

Abstract
The respiratory syncytial virus (RSV) G and F glycoproteins are the neutralization antigens, and G also is expressed in a soluble form (sG). Previously, sG was demonstrated to reduce the efficiency of RSV antibody-mediated neutralization by serving as an antigen decoy and to inhibit the antibody-mediated antiviral effects of Fc receptor-bearing leukocytes. The present study demonstrated that effective antibody-mediated restriction in vivo, and the evasion of this restriction by sG, involves pulmonary macrophages and complement, but not neutrophils.
TEXT
Respiratory syncytial virus (RSV) is the leading cause of severe respiratory disease in infants and children worldwide and is a major cause of severe respiratory disease in the elderly (reviewed in reference 3). Specific antiviral therapy or vaccines are not available, although infants and young children at high risk for severe RSV disease due to underlying conditions can be substantially protected by passive antibody immunoprophylaxis. Important and unusual features of RSV include (i) its ability to frequently infect and cause severe disease in young infants, suggesting that RSV is less sensitive to restriction by maternally derived serum antibodies than other common respiratory tract viruses (4, 6), and (ii) its ability to reinfect symptomatically (but usually with reduced disease) throughout life without antigenic change, which also is suggestive of inefficient restriction by host immunity (5). The virus has two viral neutralization antigens, the G and F surface glycoproteins, and the former is expressed as both the membrane-bound (mG) and secreted (sG) forms due to the use of two different translational start codons, ATG-1 and ATG-48 (7).
We previously compared antibody-mediated neutralization in vitro and in vivo of a recombinant wild-type (wt) RSV versus a recombinant mutant virus (mG RSV) in which the expression of sG was disabled by changing its ATG-48 start codon to ATT (encoding Ile) (16). We found that the expression of sG by wt RSV reduced the effectiveness of antibody-mediated neutralization by acting as an antigen decoy and by inhibiting the antibody-mediated antiviral effects of Fc receptor-bearing leukocytes (2). These effects likely help the virus evade antibodies present from maternal transfer or from prior infection and thus likely contribute to the ability of the virus to infect early in life and to reinfect without significant antigenic change. We hypothesized that the effect of sG on Fc receptor-bearing leukocytes involved interference with the influx and/or functioning of Fc-bearing cells, involving cell types such as macrophages, neutrophils, and natural killer cells that otherwise contribute to viral clearance by phagocytosis of antibody-antigen complexes or by antibody-dependent cell-mediated cytotoxicity (ADCC) (2). Interference with complement-mediated antiviral effects also was possible. The goals of the present study were to investigate the possible involvement of macrophages, neutrophils, and the complement system in (i) the antibody-mediated restriction of RSV replication in vivo, (ii) the sG-mediated evasion of this restriction, and (iii) the restriction of RSV replication in RSV-naïve animals.
In preliminary studies (data not shown), we investigated whether sG produced by wt RSV had the ability to inhibit phagocytosis. This was evaluated in vitro using the J774 and RAW264.7 murine macrophage cell lines and the U937 human leukemic monocyte cell line. Cells were infected for 24 h or 48 h with wt or mG RSV and evaluated for the ability to phagocytize antibody-bound phycoerythrin-labeled pHrodo Escherichia coli BioParticles (Invitrogen), as measured by flow cytometry according to the manufacturer's recommendations. The infections marginally increased, rather than decreased, the level of phagocytosis, and there was no evidence that the sG protein directly suppressed phagocytic activity (data not shown). We also evaluated whether sG affected the activation of pulmonary macrophages in mice. BALB/c mice were infected intranasally with wt RSV or mG RSV. On days 2 and 4, the animals were sacrificed and pulmonary mononuclear cells (PMC) were isolated as described previously (1). PMC were cultured overnight in the presence of monensin and stained with antibodies specific for the surface markers CD45b (a B-cell marker), CD11b, and CD11c and for intracellular accumulation of the cytokines interleukin-12 (IL-12), tumor necrosis factor alpha (TNF-α), IL-6, and alpha interferon (IFN-α), which are markers for activation, and for IL-10, a marker for suppression. Cell counting and flow cytometry analysis indicated that the total number of PMC was increased 3- to 5-fold in response to infection with either virus. However, the percentages of total macrophages (defined as CD45b− CD11c− CD11b+ [8]) and of macrophages secreting the individual cytokines, and the mean intensity of fluorescence staining for the cytokines, were comparable in mice infected with either virus and in mice that were mock infected (data not shown). Thus, there was no evidence that sG affected macrophage activation.
Next, we investigated the effect of macrophages on RSV replication in RSV-naïve BALB/c mice in the presence and absence of normal serum containing natural antibodies. Natural antibodies are “background” antibodies that are produced spontaneously without antigen stimulation and can react with pathogens. In naïve animals, natural antibodies have been shown to play a central role in restricting the early stages of several viral infections (11). To investigate a role of natural antibodies in macrophage-mediated restriction in RSV-naïve mice, we used JHD mice, which are deficient in B cells and have the BALB/c background (Taconic, Germantown, NY), and control BALB/c mice of the same age. Pulmonary macrophages were depleted by intranasally delivered clodronate liposomes, which are taken up by macrophages and induce apoptosis (10, 18). Specifically, mice were treated with clondronate liposomes (Clodrosomes) or, as a control, empty liposomes (both from Encapsula Nano Sciences, Nashville, TN) by the intranasal route at 100 μl per animal under methoxyflurane anesthesia on days −3 and −1. This resulted in a reduction in pulmonary macrophages of 80 to 85% (not shown). The mice were infected with 5 × 106 PFU of mG RSV (day 0), and the animals also received serum from normal RSV-naïve BALB/c mice (Innovative Research, Novi, MI) or phosphate-buffered saline (PBS) at 0.8 ml per mouse by the intraperitoneal (i.p.) route on days −1 and 3 (Fig. 1A). We used mG RSV, rather than wt RSV, in this experiment and other experiments that did not involve comparison of the two viruses in order to avoid any possible interference from the sG protein. In control BALB/c mice, depletion of pulmonary macrophages resulted in a 51-fold increase in pulmonary RSV replication (Fig. 1A, group B versus group A). This is consistent with the recent study of Pribul et al., showing that pulmonary macrophages play an important role in the early response to RSV (13). In JHD mice, the titers were also increased with macrophage depletion, although the increase was somewhat less (16-fold; group E versus group C). In JHD mice not treated with clodronate (Fig. 1A, group C), the viral titers were 3-fold lower, rather than greater, than in control BALB/c mice (group A), suggesting that the lack of natural antibodies was not associated with increased RSV replication. In addition, the injections of normal BALB/c mouse serum into JHD mice did not confer the increased restriction of RSV replication (Fig. 1A, compare group D versus group C), suggesting that this serum lacked natural antibodies capable of restricting RSV replication. These data suggest that BALB/c mice lack significant amounts of natural antibodies able to restrict RSV, indicating that they did not contribute to the observed macrophage-mediated restriction of RSV replication and hence would not be relevant to sG-mediated evasion.
Fig 1.
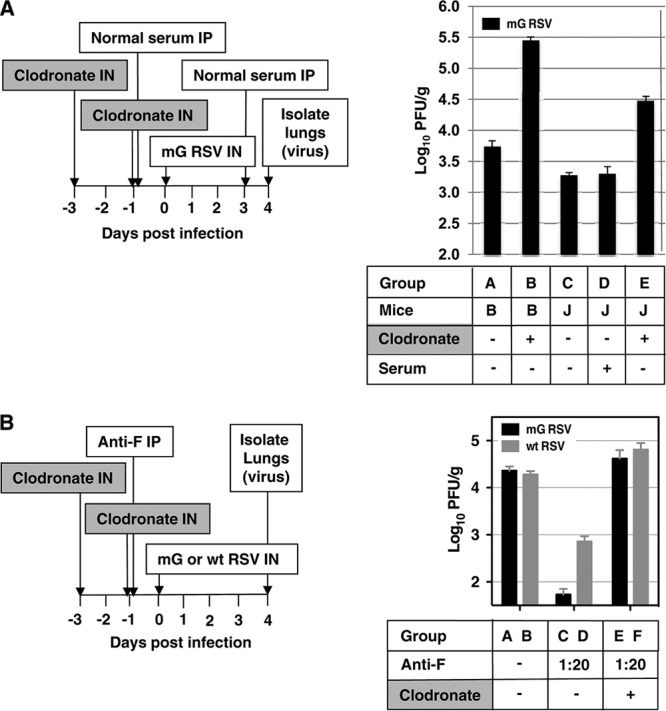
Role of pulmonary macrophages in restricting RSV replication in naïve mice in the restriction of RSV replication in vivo by RSV-specific antibodies and in the alleviation of that restriction by sG expressed by wt RSV. The plan of each experiment is shown on the left, and the results are shown on the right. (A) Pulmonary macrophages, but not natural antibodies, restrict pulmonary RSV replication in RSV-naïve mice. Groups of mice (B, BALB/c; J, JHD; 4 animals per group, except for 5 in group B) were treated intranasally with clodronate to deplete pulmonary macrophages or mock treated, treated with normal BALB/c serum or control serum, and infected with mG RSV. The animals were sacrificed on day 4, and the pulmonary RSV titers were determined (PFU per gram of lung tissue [PFU/g], mean ± standard error [SE]). Statistical significance by Student's unpaired t test: groups AB, AE, BC, BD, BE, CE, and DE, P < 0.001; groups AC, P = 0.003; groups AD, P = 0.026. (B) Pulmonary macrophages are involved in the restriction of RSV replication by RSV-specific antibodies and in the alleviation of that restriction by sG expressed by wt RSV. BALB/c mice in groups of 5 were treated with clodronate or mock treated, treated with RSV F-specific antiserum or PBS, and infected with mG or wt RSV. The animals were sacrificed on day 4, and the pulmonary RSV titers were determined. Statistical significance: groups AC, BD, CE, and DF, P < 0.001; group BF, P = 0.010. The experiment shown in panel B was performed three times with similar results.
Next, we investigated the role of pulmonary macrophages in the restriction of RSV replication conferred by RSV-specific antibodies and in the ability of sG to evade this restriction. BALB/c mice were treated (days −3 and −1) with clodronate or mock treated as described above and then injected (day −1) with a control nonimmune serum or with a monovalent mouse antiserum specific for the F protein of RSV, which had been raised by infection of donor animals with a vaccinia virus recombinant expressing the RSV F protein as described previously (2). This antiserum was administered at a 1:20 dilution, which was shown in preliminary experiments to confer the partial restriction of RSV replication. The next day (day 0), the mice were infected with 3 × 106 PFU of mG RSV or wt RSV, and on day 4, the animals were sacrificed and the pulmonary viral titers were determined. We observed three effects. First, consistent with our previous study (2), in animals that were not treated with clodronate, the administration of the RSV-specific antiserum restricted virus replication for both mG (Fig. 1B, compare groups C and A, reduction of 424-fold) and wt RSV (compare groups D and B, reduction of 26-fold). Second, also consistent with our previous study (2), this antibody-mediated restriction was greater for mG RSV, which does not express sG, than for wt RSV, which does express sG (compare groups C and D, 13-fold difference). Third, the antibody-mediated restriction of each virus was ablated by depletion of pulmonary macrophages, and indeed, replication of each virus was increased beyond the level observed in the untreated, RSV-infected controls (mG, compare groups A, C, and E; wt RSV, compare groups B, D, and F). In summary, these results showed that the restriction of RSV replication mediated by the RSV-F-specific antibodies depended in large part on pulmonary macrophages.
Next, we investigated the role of neutrophils in restricting RSV replication in RSV-naïve BALB/c mice, using animals depleted of neutrophils by treatment with neutrophil-specific antibodies. The mice were injected on day −1 with 0.2 ml of rabbit antiserum specific for mouse neutrophils (Accurate Chemical, Westbury, NY) or, in control animals, with an equal volume of normal rabbit serum (Innovative Research). The mice were infected on day 0 with 106 PFU of mG RSV and, on day 2, were then injected again with neutrophil-specific or control serum. On day 3, two mice from each group were sacrificed, and PMC were isolated for flow cytometry analysis using the following antibody stains: CD11c labeled with fluorescein, CD11b labeled with phycoerythrin, and Ly-6C (clone RB6-8C5) labeled with allophycocyanin. The neutrophil population was defined as described previously (9, 12, 14, 15), i.e., high side scatter and forward scatter, high to intermediate for CD11b, negative for CD11c, and high for Ly6G/C (macrophages have slightly lower levels of Ly6G/C). We found that injection of the antineutrophil antiserum resulted in a mean 62% reduction in the number of neutrophils. To determine the effect of neutrophil depletion on RSV replication, the remaining mice in each group were euthanized on day 4 postinfection, the lungs were isolated, and the virus titers were determined. We found no significant effect of neutrophil depletion on pulmonary replication of mG RSV (Fig. 2A). In a separate experiment, the effect of neutrophil depletion was compared for mG RSV and wt RSV: no difference in the level of replication was observed (data not shown). We then investigated the effect of neutrophil depletion on the efficiency of restriction mediated by RSV-specific antibodies. Groups of mice were injected with neutrophil-specific antiserum as above (days −1 and 2). In addition, the animals were injected with 0.5 ml of a 1:30 dilution of the previously described RSV F-specific antiserum or normal mouse serum on day −1. On day 0, the animals were infected with 106 PFU of mG RSV, and on day 4, the animals were sacrificed, the lungs were isolated, and the viral titers were determined (Fig. 2B). Depletion of neutrophils did not significantly affect the level of the restriction of RSV replication conferred by the anti-RSV F antibodies (Fig. 2B, compare group C and group B). These data suggest that neutrophils are not involved in the direct or antibody-mediated restriction of RSV replication and thus are not involved in the RSV sG-mediated evasion of antibody-mediated restriction.
Fig 2.
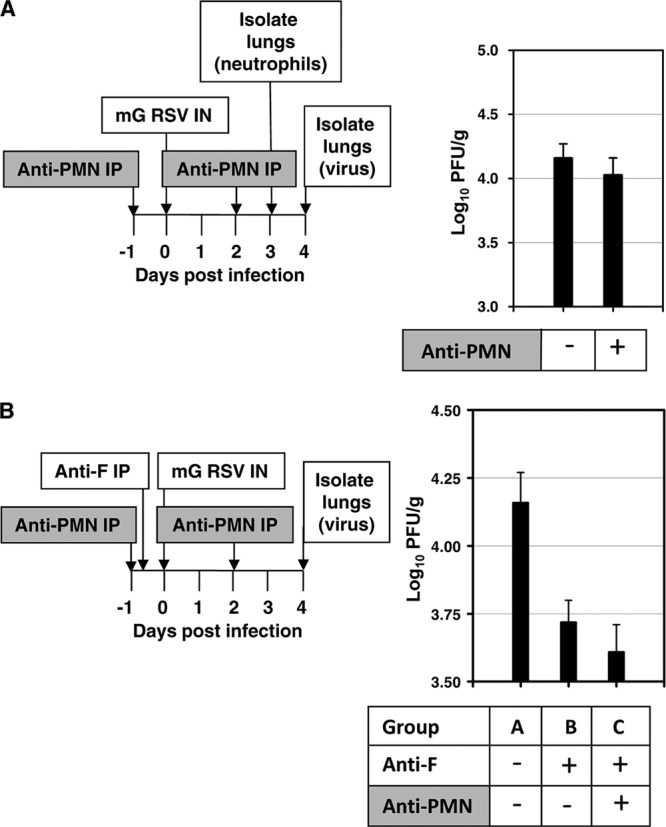
Neutrophils do not play a role in restricting RSV replication in naïve mice or in the restriction of RSV in vivo by RSV-specific antibodies. The plan of each experiment is shown on the left, and the results are shown on the right. (A) Neutrophils do not restrict pulmonary RSV replication in RSV-naïve mice. BALB/c mice in groups of 5 were treated with neutrophil-specific antiserum (anti-PMN IP) or normal serum and infected with mG RSV. Five animals from each group were sacrificed on day 3 to quantify pulmonary neutrophils by flow cytometry. The remaining animals were sacrificed on day 4, and the pulmonary RSV titers were determined (PFU/g, mean ± SE). (B) Neutrophils do not affect the ability of RSV-specific antibodies to restrict RSV in vivo. Mice in groups of 5 were treated with anti-PMN antiserum or mock treated, treated with RSV F-specific antiserum or mock treated, and infected with mG RSV. The animals were sacrificed on day 4, and the pulmonary RSV titers were determined (PFU/g, mean ± SE). Statistical significances by Student's unpaired t test: groups AB, P = 0.012; groups AC, P = 0.006.
Next, we investigated the effect of the complement system in restricting RSV replication in RSV-naïve BALB/c mice. The complement system was inactivated by i.p. injections of cobra venom factor (CVF) (Sigma-Aldrich, St. Louis, MO). CVF selectively depletes two central components of the complement system, C3 and C5, which results in disabling the complement system but otherwise is nontoxic (17). We injected mice by the i.p. route with 20 μg (or approximately 1 unit) of CVF in 500 μl of PBS or PBS without CVF on day −1 prior to infection. On day 0, the mice were inoculated with 3 × 106 PFU of mG RSV. On day 3, the mice received another injection of CVF. On days 0, 1, 2, and 4, blood samples were collected, and the efficiency of disabling the complement system was determined by analysis of serum C3 by enzyme-linked immunosorbent assay (ELISA; GenWay, San Diego, CA). This demonstrated a mean reduction of C3 of 89%, 93%, and 91% on days 0, 2, and 4, respectively. To investigate the effect of disabling the complement system on RSV replication, mice were injected with CVF (days −1 and 3) and infected with mG RSV (day 0) as described above, and on day 4, the animals were euthanized, the lungs were isolated, and the RSV titers were determined. We found that, unlike the effect of depleting pulmonary macrophages, disabling the complement system resulted in only a marginal, nonsignificant increase of the viral titers (Fig. 3A), suggesting that the complement system is not significantly involved in restricting RSV in virus-naïve animals.
Fig 3.
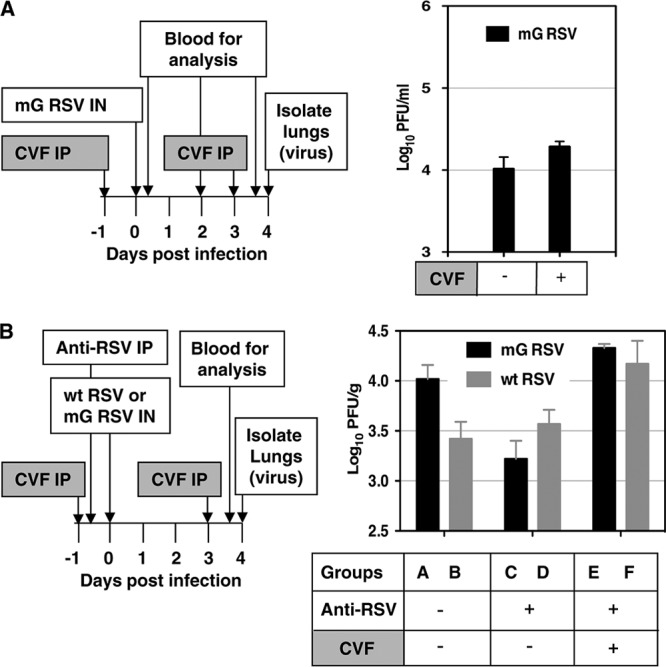
The complement system does not play a role in restricting RSV replication in naïve mice but does contribute to the restriction of RSV in vivo by RSV-specific antibodies. The plan of each experiment is shown on the left, and the results are shown on the right. (A) The complement system does not restrict RSV replication in RSV-naïve mice. BALB/c mice in groups of 4 were treated with cobra venom factor (CVF) and infected with mG RSV. Serum samples were taken on days 2 and 4 to measure depletion of factor C3. The animals were sacrificed on day 4, and the pulmonary RSV titers were determined (PFU/g, mean ± SE). (B) The complement system is required for the effective restriction of RSV in vivo by RSV-specific antibodies. BALB/c mice in groups of 4 were treated with CVF, treated with anti-RSV antiserum, and infected with mG RSV or wt RSV. The animals were sacrificed on day 4, and the pulmonary RSV titers were determined (PFU/g, mean ± SE). Statistical significances by Student's unpaired t test: groups AC, P = 0.013; groups BF, P = 0.040; groups CE, P = 0.001. The experiment shown in panel B was performed three times with essentially similar results.
To determine the role of the complement system in restriction mediated by RSV-specific antibodies, and in the sG-mediated evasion of this restriction, we disabled the complement system in groups of mice as described above and injected them (day −1) with 0.5 ml per animal of a 1:30 dilution of antiserum from donor mice that had been infected with RSV, as described previously (2), and that, in preliminary experiments, was demonstrated to be sufficient for a partial restriction of the administered dose of mG RSV in BALB/c mice. On day 0, the mice were infected with 3 × 106 PFU of either mG RSV or wt RSV, and on day 4, the animals were euthanized and the viral titers in the lungs were determined (Fig. 3B). In the experiment shown in Fig. 3B, the inoculum of mG RSV happened to be somewhat higher than that of wt RSV, resulting in higher levels of replication of this virus in the lungs, compared to wt RSV (group A versus group B). In this particular experiment, the passively transferred antibodies reduced the pulmonary titers of mG RSV by 6.3-fold, but no reduction of wt RSV titers was observed (groups C and D). Disabling the complement system prevented this restriction (group E). Moreover, the titers of both viruses were marginally increased compared to mice that did not receive antibodies, with the increase insignificant for mG RSV, and marginally significant for wt RSV (Fig. 3B, group F versus group B, P = 0.013). These observations were confirmed in two additional experiments; specifically, the observed greater restriction of mG replication by the RSV-specific antiserum was ablated when the complement system was inactivated, and the resulting titers of the two viruses were equal. These data suggest that the complement system is required for the effective antibody-mediated restriction of RSV replication and also is involved in the sG-mediated evasion of this restriction.
Taken together, the data presented in this study resulted in three major conclusions. First, macrophages, but not neutrophils or the complement system, significantly contribute to the restriction of RSV replication in RSV-naïve animals. Second, pulmonary macrophages and the complement system, but not neutrophils, are required for the effective antibody-mediated restriction of RSV in vivo. Third, the sG-mediated evasion of this restriction (evident by the greater restriction of mG versus wt RSV) was no longer observed following depletion of the pulmonary macrophages or inactivation of the complement system, while depleting neutrophils had no effect. These data suggest an interplay between the innate and adaptive immune systems during RSV infections, which likely involves various mechanisms. For example, the involvement of macrophages and complement in antibody-mediated neutralization suggest roles for opsonization, ADCC, and complement-mediated cytotoxicity. The data also suggest that the presence of the unmodified G gene, producing both the membrane and the secreted forms of the G protein in live attenuated vaccines (19), may be beneficial from the standpoint of partially preventing restriction and overattenuation of the vaccine by maternally derived antibodies in infants.
ACKNOWLEDGMENTS
All experiments with mice were approved by and performed according to the guidelines of the National Institutes of Health.
The study was funded by the NIAID Intramural Research Program.
Footnotes
Published ahead of print 25 July 2012
REFERENCES
- 1. Bukreyev A, Belyakov IM, Berzofsky JA, Murphy BR, Collins PL. 2001. Granulocyte-macrophage colony-stimulating factor expressed by recombinant respiratory syncytial virus attenuates viral replication and increases the level of pulmonary antigen-presenting cells. J. Virol. 75:12128–12140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bukreyev A, et al. 2008. The secreted form of respiratory syncytial virus G glycoprotein helps the virus evade antibody-mediated restriction of replication by acting as an antigen decoy and through effects on Fc receptor-bearing leukocytes. J. Virol. 82:12191–12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Collins PL, Crowe JEJ. 2007. Respiratory syncytial virus and metapneumovirus, p 1601–1646 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5 ed, vol 2 Lippincott-Raven Publishers, Philadelphia, PA [Google Scholar]
- 4. Hall CB, Kopelman AE, Douglas RG, Jr, Geiman JM, Meagher MP. 1979. Neonatal respiratory syncytial virus infection. N. Engl. J. Med. 300:393–396 [DOI] [PubMed] [Google Scholar]
- 5. Hall CB, Walsh EE, Long CE, Schnabel KC. 1991. Immunity to and frequency of reinfection with respiratory syncytial virus. J. Infect. Dis. 163:693–698 [DOI] [PubMed] [Google Scholar]
- 6. Henderson FW, Collier AM, Clyde WA, Jr, Denny FW. 1979. Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N. Engl. J. Med. 300:530–534 [DOI] [PubMed] [Google Scholar]
- 7. Hendricks DA, Baradaran K, McIntosh K, Patterson JL. 1987. Appearance of a soluble form of the G protein of respiratory syncytial virus in fluids of infected cells. J. Gen. Virol. 68(Pt 6):1705–1714 [DOI] [PubMed] [Google Scholar]
- 8. Kumagai Y, et al. 2007. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 27:240–252 [DOI] [PubMed] [Google Scholar]
- 9. Lagasse E, Weissman IL. 1996. Flow cytometric identification of murine neutrophils and monocytes. J. Immunol. Methods 197:139–150 [DOI] [PubMed] [Google Scholar]
- 10. Nénan S, et al. 2008. Effects of depletion of neutrophils or macrophages on the inflammatory response induced by metalloelastase (MMP-12) in mice airways. Eur. J. Pharmacol. 579:374–381 [DOI] [PubMed] [Google Scholar]
- 11. Ochsenbein AF, et al. 1999. Control of early viral and bacterial distribution and disease by natural antibodies. Science 286:2156–2159 [DOI] [PubMed] [Google Scholar]
- 12. Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. 2008. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 4:e1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pribul PK, et al. 2008. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J. Virol. 82:4441–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shiratsuchi Y, et al. 2007. Infiltrating neutrophils induce allospecific CTL in response to immunization with apoptotic cells via MCP-1 production. J. Leukoc. Biol. 81:412–420 [DOI] [PubMed] [Google Scholar]
- 15. Spörri R, Joller N, Hilbi H, Oxenius A. 2008. A novel role for neutrophils as critical activators of NK cells. J. Immunol. 181:7121–7130 [DOI] [PubMed] [Google Scholar]
- 16. Teng MN, Whitehead SS, Collins PL. 2001. Contribution of the respiratory syncytial virus G glycoprotein and its secreted and membrane-bound forms to virus replication in vitro and in vivo. Virology 289:283–296 [DOI] [PubMed] [Google Scholar]
- 17. Van den Berg CW, Aerts PC, Van Dijk H. 1991. In vivo anti-complementary activities of the cobra venom factors from Naja naja and Naja haje. J. Immunol. Methods 136:287–294 [DOI] [PubMed] [Google Scholar]
- 18. van Rooijen N, Sanders A, van den Berg TK. 1996. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J. Immunol. Methods 193:93–99 [DOI] [PubMed] [Google Scholar]
- 19. Wright PF, et al. 2007. The absence of enhanced disease with wild type respiratory syncytial virus infection occurring after receipt of live, attenuated, respiratory syncytial virus vaccines. Vaccine 25:7372–7378 [DOI] [PMC free article] [PubMed] [Google Scholar]