarota M, Bevilaqua LR, Dunkley PR, Rostas JA. 2001. Angiotensin II promotes the phosphorylation of cyclic AMP-responsive element binding protein (CREB) at Ser133 through an ERK1/2-dependent mechanism. J. Neurochem. 79:1122–1128 [DOI] [PubMed] [Google Scholar]
- 5. Clatch RJ, Miller SD, Metzner R, Dal Canto MC, Lipton HL. 1990. Monocytes/macrophages isolated from the mouse central nervous system contain infectious Theiler's murine encephalomyelitis virus (TMEV). Virology 176:244–254 [DOI] [PubMed] [Google Scholar]
- 6. Dhamija S, et al. 2011. Interleukin-1 activates synthesis of interleukin-6 by interfering with a KH-type splicing regulatory protein (KSRP)-dependent translational silencing mechanism. J. Biol. Chem. 286:33279–33288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dong C, Davis RJ, Flavell RA. 2002. MAP kinases in the immune response. Annu. Rev. Immunol. 20:55–72 [DOI] [PubMed] [Google Scholar]
- 8. Droogmans L, Cludts I, Cleuter Y, Kettmann R, Burny A. 1992. Nucleotide sequence of the bovine interleukin-6 gene promoter. DNA Seq. 3:115–117 [DOI] [PubMed] [Google Scholar]
- 9. Gadient RA, Otten U. 1994. Identification of interleukin-6 (IL-6)-expressing neurons in the cerebellum and hippocampus of normal adult rats. Neurosci. Lett. 182:243–246 [DOI] [PubMed] [Google Scholar]
- 10. Grassl C, Luckow B, Schlondorff D, Dendorfer U. 1999. Transcriptional regulation of the interleukin-6 gene in mesangial cells. J. Am. Soc. Nephrol. 10:1466–1477 [DOI] [PubMed] [Google Scholar]
- 11. Hama T, Miyamoto M, Tsukui H, Nishio C, Hatanaka H. 1989. Interleukin-6 as a neurotrophic factor for promoting the survival of cultured basal forebrain cholinergic neurons from postnatal rats. Neurosci. Lett. 104:340–344 [DOI] [PubMed] [Google Scholar]
- 12. Harker JA, Lewis GM, Mack L, Zuniga EI. 2011. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science 334:825–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hemmann U, et al. 1996. Differential activation of acute phase response factor/Stat3 and Stat1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. J. Biol. Chem. 271:12999–13007 [DOI] [PubMed] [Google Scholar]
- 14. Hou W, Kang HS, Kim BS. 2009. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J. Exp. Med. 206:313–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsieh CS, et al. 1993. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 260:547–549 [DOI] [PubMed] [Google Scholar]
- 16. Ishihara K, Hirano T. 2002. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev. 13:357–368 [DOI] [PubMed] [Google Scholar]
- 17. Jin Y-H, et al. 2007. Differential virus replication, cytokine production, and antigen-presenting function by microglia from susceptible and resistant mice infected with Theiler's virus. J. Virol. 81:11690–11702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kapil P, et al. 2009. Interleukin-12 (IL-12), but not IL-23, deficiency ameliorates viral encephalitis without affecting viral control. J. Virol. 83:5978–5986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim SH, Kim J, Sharma RP. 2004. Inhibition of p38 and ERK MAP kinases blocks endotoxin-induced nitric oxide production and differentially modulates cytokine expression. Pharmacol. Res. 49:433–439 [DOI] [PubMed] [Google Scholar]
- 20. Kondadasula SV, et al. 2008. Colocalization of the IL-12 receptor and FcγRIIIa to natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-γ. Blood 111:4173–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leonard M, Ryan MP, Watson AJ, Schramek H, Healy E. 1999. Role of MAP kinase pathways in mediating IL-6 production in human primary mesangial and proximal tubular cells. Kidney Int. 56:1366–1377 [DOI] [PubMed] [Google Scholar]
- 22. Li X, Leung S, Qureshi S, Darnell JE, Jr, Stark GR. 1996. Formation of STAT1-STAT2 heterodimers and their role in the activation of IRF-1 gene transcription by interferon-alpha. J. Biol. Chem. 271:5790–5794 [DOI] [PubMed] [Google Scholar]
- 23. Marie I, Durbin JE, Levy DE. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17:6660–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. 2011. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. (Lond.) 122:143–159 [DOI] [PubMed] [Google Scholar]
- 25. Monteyne P, Bihl F, Levillayer F, Brahic M, Bureau JF. 1999. The Th1/Th2 balance does not account for the difference of susceptibility of mouse strains to Theiler's virus persistent infection. J. Immunol. 162:7330–7334 [PubMed] [Google Scholar]
- 26. Moore TC, Al-Salleeh FM, Brown DM, Petro TM. 2011. IRF3 polymorphisms induce different innate anti-Theiler's virus immune responses in RAW264.7 macrophages. Virology 418:40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moore WM, et al. 1994. L-N6-(1-Iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J. Med. Chem. 37:3886–3888 [DOI] [PubMed] [Google Scholar]
- 28. Mott KR, Underhill D, Wechsler SL, Town T, Ghiasi H. 2009. A role for the JAK-STAT1 pathway in blocking replication of HSV-1 in dendritic cells and macrophages. Virol. J. 6:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nishiya T, et al. 1997. Activation of Stat1 and subsequent transcription of inducible nitric oxide synthase gene in C6 glioma cells is independent of interferon-γ-induced MAPK activation that is mediated by p21ras. FEBS Lett. 408:33–38 [DOI] [PubMed] [Google Scholar]
- 30. O'Gorman WE, et al. 2010. Alternate mechanisms of initial pattern recognition drive differential immune responses to related poxviruses. Cell Host Microbe 8:174–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oh YS, Lee Y-J, Park EY, Jun H-S. 2011. Interleukin-6 treatment induces beta-cell apoptosis via STAT-3-mediated nitric oxide production. Diabetes Metab. Res. Rev. 27:813–819 [DOI] [PubMed] [Google Scholar]
- 32. Okuda Y, et al. 1998. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int. Immunol. 10:703–708 [DOI] [PubMed] [Google Scholar]
- 33. Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. 2004. Theiler's virus infection: a model for multiple sclerosis. Clin. Microbiol. Rev. 17:174–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Palma JP, Kwon D, Clipstone NA, Kim BS. 2003. Infection with Theiler's murine encephalomyelitis virus directly induces proinflammatory cytokines in primary astrocytes via NF-{kappa}B activation: potential role for the initiation of demyelinating disease. J. Virol. 77:6322–6331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pavelko KD, et al. 2003. Interleukin-6 protects anterior horn neurons from lethal virus-induced injury. J. Neurosci. 23:481–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Persson E, Voznesensky OS, Huang YF, Lerner UH. 2005. Increased expression of interleukin-6 by vasoactive intestinal peptide is associated with regulation of CREB, AP-1 and C/EBP, but not NF-kappaB, in mouse calvarial osteoblasts. Bone 37:513–529 [DOI] [PubMed] [Google Scholar]
- 37. Petro TM. 2005. Disparate expression of IL-12 by SJL/J and B10.S macrophages during Theiler's virus infection is associated with activity of TLR7 and mitogen-activated protein kinases. Microbes Infect. 7:224–232 [DOI] [PubMed] [Google Scholar]
- 38. Petro TM. 2005. ERK-MAP-kinases differentially regulate expression of IL-23 p19 compared with p40 and IFN-beta in Theiler's virus-infected RAW264.7 cells. Immunol. Lett. 97:47–53 [DOI] [PubMed] [Google Scholar]
- 39. Rodriguez M, Pavelko KD, McKinney CW, Leibowitz JL. 1994. Recombinant human IL-6 suppresses demyelination in a viral model of multiple sclerosis. J. Immunol. 153:3811–3821 [PubMed] [Google Scholar]
- 40. Samoilova EB, Horton JL, Hilliard B, Liu T-ST, Chen Y. 1998. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J. Immunol. 161:6480–6486 [PubMed] [Google Scholar]
- 41. Sanders SP, Siekierski ES, Porter JD, Richards SM, Proud D. 1998. Nitric oxide inhibits rhinovirus-induced cytokine production and viral replication in a human respiratory epithelial cell line. J. Virol. 72:934–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sawada T, Falk LA, Rao P, Murphy WJ, Pluznik DH. 1997. IL-6 induction of protein-DNA complexes via a novel regulatory region of the inducible nitric oxide synthase gene promoter: role of octamer binding proteins. J. Immunol. 158:5267–5276 [PubMed] [Google Scholar]
- 43. Schobitz B, de Kloet ER, Sutanto W, Holsboer F. 1993. Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur. J. Neurosci. 5:1426–1435 [DOI] [PubMed] [Google Scholar]
- 44. Sehgal PB, et al. 1988. Regulation of the acute phase and immune responses in viral disease. Enhanced expression of the beta 2-interferon/hepatocyte-stimulating factor/interleukin 6 gene in virus-infected human fibroblasts. J. Exp. Med. 167:1951–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shaywitz AJ, Greenberg ME. 1999. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68:821–861 [DOI] [PubMed] [Google Scholar]
- 46. Shresta S, et al. 2005. Critical roles for both STAT1-dependent and STAT1-independent pathways in the control of primary Dengue virus infection in mice. J. Immunol. 175:3946–3954 [DOI] [PubMed] [Google Scholar]
- 47. Spooren A, et al. 2011. Interleukin-6, a mental cytokine. Brain Res. Rev. 67:157–183 [DOI] [PubMed] [Google Scholar]
- 48. Starnes HF, Jr, et al. 1990. Anti-IL-6 monoclonal antibodies protect against lethal Escherichia coli infection and lethal tumor necrosis factor-alpha challenge in mice. J. Immunol. 145:4185–4191 [PubMed] [Google Scholar]
- 49. Tuyt LM, et al. 1999. Extracellular-regulated kinase 1/2, Jun N-terminal kinase, and c-Jun are involved in NF-kappa B-dependent IL-6 expression in human monocytes. J. Immunol. 162:4893–4902 [PubMed] [Google Scholar]
- 50. Xu Z, et al. 2011. miR-365, a novel negative regulator of interleukin-6 gene expression, is cooperatively regulated by Sp1 and NF-kB. J. Biol. Chem. 286:21401–21412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zanassi P, et al. 2001. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J. Biol. Chem. 276:11487–11495 [DOI] [PubMed] [Google Scholar]
- 52. Zhang P-L, et al. 2006. Increased myelinating capacity of embryonic stem cell derived oligodendrocyte precursors after treatment by interleukin-6/soluble interleukin-6 receptor fusion protein. Mol. Cell Neurosci. 31:387–398 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.